一种核酸扩增方法及其应用
文献发布时间:2024-04-18 19:58:21
技术领域
本发明属于生物技术领域,具体涉及一种核酸扩增方法及其应用,所述方法是一种专门针对富含AT碱基的序列进行核酸扩增的方法。
背景技术
核酸扩增技术(nucleic acid amplification technology,NAAT)是一类分子生物学技术的总称,这类技术通过引物、DNA聚合酶和其它试剂在特定温度下进行反应,实现微量核酸的快速、特异性扩增,可广泛应用于疾病诊断、病原微生物检测、食品安全检测、动植物检疫以及涉及分子克隆的各项应用,如测序、基因克隆、基因操作、等位基因分析、突变检测等。其中聚合酶链式反应(polymerase chain reaction,PCR)是最早实现应用的核酸扩增技术,它模仿DNA在体内复制的过程,通过一对特异的寡核苷酸引物与待扩增DNA片段互补,再经过若干个循环的变性、退火、延伸过程,实现DNA片段的指数级扩增。PCR技术由Cetus公司的Mullis于1985年发明,随后Saiki等人将热稳定DNA聚合酶引入PCR反应体系,解决了早期PCR技术因热变性循环过程中DNA聚合酶钝化导致需要不断人工添加聚合酶的缺点,大大提高了核酸扩增效率并使该技术可以实现自动化,自此以后,以PCR为代表的核酸扩增技术得到广泛的推广和应用,也极大的推动了分子生物学的发展。
众所周知,现有核酸扩增技术通常会避开富含GC或AT的区域,其原因在于,这些区域易通过自身互补配对形成发夹环二级结构,从而阻碍引物与模板的结合;即使引物与模板能够勉强结合,也容易影响DNA聚合酶延模板链的延伸,从而导致DNA聚合酶沿模板扩增时“卡顿”并干扰DNA合成。因此,为了提高反应成功率,传统的核酸扩增技术对扩增区域的碱基组成具有选择偏好性,其引物设计往往集中在GC含量40%~60%的区域,以45%~55%为最适宜,不同扩增技术略有差别,如PCR的引物设计建议G+C含量为40%~60%(见分子克隆实验指南第四版,表7-1引物设计),LAMP引物设计建议G+C含量为40%~65%(见AGuide to LAMP primer designing(PrimerExplorer V3))。
然而,自然界中生物体的核酸碱基组成差别巨大,如疟原虫基因组AT碱基含量约为82%,周慧琦等研究了2670株细菌和古细菌,发现其基因组GC含量变化范围从14%到75%(周慧琦,2014)。从检测角度讲,现有核酸扩增技术的碱基组成选择偏好性忽略了大量潜在的靶序列,放大了目标对象被成功检出的难度;从基因操作角度讲,该选择偏好性导致相当多的目标序列难以被顺利扩增,从而难以开展进一步分子操作。如能针对传统核酸扩增方法所回避的GC碱基含量、AT碱基含量非均衡域设计适当的引物,避免形成二级结构,同时设置适宜的反应条件,顺利开展核酸扩增实验,将显著增加目标对象被检出的成功率,同时使得更多序列能够被扩增进而方便地实施下游分子操作。
针对GC碱基含量、AT碱基含量非均衡区域开展核酸扩增反应,其关键步骤是引物设计环节。鉴于以上分析,领域内公开发布的种属特异基因(或序列)通常来自于GC含量40%~60%甚至45%~55%的区域,因而无法应用于针对GC、AT碱基含量非均衡区域的引物设计。另外,此类序列中连续存在的GC-rich或AT-rich区,其碱基组成多样性相比其它区域偏弱,又为特异性引物的设计带来额外的困难。因此首先需要提出一套能够针对GC、AT碱基含量非均衡序列进行高通量、自动化、高效率的引物设计算法流程。
除了扩增区域选择和引物设计外,扩增区域和引物的Tm值计算也是扩增过程能否顺利完成的重要影响因素。扩增区域和引物的Tm值通常按照最近邻二态模型进行计算,但是不同研究者及引物生产公司采用的具体计算公式各有不同,如分子克隆实验指南第四版中建议的引物Tm计算公式为Tm=4×(G,C数)+2×(A,T数),TaKaRa公司建议的引物Tm计算公式为Tm=4×(G,C数)+2×(A,T数)+32-2×(总碱基数);生工生物的引物Tm计算公式为Tm(0.05MNa
综上所述,领域内亟需发展一种针对传统核酸扩增技术难以涉及的区域的核酸扩增技术,能够高通量、自动化地设计特异性引物,计算其Tm值,并设置适宜的变性温度、退火温度等反应条件,拓展核酸扩增技术的应用范围,提升扩增的特异性,满足核酸检测、分子遗传学研究等多方面的需求。
发明内容
本发明提出了一种创新的核酸扩增方法。该方法以富含AT的核酸序列为扩增目标,进行目标序列识别、引物设计、变性温度及退火温度计算、模板局部解链的核酸扩增等。首先,采用自动化的引物设计流程,充分利用公共数据资源中丰富的基因组序列信息,高通量地设计针对富含AT的目标序列的特异性引物对。其次,拟合了符合富含AT序列特点的Tm值计算公式,可根据理论扩增产物序列及引物的序列长度、碱基组成等因素计算变性温度和退火温度;考虑到富含AT的核酸序列变性温度显著低于GC含量40%~60%以及高GC的序列,因此将计算所得的理论扩增产物序列Tm值作为最低变性温度,在该温度下,能够在不添加任何化学变性剂的前提下,对双链DNA的富含AT区域进行局部解链,进而启动核酸扩增反应。
本发明一方面在引物设计环节引入公共数据资源中的海量基因组数据,进行序列特异性排查;另一方面,在变性温度设置环节有别于传统核酸扩增方法在93~95℃变性温度下打开所有双链结构,而是通过降低变性温度仅打开富含AT序列,从而在源头上极大地减少了非靶向区域发生非特异扩增的可能性。这两方面的设计策略显著降低了核酸扩增反应中常见的非特异性扩增的可能性,在此基础上形成了针对富含AT序列的核酸扩增方法。该方法的引物设计步骤采用C语言和Perl语言编程实现,具有高通量、自动化的特点,整个扩增方法特异性强、成功率高,有效地拓展了传统核酸扩增技术的应用范围,显著提升了反应性能。
本发明中,所述核酸扩增方法包括识别待扩增核酸序列中富含AT碱基的序列作为目标序列;针对目标序列自动化、高通量地设计特异性引物;设计特定的Tm值计算公式计算反应变性及退火温度,并据此设置核酸扩增反应条件;在模板局部解链的条件下进行核酸扩增反应。
具体包括如下步骤:
(1)筛选待扩增核酸序列中富含AT碱基的目标序列;
(2)针对所述目标序列自动化、高通量地设计兼具通用性和特异性的引物;
(3)通过特定公式计算反应变性温度及退火温度,并设置核酸扩增反应条件;即,基于GC的百分含量、引物序列长度、引物对的理论扩增产物序列长度计算反应变性温度及退火温度,并设置核酸扩增反应条件;
(4)在模板局部解链的条件下进行核酸扩增反应,得到扩增产物。
本发明步骤(1)中,对于待扩增核酸序列,从第一个碱基开始滑动宽度为1000bp的窗口,步长为5~100bp。针对每次窗口所在位置包含的序列计算AT碱基含量,保留序列AT碱基含量大于60%的区域作为目标序列。优选地,保留序列AT碱基含量在60-80%的区域作为目标序列。
本发明步骤(2)中,所述引物的设计方法,包括:(2.1)针对一条目标序列进行单条引物设计,得到候选引物;(2.2)对所述候选引物进行理化性质判定,筛选符合条件的单条引物;(2.3)将步骤(2.2)筛选得到的单条引物组合成引物对;(2.4)对所述引物对进行通用性、特异性的判定;(2.5)输出符合条件的引物对,得到所述特异性引物。
本发明中,可以采用任何能实现高通量的引物设计的编程语言来实现,如可操作性强、速度较快的C、Perl等编程语言实现。
步骤(2.1)中,针对目标序列设计候选引物,候选引物需满足以下条件:a)引物序列长度在20bp~36bp之间;b)AT碱基含量在55%~80%,c)连续GC数≤5;同时记录所述候选引物匹配到目标序列上的位置信息、正负链信息。
其中,优选地,所述AT碱基含量为60%~75%。
其中,所述“连续GC数”是指引物序列中连续碱基G或者连续碱基C的数量,如引物序列AAGGGGGTTCCAGGCATTA中连续GC数为5/2/2。
步骤(2.2)中,对满足上述(2.1)条件的单条引物进行理化性质判定,包含但不限于3’端稳定性、5’端稳定性和/或二级结构稳定性等物理化学性质,保留符合要求的单条引物。
其中,所述“符合要求的单条引物”是指具备3’端稳定性、5’端稳定性和二级结构稳定性的单条引物。
步骤(2.3)中,对上述步骤(2.2)筛选得到的单条引物,根据其匹配到目标序列上的位置信息、正负链信息,将单条引物组合成引物对;引物对的理论扩增产物序列的长度应在200bp~600bp之间。根据公式0.466×(GC的百分含量)×100+66.04-(450/引物序列长度)计算单条引物的Tm值;以引物对Tm差值≤3℃,以及引物之间不能发生相互作用为条件,对引物对进行筛选,得到候选引物对。其中,GC的百分含量是指引物中的碱基G和C的个数占引物总碱基个数的百分比;引物序列长度是指引物的碱基个数。
步骤(2.4)中,对步骤(3)得到的候选引物对中的每一条引物做通用性、特异性判定。其中通用性判定指检查引物对是否严格匹配到所有的目标序列(如某待测物种的多个品系,例如SARS-CoV-2的多个病毒株基因组);
特异性判定指检查单条引物是否不能与所有目标序列之外的非目标序列发生特异性匹配,所述非目标序列指除待扩增核酸序列之外的其它核酸序列,所述特异性匹配指不大于2个错配的匹配。
通过通用性和特异性判定的候选引物可进入下一步骤。
步骤(2.5)中,输出符合条件的引物对,即得到所述核酸扩增反应的引物对。
本发明所述步骤(3)中,所述反应变性温度的计算公式为0.357×(GC的百分含量)×100+70.582-(990/引物对的理论扩增产物序列长度),记为Tma;其中,GC的百分含量是指引物对的理论扩增产物序列中的碱基G和C的个数占引物对的理论扩增产物序列总碱基个数的百分比。
本发明所述步骤(3)中,所述反应退火温度为上述步骤(2.3)中两条引物的Tm值的平均值,记为Tmb。即,根据公式0.466×(GC的百分含量)×100+66.04-(450/引物序列长度)计算单条引物的Tm值,将所述Tm值取平均值即为所述反应退火温度,记为Tmb;其中,GC的百分含量是指引物中的碱基G和C的个数占引物总碱基个数的百分比。
本发明所述步骤(3)中,所述核酸扩增反应条件包括:在变性温度下反应5秒,在退火温度下反应5秒,在延伸温度下反应20秒,以上过程重复30-40次。其中变性温度根据需要可在Tma±5℃之间调整;退火温度根据需要可在Tmb±2℃之间调整;延伸温度为72℃。
本发明还提供了所述的方法在用于富含AT区的目标核酸序列进行核酸扩增中的应用。
本发明还提供了如上所述设计方法获得的引物对。
本发明中,所述引物对为:
引物对A:
SVCV-F:5’-ATAATTTCAGACGACATTGAATATCTC-3’(SEQ ID NO:1)
SVCV-R:5’-TCAACAGAGTCAATTTGTGC-3’(SEQ ID NO:2);和/或,
引物对B:
Cro-F:5’-CGCCATAACTGCATAATCAT-3’(SEQ ID NO:3)
Cro-R:5’-ATAACGAGTTACCGTGCAGA-3’(SEQ ID NO:4)。
本发明还提供了如上所述的方法或如上所述的引物对分别在鲤春病毒血症病毒(SVCV)和/或阪崎克罗诺杆菌(Cronobactersakazakii)相关基因或区域的扩增和/或检测中的应用。
本发明还提供了如上所述的引物对在制备扩增和/或检测鲤春病毒血症病毒(SVCV)和/或阪崎克罗诺杆菌(Cronobactersakazakii)相关基因或区域的产品中的应用。
本发明还提供了一种诊断试剂,所述诊断试剂包括如上所述的引物对。
本发明还提供了一种核酸扩增设备,包括:存储器、处理器和扩增模块;所述存储器上存储有计算机程序,当所述计算机程序被所述处理器执行时,实现如上所述方法的步骤1-3;所述扩增模块用于在模板局部解链的条件下进行核酸扩增反应,得到扩增产物。
本发明的有益效果在于,本发明靶向传统核酸扩增方法通常需要规避的富含AT碱基的序列,极大地拓展了候选靶标区域;通过基于海量基因组数据的特异性排查,以及针对富含AT区域的Tm值的精确计算,从而对双链DNA进行局部解链,大大降低了核酸扩增技术中普遍存在的非特异性扩增的可能性。该方法有效地拓展了传统核酸扩增技术的应用范围,同时在引物设计通量、反应特异性等方面显著提升了反应性能,可满足核酸检测、分子遗传学研究等多方面的需求。
附图说明
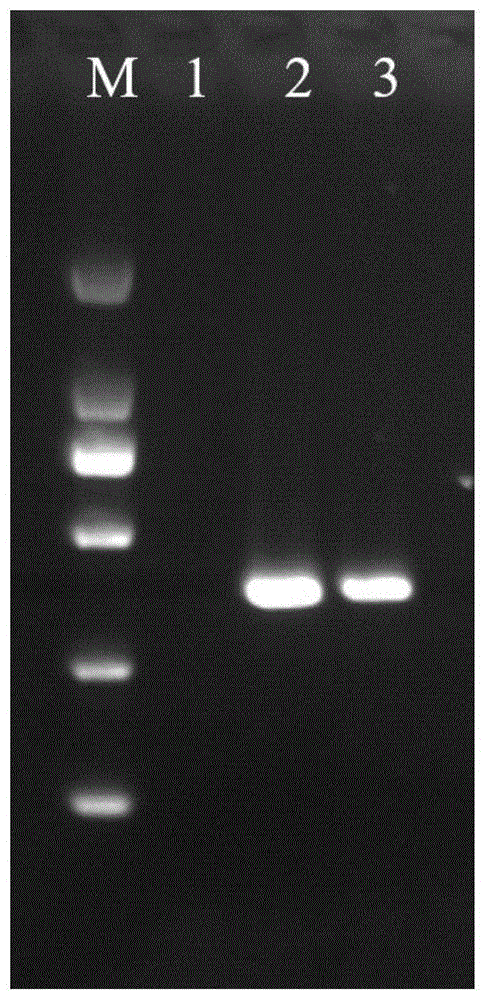
图1为基于本发明的鲤春病毒血症病毒(SVCV)核酸扩增产物的电泳结果。
图2为基于国标《GB/T 15805.5-2018鱼类检疫方法第5部分:鲤春病毒血症病毒(SVCV)》中传统PCR方法的鲤春病毒血症病毒(SVCV)核酸第一轮扩增产物的电泳结果。
图3为基于国标《GB/T 15805.5-2018鱼类检疫方法第5部分:鲤春病毒血症病毒(SVCV)》中巢式PCR方法的鲤春病毒血症病毒(SVCV)核酸第二轮扩增产物的电泳结果。
图4为基于本发明的阪崎克罗诺杆菌(Cronobactersakazakii)不同变性温度下核酸扩增特异性检测的染料显色结果。
图5为基于本发明的阪崎克罗诺杆菌(Cronobactersakazakii)94℃变性温度下核酸扩增特异性检测的扩增产物电泳结果。
图6为基于本发明的阪崎克罗诺杆菌(Cronobactersakazakii)82℃变性温度下核酸扩增特异性检测的扩增产物电泳结果。
具体实施方式
结合以下具体实施例和附图,对本发明作进一步的详细说明。实施本发明的过程、条件、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。
实施例1鲤春病毒血症病毒(SVCV)特异性序列的扩增用于SVCV的特异性检测
本发明针对鲤春病毒血症病毒(SVCV)基因组筛选富含AT的序列并设计特异性引物,根据计算的引物Tm和目标序列Tm设置反应条件进行核酸扩增,通过判断反应结果是否为阳性,确定待测样品中是否存在目标序列,进而确定待测样品中是否存在鲤春病毒血症病毒。具体步骤如下:
(1)富含AT碱基的目标序列的筛选:
使用2019年8月5号从NCBI的FTP上下载的有完整基因组序列的细菌、古菌和病毒的数据,共2896个全基因组序列。设定集合A,其包含所有鲤春病毒血症病毒基因组序列;设定集合B,其包所有含非鲤春病毒血症病毒基因组序列。以GI号为14329182的鲤春病毒血症病毒基因组序列作为参考基因组,从基因组第一个碱基开始滑动宽度为1000bp的窗口,步长为50bp;每次滑动前计算一次序列AT碱基含量,保留序列AT碱基含量大于60%的区域作为候选目标序列。以上过程采用perl脚本实现。
(2)特异性引物的设计:
根据PCR引物的特性及本发明的需求,设定引物的AT碱基含量为60%~75%、3’端稳定性ΔG<4、5’端稳定性ΔG<3及引物序列长度(20~36bp)等特性参数,同时设定单条引物不能产生发卡结构、自身不能发生相互作用等条件,以步骤(1)中的候选目标序列作为单条引物设计的候选序列,计算得出符合上述设定条件的单条引物。通过公式0.466×(GC的百分含量)×100+66.04-(450/引物序列长度)计算单条引物的Tm值,并记录每条引物在目标序列上的位置、正负链信息(即来源于正链还是负链)以及引物的长度等信息。
根据单条引物的位置信息进行引物配对,保留同时满足引物对Tm差值<3℃以及引物对扩增区域在200~600bp两个条件的引物对作为候选引物对。
使用比对软件Bowtie,将上一步设计出的候选引物对中的每一条引物分别与集合A中的目标基因组序列和集合B中的非目标基因组进行序列比对。为了保证引物的通用性,当该单条引物与集合A中的目标序列进行比对时,使用参数设置“-a-n 0”,即要求该单条引物与目标序列完全匹配;为了保证引物的特异性,当该单条引物与集合B中的非目标序列进行比对时,使用参数设置“-a-n 3”,即允许该单条引物与非目标序列有不超过3个错配。系统输出满足条件的引物对,其中引物对的数量可预先设置,在本实施例中引物对的数量预先设置为10。以上高通量、自动化引物设计流程采用C、Perl脚本实现。
程序运行后在AT富集区设计出PCR反应引物对10个,随机选择一个引物组进行有效性验证。所述引物对的序列为:
SVCV-F:5’-TAATTTCAGACGACATTGAATATCTCA-3’(SEQ ID NO:1)
SVCV-R:5’-AACAGAGTCAATTTGTGCAG-3’(SEQ ID NO:2)
所述引物的AT碱基含量分别为70%和60%,所述引物对的理论扩增产物序列的AT百分含量为60%。
(3)PCR反应变性温度和退火温度的计算:
使用公式0.466×(GC的百分含量)×100+66.04-(450/引物序列长度)计算可知,引物SVCV-F的Tm=63.4℃,SVCV-R的Tm=62.2℃,该引物对平均Tm值为62.8℃;使用公式0.357×(GC的百分含量)×100+70.582-(990/扩增产物序列长度)计算扩增区域变性温度为82.1℃。
(4)核酸扩增反应及结果检测:
核酸扩增反应体系配置如下表1所示,根据计算结果,设置核酸扩增反应条件为:42℃反应10分钟,然后以86℃反应5秒,61℃反应5秒,72℃反应20秒为一个反应循环,共进行30次循环反应。反应完成后对扩增产物进行琼脂糖凝胶电泳,根据电泳条带判断扩增结果是否为阳性,即待测样品中是否存在目标序列。
表1.鲤春病毒血症病毒(SVCV)核酸扩增反应体系
如图1所示鲤春病毒血症病毒(SVCV)核酸扩增产物的电泳图,本发明所设计的核酸扩增方法对SVCV核酸目标序列成功地进行了扩增。图1中,泳道M为DNA分子量标准DL2000;泳道1为灭菌水作阴性对照模板的扩增产物,无核酸扩增产物条带,与预期相符;泳道2~3分别为含检测目标序列的质粒DNA模板以及SVCV RNA模板的扩增产物,均呈现出核酸扩增产物条带,条带位置介于分子量标准250bp与500bp之间,符合片段理论大小358bp。
与此同时,本发明根据国标《GB/T 15805.5-2018鱼类检疫方法第5部分:鲤春病毒血症病毒(SVCV)》第6条,采用传统PCR方法对以上SVCV模板进行了两轮核酸扩增,两轮扩增产物的电泳图分别如图2、3所示。其中,泳道M为DNA分子量标准DL2000(图2、3)。泳道SV-1~SV-4为以SVCVRNA为模板的扩增产物,第一轮PCR扩增后在714bp处有微弱条带,同时存在大量非特异扩增导致的弥散条带(图2);经第二轮巢式PCR扩增后在714bp处有清晰条带,同时仍存在大量非特异扩增导致的弥散条带(图3)。泳道cell-1~cell-2为以鱼类细胞RNA为阴性对照模板的扩增产物,两轮PCR均存在大量非特异扩增导致的弥散条带(图2、3)。泳道H
与国标方法相比,本发明方法能显著抑制非特异性扩增产生,同时反应产物浓度更高,无需使用巢氏扩增策略,一步即可得到明确的扩增结果,显著加快了检测速度、提升了检测效率。
本实施例清晰地展示了选择优化适当的区域进行核酸扩增,能够大大提高反应效率、降低非特异性扩增现象。
实施例2阪崎克罗诺杆菌(Cronobactersakazakii)的检测
本发明针对阪崎克罗诺杆菌(Cronobactersakazakii)基因组筛选富含AT的序列并设计特异性引物,根据计算的引物Tm和目标序列Tm设置反应条件进行核酸扩增,通过判断反应结果是否为阳性,确定待测样品中是否存在目标序列,进而确定待测样品中是否存在阪崎克罗诺杆菌。具体步骤如下:
(1)富含AT碱基的目标序列的筛选:
使用2019年8月5号从NCBI的FTP上下载的有完整基因组序列的细菌、古菌和病毒的数据,共2896个全基因组序列。设定集合A,其包含所有阪崎克罗诺杆菌基因组序列;设定集合B,其包含所有非阪崎克罗诺杆菌基因组序列。以GI号为156932229的阪崎克罗诺杆菌基因组序列作为参考基因组,从基因组第一个碱基开始滑动宽度为1000bp的窗口,步长为50bp;每次滑动前计算一次序列AT碱基含量,保留序列AT碱基含量大于60%的区域作为候选目标序列。以上过程采用perl脚本实现。
(2)特异性引物的设计:
根据PCR引物的特性及本发明的需求,设定引物的AT碱基含量为55%~75%、3’端稳定性ΔG<4、5’端稳定性ΔG<3及引物序列长度(20~36bp)等特性参数,同时设定单条引物不能产生发卡结构、自身不能发生相互作用等条件,以步骤(1)中的候选目标序列作为单条引物设计的候选序列,计算得出符合上述设定条件的单条引物。通过公式0.466×(GC的百分含量)×100+66.04-(450/引物序列长度)计算单条引物的Tm值,并记录每条引物在目标序列上的位置、正负链信息(即来源于正链还是负链)以及引物的长度等信息。
根据单条引物的位置信息进行引物配对,保留同时满足引物对Tm差值<3℃以及引物对扩增区域在200~600bp两个条件的引物对作为候选引物对。
使用比对软件Bowtie,将上一步设计出的候选引物对中的每一条引物分别与集合A中的目标基因组序列和集合B中的非目标基因组进行序列比对。为了保证引物的通用性,当该单条引物与集合A中的目标序列进行比对时,使用参数设置“-a -n 0”,即要求该单条引物与目标序列完全匹配;为了保证引物的特异性,当该单条引物与集合B中的非目标序列进行比对时,使用参数设置“-a -n 3”,即允许该单条引物与非目标序列有不超过3个错配。系统输出满足条件的引物对,其中引物对的数量可预先设置,在本实施例中引物对的数量预先设置为20。以上高通量、自动化引物设计流程采用C、Perl脚本实现。
程序运行后在AT富集区设计出PCR反应引物对20个,随机选择一个引物组进行有效性验证。所述引物对的序列为:
Cro-F:5’-CGCCATAACTGCATAATCAT-3’(SEQ ID NO:3)
Cro-R:5’-ATAACGAGTTACCGTGCAGA-3’(SEQ ID NO:4)
所述引物的AT碱基含量分别为60%和55%,所述引物对的理论扩增产物序列AT的百分含量为69%。
(3)PCR反应变性温度和退火温度的计算:
使用公式0.466×(GC的百分含量)×100+66.04-(450/引物序列长度)计算可知引物Cro-F的Tm=62.2℃,Cro-R的Tm=64.5℃,该引物对平均Tm值为63.4℃;使用公式0.357×(GC的百分含量)×100+70.582-(990/扩增产物序列长度)计算扩增区域变性温度为78.3℃。
(4)核酸扩增反应及结果检测:
核酸扩增反应体系配置如下表2所示。根据计算结果,实验共设3组,分别测试在94℃/90℃/82℃变性温度下引物针对不同检测对象的特异性,核酸扩增反应条件为94℃/90℃/82℃变性5秒,63℃退火5秒,72℃延伸20秒,以上过程重复35次。特异性检测对象清单见表3。
反应完成后通过两种方式判定扩增结果,一是加入终浓度25x的SYBRGreen I染料,通过颜色判定扩增结果是否为阳性,即待测样品中是否存在目标序列;二是对扩增产物进行琼脂糖凝胶电泳,根据电泳条带判断扩增结果是否为阳性,即待测样品中是否存在目标序列。
表2.阪崎克罗诺杆菌(Cronobactersakazakii)核酸扩增反应体系
表3.阪崎克罗诺杆菌(Cronobactersakazakii)核酸扩增反应特异性检测对象清单
实验结果见图4、5、6,其中1-22分别为金黄色葡萄球菌、金黄色葡萄球菌金黄亚种、表皮葡萄球菌、马红球菌、蜡样芽孢杆菌、蕈样芽孢杆菌、单核增生李斯特氏菌、英诺克李斯特氏菌、伊氏李斯特氏菌、肠沙门氏菌肠亚种、肠炎沙门氏菌、鼠伤寒沙门氏菌、乙型副伤寒沙门氏菌、痢疾志贺氏菌、鲍氏志贺氏菌、福氏志贺氏菌、大肠埃希氏菌(含肉毒梭菌A型基因)、致病性大肠埃希氏菌、致泻大肠埃希氏菌、产肠毒素大肠埃希氏菌、肠产毒性大肠埃希氏菌、出血性大肠埃希氏菌,24-30分别为小肠结肠炎耶尔森氏菌、假结核耶尔森氏菌、创伤弧菌、副溶血弧菌、弗氏弧菌、霍乱弧菌和福式志贺氏菌,N:阴性对照,P:阳性对照(含目的序列的质粒);23为阪崎克罗诺杆菌。
图4展示了本发明针对阪崎克罗诺杆菌(Cronobactersakazakii)的核酸扩增反应在不同变性温度下的特异性检测的SYBRGreen I染料显色结果。若扩增产物呈亮绿色,则为阳性;若扩增产物呈橙色,则为阴性。图4中,当变性温度为94℃/90℃,阴性对照(N)呈现橙色,为阴性结果,符合预期;阳性对照(P)和23号阪崎克罗诺杆菌呈现亮绿色,为阳性结果,符合预期。说明整个反应体系能正常工作。然而,其它细菌基因组DNA模板扩增产物出现大量阳性结果,如变性温度94℃条件下的3、4、10~18、20~22、24、26、27、29、30号,变性温度90℃条件下的3、7、10~18、20~22、24、26、27、29、30号管所示,与预期不符,提示在94℃/90℃变性温度下易产生非特异性扩增,导致结果呈假阳性。当变性温度为82℃,阴性对照(N)呈现橙色,为阴性结果,符合预期;阳性对照(P)和23号阪崎克罗诺杆菌呈现亮绿色,为阳性结果,符合预期。说明整个反应体系能正常工作。同时,其它细菌基因组DNA模板检测结果均呈阴性,如第1~22、24~30号管所示。所有结果符合预期,提示在82℃变性温度下未产生非特异性扩增或即使产生微量非特异扩增但不足以影响染料法结果判定。
图5展示了本发明针对阪崎克罗诺杆菌(Cronobactersakazakii)的核酸扩增反应在94℃变性温度下的特异性检测的扩增产物电泳结果。若扩增产物电泳后在291bp处有一条单一的条带,则为阳性;若扩增产物电泳后无条带,则为阴性;若扩增反应产物电泳后在291bp之外出现一条或多条条带,则为非特异扩增所导致的假阳性。图5中,阴性对照(N)无条带,符合预期;阳性对照(P)和23号阪崎克罗诺杆菌在291bp处均有一条清晰条带,符合预期。说明整个反应体系能正常工作。然而,其它细菌基因组DNA模板扩增产物在500bp~2000bp范围内出现多条条带,与预期不符,提示在94℃变性温度下反应体系产生大量非特异性扩增,该结果与图4中染料显色结果一致。
图6展示了本发明针对阪崎克罗诺杆菌(Cronobactersakazakii)的核酸扩增反应在82℃变性温度下的特异性检测的扩增产物电泳结果。若扩增产物电泳后在291bp处有一条单一的条带,则为阳性;若扩增反应产物电泳后无条带,则为阴性;若扩增反应产物电泳后在291bp之外出现一条或多条条带,则为非特异扩增所导致的假阳性。图6中,阴性对照(N)无条带,符合预期;阳性对照(P)和23号阪崎克罗诺杆菌在291bp处均有一条清晰条带,符合预期。说明整个反应体系能正常工作。同时,其它细菌基因组DNA模板扩增反应产物大部分无条带,呈阴性结果,如2~6、10、12、13、15~22、26~30号泳道所示;少数模板扩增反应产物电泳后出现极微弱条带,易于与阳性结果区分,如1、7、8、9、11、14、24、25管所示,提示在82℃变性温度下不产生或产生在电泳结果中易于区分的微量非特异性扩增。
综合图4~6及表3可看出,本发明核酸扩增方法针对阪崎克罗诺杆菌的应用具有良好的菌株特异性,即,在本发明提出的反应体系和反应条件下,仅阪崎克罗诺杆菌呈现阳性结果,而非阪崎克罗诺杆菌为阴性结果。值得注意的是,当使用传统PCR方法的变性温度(90℃或者94℃)进行反应时,尽管电泳结果(图5)显示目标检测对象(阪崎克罗诺杆菌,第23泳道)有单一清晰的条带,然而非目标检测对象普遍出现大量非特异性扩增,如第3泳道的表皮葡萄球菌;若使用染料显色法判定检测结果,如图4所示,在90℃和94℃变性条件下,呈现大量假阳性结果,从而无法区分检测对象和非检测对象。在同样的反应体系下,当使用本发明计算生成的变性温度82℃进行反应时,电泳结果(图6)显示目标检测对象(阪崎克罗诺杆菌,第23泳道)有单一清晰的条带,而非目标检测对象少有非特异性扩增发生,即使有,非特异性扩增条带也很微弱,极易与阳性结果区分;若使用染料显色法判定检测结果,如图4所示,在82℃变性条件下,仅目标检测对象(阪崎克罗诺杆菌,23号管)呈阳性结果,非目标检测对象均呈阴性结果,表现出良好的区分性。
本实施例完美地验证了本方法所提出的,通过降低变性温度对模板序列进行局部解链,能够显著降低非特异性扩增。
本发明还提供了一种核酸扩增设备,包括:存储器、处理器和扩增模块;所述存储器上存储有计算机程序,当所述计算机程序被所述处理器执行时,实现如上实施例所述方法的步骤;所述扩增模块用于在模板局部解链的条件下进行核酸扩增反应,得到扩增产物。
参考文献
周慧琦.(2014).基因组GC含量与碱基、密码子和氨基酸使用偏好的关系.(mastermaster),电子科技大学,(67)
本发明的保护内容不局限于以上实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。
SEQUENCE LISTING
<110> 上海生物信息技术研究中心,上海旺旺食品集团有限公司
<120> 一种核酸扩增方法及其应用
<160> 5
<170> PatentIn version 3.3
<210> 1
<211> 27
<212> DNA
<213> 人工序列
<400> 1
ataatttcag acgacattga atatctc 27
<210> 2
<211> 20
<212> DNA
<213> 人工序列
<400> 2
tcaacagagt caatttgtgc 20
<210> 3
<211> 20
<212> DNA
<213> 人工序列
<400> 3
cgccataact gcataatcat 20
<210> 4
<211> 20
<212> DNA
<213> 人工序列
<400> 4
ataacgagtt accgtgcaga 20
<210> 5
<211> 19
<212> DNA
<213> 人工序列
<400> 5
aagggggttc caggcatta 19
- 一种变性泡介导的靶标核酸扩增方法及其专用试剂盒与应用
- 一种核酸扩增试剂所需的冻干保护体系及其制备方法
- 一种核酸提取扩增检测试剂盒及其检测方法
- 一种病原体核酸捕获扩增检测方法
- 核酸扩增方法、核酸提取用设备、核酸扩增反应用筒以及核酸扩增反应用试剂盒
- 核酸扩增方法、核酸提取用设备、核酸扩增反应用盒、以及核酸扩增反应用套件