一种吡啶类衍生物、其制备方法及用途
文献发布时间:2024-04-18 19:58:26
本申请涉及药物化学领域,具体涉及一种作为赖氨酸特异性去甲基酶1(LSD1)抑制剂的吡啶类衍生物或其药学上可接受的盐、其制备方法及用途。
组蛋白赖氨酸特异性去甲基化酶1(LSD1)是首个被报道的组蛋白去甲基化酶,在黄素腺嘌呤二核苷酸(FAD)的辅助下,可以特异性地识别H3K4和H3K9底物,并去除其单甲基或双甲基修饰。同时,LSD1可以与多种因子共同形成转录调控复合物,在转录因子的招募下到达指定基因的启动子区域,进而调控基因表达。
研究表明,LSD1与多种疾病相关,在多种肿瘤细胞中高表达,并与肿瘤不良预后密切相关,下调LSD1表达或抑制其活性可明显抑制肿瘤细胞生长。因此,开发一种高效、特异性的LSD1抑制剂,对于肿瘤等疾病的治疗具有非常重要的意义。
发明内容
本申请涉及一种吡啶类衍生物作为LSD1抑制剂,特别是涉及一种吡啶类衍生物及其制备方法和其在医药上的应用,特别是如下式I所示的吡啶类衍生物及其在制备治疗LSD1介导的疾病的药物中的用途,更具体而言,在制备适用于肿瘤药物中的用途。
本申请的一个方面提供了如下式I所示结构的吡啶类衍生物、其立体异构体或其药学上可接受的盐:
其中,
环A表示饱和环烷基、饱和杂环烷基、不饱和环烃基或不饱和杂环烃基;
环B表示单环或双环不饱和烃基、或单环或双环不饱和杂环烃基;
R
R
R
R
R
m选自0、1、2、3、4或5;
n选自0、1、2、3、4或5;
q选自0、1、2、3或4。
在一些实施方式中,环A选自C
优选地,环A选自C
更优选地,环A选自C
或者优选地,环A选自哌啶基、四氢吡啶基、哌嗪基、氮杂环丁基、氮杂环戊基、氮杂环己基、氮杂环庚基、高哌嗪基、环己烷基、环丁烷基、环戊烷基、环庚烷基、二氮杂螺壬烷基、二氮杂螺癸烷基、一氧杂一氮杂螺壬烷基、一氧杂一氮杂螺癸烷基。
在一些实施方式中,环B选自C
优选地,环B选自C
或者优选地,环B选自苯基、吡啶基、二氢吡啶基、吡嗪基、哒嗪基、嘧啶基、含有1-3个选自氮原子或氧原子的杂原子的苯并5元至6元杂环烯基(例如吲哚基、异吲哚基、异吲哚啉基、二氢吲哚基、吲唑基、二氢吲唑基、苯并噁唑基、苯并异噁唑基、喹啉基、异喹啉基、喹唑啉基、喹喔啉基、苯并二噁茂基、苯并呋喃基、苯并咪唑基、苯并二氢噁嗪基)。
在一些实施方式中,R
优选地,R
在一些实施方式中,m为0、1、2、3、4或5,R
或者在一些实施方式中,m为0、1、2或3(优选0、1或2),R
在一些实施方式中,n选自0、1、2、3、4或5,R
或者在一些实施方式中,n选自0、1、2、3或4,R
在一些实施方式中,q选自0、1或2,R
或者在一些实施方式中,q为1或2,R
优选地,在式I化合物、其立体异构体或其药学上可接受的盐中,
环A选自5元至8元饱和单环烷基、4元至10元饱和单环或螺环杂环烷基,其中所述杂环烷基含有1-3个氮原子作为杂原子;
环B选自5元至8元不饱和单环烃基、5元至14元不饱和单环或双环杂环烃基,其中所述杂环烃基含有0-4个氮原子、0-2个氧原子作为杂原子且具有氮、氧中的至少一者;
R
R
R
R
R
m选自0、1、2、3、4或5;
n选自0、1、2、3、4或5;
q选自0、1、2、3或4。
优选地,在式I化合物、其立体异构体或其药学上可接受的盐中,
环A选自4元至10元饱和单环或螺环杂环烷基,其中所述杂环烷基含有1-3个氮原子作为杂原子;优选含有1-2个氮原子作为杂原子;或者,环A选自哌啶基、哌嗪基、氮杂环丁基、氮杂环庚基、高哌嗪基、二氮杂螺壬烷基、二氮杂螺癸烷基、或一氧杂一氮杂螺癸烷基;
环B选自6元不饱和单环烃基或6元至10元不饱和双环杂环烃基,其中所述杂环烃基含有0-4个氮原子、0-2个氧原子作为杂原子且具有氮、氧中的至少一者;优选地,B环选择6元至10元的芳基或杂环芳基,其中所述杂环芳基含有1-2氮原子、0-2个氧原子作为杂原子且具有氮、氧中的至少一者;更优选地,环B选自苯基、吡啶基、吲哚基、二氢吲哚基、吲唑基、苯并异噁唑基、苯并噁唑基、苯并二噁茂基或苯并二氢噁嗪基;
R
R
R
R
R
m选自0、1、2、3、4或5;优选地,m选自0、1、2或3;
n选自0、1、2、3、4或5;优选地,n选自0、1、2、3或4;
q选自0、1、2、3或4;优选地,q选自1或2。
在一些实施方式中,在式I化合物、其立体异构体或其药学上可接受的盐中,
环A选自哌啶基、哌嗪基、氮杂环丁基、氮杂环庚基、高哌嗪基、二氮杂螺壬烷基、二氮杂螺癸烷基、或一氧杂一氮杂螺癸烷基;
环B选自苯基、吡啶基、吲哚基、二氢吲哚基、吲唑基、苯并异噁唑基、苯并噁唑基、苯并二噁茂基或苯并二氢噁嗪基;
R
R
R
R
m选自0、1、2或3;
n选自0、1、2、3或4;
q选自1或2。
优选地,式I化合物、其立体异构体或其药学上可接受的盐进一步具有式II所示的结构,
其中,
环A选自环己烷基、哌啶基、哌嗪基、氮杂环庚基或高哌嗪基;优选地,所述哌啶基、哌嗪基、氮杂环庚基或高哌嗪基通过N原子连接到吡啶环上;
环B选自苯基、吡啶基、吲哚基、二氢吲哚基或吲唑基;
R
R
R
m选自0、1、2或3;
n选自0、1、2、3或4。
或者,在一些实施方式中,在式II的化合物、其立体异构体或其药学上可接受的盐中,
环A选自环己烷基、哌啶基、哌嗪基、氮杂环丁基、氮杂环庚基、高哌嗪基、二氮杂螺壬烷基或二氮杂螺癸烷基;优选地,所述哌啶基、哌嗪基、氮杂环丁基、氮杂环庚基、高哌嗪基、二氮杂螺壬烷基或二氮杂螺癸烷基通过N原子连接到吡啶环上;
环B选自苯基、吡啶基、吲哚基、二氢吲哚基、吲唑基、苯并异噁唑基、苯并噁唑基或苯并二噁茂基;
R
R
R
m选自0、1、2或3;
n选自0、1、2、3或4。
更优选地,式I所示结构的杂环类衍生物、其立体异构体或其药学上可接受的盐选自如下的化合物、 其立体异构体或其药学上可接受的盐:
本申请还涵盖通过对上述各实施方式和优选方案的任意组合、删减或调换而得到的方案。
本申请的另一方面提供了制备上述式I所示结构的吡啶类衍生物的制备方法,所述方法包括以下步骤:
方法一:
其中,LG表示离去基团,所述的离去基团包括但不限于卤素原子、甲磺酰基氧基、对甲苯磺酰基氧基等。R
(1)使化合物I-1与化合物I-2反应得到化合物I-3
所述反应优选在合适的有机溶剂中进行。所述有机溶剂可选自四氢呋喃、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、1,4-二氧六环及其任意组合,优选N,N-二甲基甲酰胺。所述反应优选在适合的碱存在下进行;和/或在过渡金属催化剂及配体存在下进行。所述碱可选自三乙胺、吡啶、4-二甲胺基吡啶、二异丙基乙胺、碳酸钾、碳酸铯、碳酸钠或叔丁醇钾,优选碳酸钾。所述过渡金属催化剂可选自Pd(dppf)Cl
(2)使化合物I-3经取代反应得到化合物I-4
所述反应优选在适合的有机溶剂中进行。所述有机溶剂可选自乙腈、甲苯、氯仿、四氢呋喃及其任意组合,优选乙腈。所述反应优选在适合的溴代试剂存在下进行。所述溴代试剂可选自N-溴代丁二酰亚胺、液溴、吡啶溴鎓盐,优选N-溴代丁二酰亚胺。所述反应优选在适合的温度下进行,所述温度优选为20-50℃。所述反应优选进行合适的时间,例如2-6小时。
(3)使化合物I-4与化合物I-5的硼酸或硼酸酯经偶联反应得到化合物I-6
所述反应优选在适合的有机溶剂中进行。所述有机溶剂可选自四氢呋喃、1,4-二氧六环、甲苯、乙腈、乙醇、水及其任意组合,优选1,4-二氧六环与水组合。所述反应优选在适合的催化剂存在下进行。所述催 化剂可选自Pd(dppf)Cl
(4)使化合物I-6与化合物I-7经偶联反应得到式I所示结构的化合物
所述反应优选在适合的有机溶剂中进行。所述有机溶剂可选自四氢呋喃、1,4-二氧六环、甲苯、乙腈、乙醇、水及其任意组合,优选1,4-二氧六环与水组合。所述反应优选在适合的催化剂存在下进行。所述催化剂可选自Pd(dppf)Cl
方法二:
其中,LG表示离去基团,所述的离去基团包括但不限于卤素原子、甲磺酰基氧基、对甲苯磺酰基氧基等。R
(1)使化合物I-1经取代反应得到化合物I-8
所述反应优选在适合的有机溶剂中进行。所述有机溶剂可选自乙腈、甲苯、氯仿、四氢呋喃及其任意组合,优选乙腈。所述反应优选在适合的溴代试剂存在下进行。所述溴代试剂可选自N-溴代丁二酰亚胺、液溴、吡啶溴鎓盐,优选N-溴代丁二酰亚胺。所述反应优选在适合的温度下进行,所述温度优选为20-50℃。所述反应优选进行合适的时间,例如2-6小时。
(2)使化合物I-8与化合物I-5的硼酸或硼酸酯经偶联反应得到化合物I-9
所述反应优选在适合的有机溶剂中进行。所述有机溶剂可选自四氢呋喃、1,4-二氧六环、甲苯、乙腈、乙醇、水及其任意组合,优选1,4-二氧六环与水组合。所述反应优选在适合的催化剂存在下进行。所述催化剂可选自Pd(dppf)Cl
(3)使化合物I-9与化合物I-7经偶联反应得到化合物I-10
所述反应优选在适合的有机溶剂中进行。所述有机溶剂可选自四氢呋喃、1,4-二氧六环、甲苯、乙腈、乙醇、水及其任意组合,优选1,4-二氧六环与水组合。所述反应优选在适合的催化剂存在下进行。所述催化剂可选自Pd(dppf)Cl
(4)使化合物I-10与化合物I-2反应得到式I所示结构的化合物
所述反应优选在合适的有机溶剂中进行。所述有机溶剂可选自四氢呋喃、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、1,4-二氧六环及其任意组合,优选N,N-二甲基甲酰胺。所述反应优选在适合的碱存在下进行;和/或在过渡金属催化剂及配体存在下进行。所述碱可选自三乙胺、吡啶、4-二甲胺基吡啶、二异丙基乙胺、碳酸钾、碳酸铯、碳酸钠或叔丁醇钾,优选碳酸钾。所述过渡金属催化剂可选自Pd(dppf)Cl
方法三:
其中,LG表示离去基团,所述的离去基团包括但不限于卤素原子、甲磺酰基氧基、对甲苯磺酰基氧基等。R
(1)使化合物I-3与化合物I-7经偶联反应得到化合物I-11
所述反应优选在适合的有机溶剂中进行。所述有机溶剂可选自四氢呋喃、1,4-二氧六环、甲苯、乙腈、乙醇、水及其任意组合,优选1,4-二氧六环与水组合。所述反应优选在适合的催化剂存在下进行。所述催化剂可选自Pd(dppf)Cl
(2)使化合物I-11经取代反应得到化合物I-12
所述反应优选在适合的有机溶剂中进行。所述有机溶剂可选自乙腈、甲苯、氯仿、四氢呋喃及其任意组合,优选乙腈。所述反应优选在适合的溴代试剂存在下进行。所述溴代试剂可选自N-溴代丁二酰亚胺、液溴、吡啶溴鎓盐,优选N-溴代丁二酰亚胺。所述反应优选在适合的温度下进行,所述温度优选为20-50℃。所述反应优选进行合适的时间,例如2-6小时。
(3)使化合物I-12与化合物I-5的硼酸或硼酸酯经偶联反应得到式I化合物
所述反应优选在适合的有机溶剂中进行。所述有机溶剂可选自四氢呋喃、1,4-二氧六环、甲苯、乙腈、乙醇、水及其任意组合,优选1,4-二氧六环与水组合。所述反应优选在适合的催化剂存在下进行。所述催化剂可选自Pd(dppf)Cl
上述各反应步骤的具体条件为本领域公知,对此本申请不做具体限定。如本申请的教导结合本领域公知常识,本领域技术人员可以对通式中的各取代基进行选择替换以制备得到不同的化合物,这些选择和替换均在本申请的保护范围之内。
本申请还涉及一种药物组合物,其包含上述的式I化合物、其立体异构体或其药学上可接受的盐,以及药学上可以接受的辅料。
本申请还涉及上述式I化合物、其立体异构体或其药学上可接受的盐、或者上述的药物组合物在制备 预防或治疗与LSD1相关疾病的药物中的用途。或者,本申请还涉及用于预防或治疗与LSD1相关疾病的式I化合物、其立体异构体或其药学上可接受的盐、或者上述的药物组合物。或者,本申请还涉及一种预防或治疗与LSD1相关疾病的方法,包括向有需要的受试者给予上述式I化合物、其立体异构体或其药学上可接受的盐、或者上述的药物组合物。
在一些实施例方案中,所述与LSD1相关疾病选自肿瘤或癌症,例如急性髓系白血病(AML)、慢性髓系白血病(CML)、急性淋巴细胞白血病(ALL)、慢性淋巴细胞白血病(CLL)、小细胞肺癌(SCLC)、非小细胞肺癌(NSCLC)、淋巴瘤、恶性肉瘤、乳腺癌、宫颈癌、结肠癌、肺癌、口腔癌、脑癌、胃癌、肝癌、结肠直肠癌、胰腺癌、皮肤癌、前列腺癌、骨癌、肾癌、卵巢癌、膀胱癌、输卵管肿瘤、腹膜肿瘤、黑色素瘤、神经胶质瘤、神经胶母细胞瘤、乳突状恶性瘤、头颈部肿瘤、或骨髓瘤。
本申请还涉及上述式I化合物、其立体异构体或其药学上可接受的盐、或者上述的药物组合物在制备LSD1抑制剂中的用途。或者本申请还涉及用作LSD1抑制剂的上述式I化合物、其立体异构体或其药学上可接受的盐、或者上述的药物组合物。或者,本申请还涉及一种抑制LSD1的方法,包括向有需要的受试者给予上述式I化合物、其立体异构体或其药学上可接受的盐、或者上述的药物组合物。
本申请发现一类结构全新的具有如式I所示结构的LSD1抑制剂,其具有较好的抑制活性。
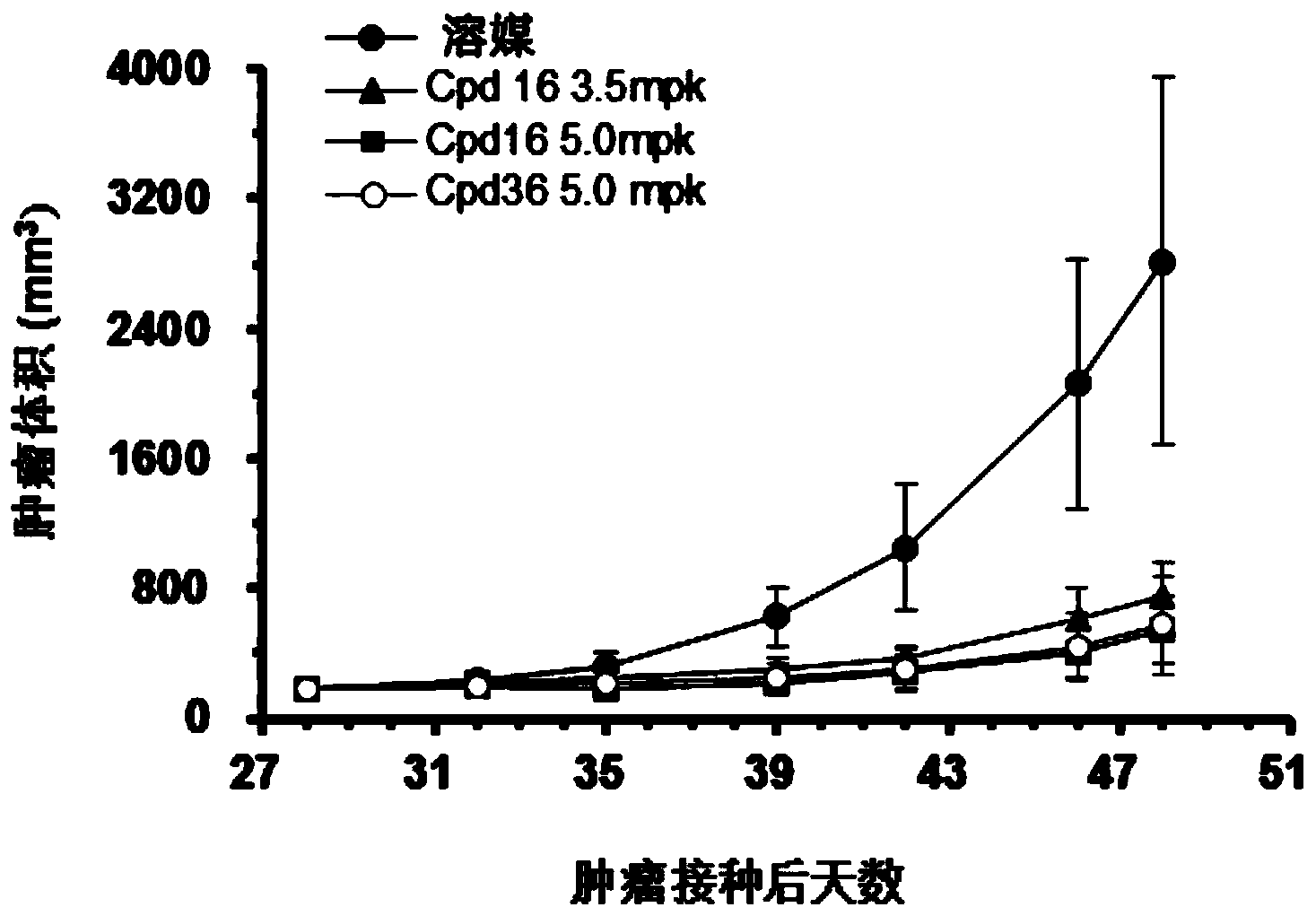
图1示出了受试化合物组和溶媒组的小鼠体内的肿瘤体积变化情况,其中,Cpd表示化合物。
图2示出了受试化合物组和溶媒组的瘤重变化情况。
图3示出了受试化合物组和溶媒组的小鼠的体重变化率。
图4示出了受试化合物组和溶媒组的小鼠体内的肿瘤体积变化情况。
图5示出了受试合物组和溶媒组的小鼠的体重变化率。
为了使本申请的方面和技术方案更加清楚,以下结合具体实施例进一步阐述本申请。应理解,这些实施例仅用于说明本申请而不用于限制本申请范围。并且,下列实施例中未提及的具体实验方法,均按照常规实验方法进行。
定义和说明
非另有说明,本申请中所用的术语具有下列含义。一个特定的术语在没有特别定义的情况下不应该被认为是不确定的或不清楚的,而应该按照本领域普通的含义去理解。
本申请中的某些结构单元或者基团中的共价键未与具体的原子连接时,表示该共价键可以与该结构单元或者基团中的任意原子连接,只要不违背价键连接规则。
在本文中,除非另有说明,“烷氧基”指-O-烷基。
在本文中,除非另有说明,“卤素”是指氟、氯、溴或碘。
在本文中,除非另有说明,“环烷基氧”基指-O-环烷基。
在本文中,除非另有说明,“饱和环烷基”涵盖单环、双环形式的桥环、双环形式的螺环等,是指完全饱和的脂环烃。例如,本文中的环烷基可为C
在本文中,除非另有说明,“饱和杂环烷基”是指其中的1个或多个(例如1-6个、1-5个、1-3个、1个或2个)碳原子被选自N、O或S中的杂原子取代的环烷基。
在本文中,除非另有说明,“环烃基”是指碳原子成环的烃基,包括饱和的环烃基和不饱和的环烃基(例如芳香基)。在本文中,不饱和环烃基有时也被称为不饱和环烷基。在本文中,环烃基可为单环、双环形式的桥环、双环形式的螺环、双环形式的稠环,例如可为C
在本文中,除非另有说明,“取代或未被取代的氨基”是涵盖未被取代的氨基或被选自如下的基团取代的氨基:C
在本文中,除非另有说明,使用的术语“Cm-Cn”是指由该术语修饰的该部分中具有m-n个碳原子(n大于m,且二者为整数)。例如,C
术语“受试者”与“患者”和“个体”等同,并且表示人或非人动物(哺乳动物,例如灵长类动物、啮齿动物等)。“哺乳动物”包括人和家畜(如实验室哺乳动物与家庭宠物,例如猫、狗、猪、羊、牛、绵羊、山羊、马、家兔),及非驯养哺乳动物,如野生哺乳动物等。
术语“治疗”意为将本申请所述化合物或制剂进行给药以改善或消除疾病或与所述疾病相关的一个或多个症状,且包括抑制疾病或病症的进展、缓解疾病或病症。
术语“药学上可接受的”是指某载体、运载物、稀释剂、辅料、和/或所形成的盐通常在化学上或物理上与构成某药物剂型的其它成分相兼容,并在生理上与受体相兼容,而没有过多的毒性、刺激性、过敏性反应或其它问题或并发症,与合理的利益/风险比相称。
在本申请中,术语“包括”、“包含”和“含有”及其等同物应理解为开放的、非排他性的意义,即“包括但不限于”,意味着除所列出的要素、组分和步骤外,还可涵盖其它未指明的要素、组分和步骤。在本文中,除非上下文另有明确规定,否则单数术语涵盖复数指代物,反之亦然。类似地,除非上下文另有明确指示,词语“或”意在包括“和”。
除非另有说明,在本文中,代表成分的量或理化性质或者反应条件等的参数值应当被理解为在所有情况下均由术语“约”修饰。当用术语“约”描述本申请时,术语“约”表示存在的误差值,例如表示在某一特定值的±5%、例如±1%或±0.1%的范围内变化。
在本申请中,当化学名称和结构式不一致时,应当以结构式所示为准,除非如上下文可以推断化学名称而非结构式是正确的。
本文中的缩写具有以下含义:
化合物的结构是通过质谱(MS)或者核磁共振(
在本申请未给出特殊说明的情况下,本申请中所提及的反应均在氮气氛围下进行。在本申请的术语“氮气氛围”是指例如将反应瓶连接一个1L容积的氮气气球。
在本申请的术语“氢气氛围”是指例如将反应瓶连接一个1L容积的氢气气球。
在本申请未给出特殊说明的情况下,本申请反应中提及的溶液是水溶液。
在本申请的术语“室温”是指温度处于10℃-25℃之间。
为了描述和公开的目的,以引用的方式将所有的专利、专利申请和其它已确定的出版物在此明确地并 入本文。这些出版物仅因为它们的公开早于本申请的申请日而提供。所有关于这些文件的日期的声明或这些文件的内容的表述是基于申请者可得的信息,并且不构成任何关于这些文件的日期或这些文件的内容的正确性的承认。而且,在任何国家,在本文中对这些出版物的任何引用并不构成关于该出版物成为本领域的公知常识的一部分的认可。
如下将通过实施例对本申请进行详细说明,但是本领域技术人员可以理解的是,本申请的保护范围不仅限于此。本领域技术人员可以在不脱离本申请的精神或者范围的情况下,对本申请的实施方式和实施例进行各种修饰、变化、组合等,由此得到调整后的方案也落在本申请的保护范围内。
实施例1 6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(1)的制备
第一步:(1-(6-氯-4-氰基吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(1b)的制备
将化合物1a(550mg,3.18mmol)、4-叔丁氧羰基氨基哌啶(643mg,3.21mmol)、K
第二步:(1-(5-溴-6-氯-4-氰基吡啶-2-基)哌啶-4-基)氨基甲酸酯叔丁酯(1c)的制备
将化合物1b(538mg,1.60mmol)溶于乙腈(15mL)中,在冰浴下向体系中缓慢加入NBS(288mg,1.62mmol),反应体系置于室温下搅拌反应2小时。将反应体系减压浓缩,粗品经硅胶柱层析纯化得到标题化合物542mg,收率:82%。
第三步:[1-(6-氯-4-氰基-5-(3-氟-4-甲氧基苯基)吡啶-2-基)哌啶-4-基]氨基甲酸叔丁酯(1d)的制备
将化合物1c(231mg,0.56mmol)、3-氟-4-甲氧基苯硼酸(97mg,0.57mmol)、K
第四步:(1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(1e)的制备
将化合物1d(47mg,0.10mmol)、3-氟-4-氰基苯硼酸(25mg,0.15mmol)、K
第五步:6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(1)的制备
将化合物1e(27mg,0.05mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物16mg,收率:73%。
LC-MS(ESI)m/z(M+H)
1
实施例2 6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟酰胺(2)的制备
第一步:[1-(4-氨基甲酰基-6-氯-5-(3-氟-4-甲氧基苯基)吡啶-2-基)哌啶-4-基]氨基甲酸叔丁酯(2a)的制备
将化合物1d(125mg,0.27mmol)溶于甲醇溶液(1mL)中,随后依次加入二甲基亚砜(1mL)、NaOH(32mg,0.80mmol),反应体系在搅拌下缓慢滴入H
第二步:(1-(4-氨基甲酰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(2b)的制备
将化合物2a(55mg,0.11mmol)、3-氟-4-氰基苯硼酸(28mg,0.17mmol)、K
第三步:6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟酰胺(2)的制备
将化合物2b(48mg,0.09mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物31mg,收率:79%。
LC-MS(ESI)m/z(M+H)
1
实施例3 4-(4-(4-氨基哌啶-1-基)-7-(3-氟-4-甲氧基苯基)-3-氧代-2,3-二氢-1H-吡咯[3,4-c]吡啶-6-基)-2-氟苯腈(3)的制备
第一步:2,6-二氯-4-碘烟酸(3b)的制备
将化合物3a(10.0g,36.51mmol)溶于干燥的四氢呋喃(110mL),置换氮气,将体系置于-50℃,向体系中缓慢滴加LDA(20mL,40.00mmol,2mol/L in THF/Heptane/Ethylbenzene),滴加完毕,体系维持-50℃继续搅拌反应2小时。将反应液快速倒入装有碎干冰的量杯中并搅拌,缓慢恢复至室温,反应液倾入水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品未经进一步纯化,直接用于下一步反应。
第二步:2,6-二氯-4-碘烟酸甲酯(3c)的制备
将化合物3b(12.0g,37.75mmol)溶于N,N-二甲基甲酰胺(130mL)中,然后依次加入K
第三步:2,6-二氯-4-氰基烟酸甲酯(3d)的制备
将化合物3c(4.3g,12.95mmol)、Zn(CN)
第四步:2-(4-((叔丁氧羰基)氨基)哌啶-1-基)-6-氯-4-氰基烟酸甲酯(3e)的制备
将化合物3d(1.0g,4.33mmol)、4-叔丁氧羰基氨基哌啶(1.3g,6.49mmol)、三乙胺(900mg,8.89mmol)溶于四氢呋喃(10mL)中。将体系加热至50℃搅拌反应3小时。反应体系冷却至室温,反应液倾入水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物647mg,收率:38%。
第五步:(1-(6-氯-3-氧代-2,3-二氢-1H-吡咯[3,4-c]吡啶-4-基)哌啶-4-基)氨基甲酸叔丁酯(3f)的制备
向反应瓶中加入化合物3e(647mg,1.64mmol)溶于甲醇(10mL)中,然后依次加入氨水(10滴)、Raney-Ni(267mg),置换氢气,反应体系在氢气氛围下置于室温反应过夜。将反应液过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物218mg,收率:36%。
第六步:(1-(7-溴-6-氯-3-氧-2,3-二氢-1H-吡咯[3,4-c]吡啶-4-基)哌啶-4-基)氨基甲酸叔丁酯(3g)的制备
将化合物3f(175mg,0.48mmol)溶于乙腈(4mL)中,冰浴条件下向体系中缓慢加入NBS(128mg,0.72mmol),反应体系维持冰浴条件下搅拌反应1小时。反应液倾入水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物210mg,收率:99%。
第七步:(1-(6-氯-7-(3-氟-4-甲氧基苯基)-3-氧代-2,3-二氢-1H-吡咯并[3,4-c]吡啶-4-基)哌啶-4-基)氨基甲酸叔丁酯(3h)的制备
将化合物3g(120mg,0.27mmol)、3-氟-4-甲氧基苯硼酸(46mg,0.27mmol)、K
第八步:(1-(6-(4-氰基-3-氟苯基)-7-(3-氟-4-甲氧基苯基)-3-氧代-2,3-二氢-1H-吡咯并[3,4-c]吡啶-4-基)哌啶-4-基)氨基甲酸叔丁酯(3i)的制备
将化合物3h(33mg,0.07mmol)、3-氟-4-氰基苯硼酸(16mg,0.01mmol)、K
第九步:4-(4-(4-氨基哌啶-1-基)-7-(3-氟-4-甲氧基苯基)-3-氧代-2,3-二氢-1H-吡咯[3,4-c]吡啶-6-基)-2-氟苯腈(3)的制备
将化合物3i(34mg,0.06mmol)溶于1,4-二氧六环(2mL)中,然后加入盐酸-1,4-二氧六环溶液(2mL,4M in 1,4-dioxane),反应体系在室温下搅拌反应2小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物12mg,收率:43%。
LC-MS(ESI)m/z(M+H)
1
实施例4 6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(1-甲基-1H-吲唑-5-基)异烟腈(4)的制备
第一步:(1-(6-氯-4-氰基-5-(1-甲基-1H-吲唑-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(4a)的制备
将化合物1c(130mg,0.31mmol)、1-甲基吲唑-5-硼酸(56mg,0.32mmol)、K
第二步:(1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(1-甲基-1H-吲唑-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(4b)的制备
向反应瓶中加入化合物4a(50mg,0.11mmol)、3-氟-4-氰基苯硼酸(28mg,0.17mmol)、K
第三步:6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(1-甲基-1H-吲唑-5-基)异烟腈(4)的制备
将化合物4b(20mg,0.04mmol)溶于乙酸乙酯(0.5mL)中,然后加入盐酸-乙酸乙酯溶液(0.5mL,3M),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物15mg,收率:92%。
LC-MS(ESI)m/z(M+H)
1
实施例5 6-(4-氨基-3-羟基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(5)的制备
第一步:4-氨基-3-羟基哌啶-1-羧酸苄酯(5b)的制备
将化合物5a(1.0g,4.29mmol)溶于乙醇(5mL)中,再加入氨水(5mL),将反应体系密封后加热至70℃搅拌反应过夜。将反应体系冷却至室温,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品未经纯化,直接用于下一步反应。
第二步:4-((叔丁氧羰基)氨基)-3-羟基哌啶-1-羧酸苄酯(5c)的制备
将化合物5b(700mg,2.80mmol)溶于二氯甲烷(10mL)中,在冰浴下向体系中依次加入三乙胺(424mg,4.19mmol)、二碳酸二叔丁酯(610mg,2.80mmol),反应体系置于室温下搅拌反应过夜。向体系中加入水淬灭反应,用二氯甲烷萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物500mg,两步合计收率:33%。
第三步:(3-羟基哌啶-4-基)氨基甲酸叔丁酯(5d)的制备
将化合物5c(500mg,1.43mmol)溶于异丙醇(5mL)中,然后加入Pd/C(50mg,10%),用氢气置换体系中的空气,然后体系在氢气氛围下室温搅拌过夜反应4小时。将反应体系过滤并减压浓缩,粗品经硅胶柱层析纯化得到标题化合物200mg,收率:65%。
第四步:2-氯-6-((4-甲氧基苄基)氨基)异烟腈(5e)的制备
将化合物1a(7.0g,40.46mmol)溶于DMSO(70mL)中,然后依次加入DIEA(5.3g,41.09mmol)、对甲氧基苄胺(5.7g,41.55mmol),反应体系置于100℃搅拌反应过夜。反应体系冷却至室温,反应液倾入水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品未经纯化,直接用于下一步。
第五步:2-氨基-6-氯异烟腈(5f)的制备
将化合物5e(9.0g,32.88mmol)溶于二氯甲烷(50mL)中,然后加入三氟乙酸(50mL),反应体系置于室温搅拌反应6小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,所得粗品经薄层硅胶板纯化得到标题化合物5.0g,两步合计收率:81%。
第六步:6-氨基-3-溴-2-氯异烟腈(5g)的制备
将化合物5f(5.0g,32.56mmol)溶于乙腈(50mL)中,在冰浴下向体系中缓慢滴加溴素(5.3g,33.13mmol),反应体系置于室温搅拌反应6小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物4.0g,收率:53%。
第七步:6-氨基-2-氯-3-(3-氟-4-甲氧基苯基)异烟腈(5h)的制备
将化合物5g(2.0g,8.60mmol)溶于1,4-二氧六环(20mL)和水(4mL)中,然后依次加入3-氟-4-甲氧基苯硼酸(1.5g,8.83mmol)、K
第八步:6-氨基-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(5i)的制备
将化合物5h(1.5g,5.40mmol)溶于1,4-二氧六环(15mL)和水(3mL)中,然后依次加入3-氟-4-氰基苯硼酸(2.7g,16.37mmol)、K
第九步:2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)-6-碘异烟腈(5j)的制备
将化合物5i(1.5g,4.14mmol)溶于乙腈(15mL)中,然后依次加入碘化钾(4.1g,24.70mmol)、亚硝酸异戊酯(2.9g,24.75mmol),将反应体系置于75℃搅拌反应36小时。反应体系冷却至室温,减压浓缩反应液,向体系中加入水稀释,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物700mg,收率:36%。
第十步:(1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)-3-羟基哌啶-4-基)氨基甲酸叔丁酯(5k)的制备
将化合物5j(50mg,0.11mmol)溶于1,4-二氧六环(3mL)中,然后依次加入化合物5d(48mg,0.22mmol)、Cs
第十一步:6-(4-氨基-3-羟基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(5)的制备
将化合物5k(20mg,0.04mmol)溶于1,4-二氧六环溶液(2mL)中,然后加入盐酸-1,4-二氧六环溶液(2mL,4M in 1,4-dioxane),反应体系置于室温搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物8mg,收率:49%。
LC-MS(ESI)m/z(M+H)
实施例6 6-(4-(氨基甲基)-4-羟基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(6)的制备
第一步:4-(氨基甲基)-1-苄基哌啶-4-醇(6b)的制备
将化合物6-1(500mg,2.46mmol)溶于甲醇(2mL)中,再加入氨水(5mL),将该体系密封后置于室温搅拌反应4小时。将反应体系冷却至室温,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品未经纯化,直接用于下一步反应。
第二步:((1-苄基-4-羟基哌啶-4-基)甲基)氨基甲酸叔丁酯(6c)的制备
将化合物6b(500mg,2.27mmol)溶于二氯甲烷(10mL)中,冰浴条件下向体系中加入二碳酸二叔丁酯(495mg,2.27mmol),反应体系置于室温下搅拌反应过夜。向体系中加入水淬灭反应,用二氯甲烷萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物600mg,两步合计收率:76%。
第三步:((4-羟基哌啶-4-基)甲基)氨基甲酸叔丁酯(6d)的制备
将化合物6c(300mg,0.94mmol)溶于异丙醇(5mL)中,然后室温下加入氢氧化钯碳(30mg,10%),用氢气置换体系中的空气,然后体系在氢气氛围下置于室温搅拌过夜。将反应体系过滤并减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物100mg,收率:46%。
第四步:((1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)-4-羟基哌啶-4-基)甲基)氨基甲酸叔丁酯(6e)的制备
将化合物5j(50mg,0.11mmol)溶于1,4-二氧六环(3mL)中,然后依次加入化合物6d(51mg,0.22mmol)、Cs
第五步:6-(4-(氨基甲基)-4-羟基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(6)的制备
将化合物6e(22mg,0.04mmol)溶于1,4-二氧六环溶液(2mL)中,然后加入盐酸-1,4-二氧六环溶液(2mL,4M in 1,4-dioxane),反应体系置于室温搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物10mg,收率:55%。
LC-MS(ESI)m/z(M+H)
1
实施例7 6-(3-氨基氮杂环丁烷-1-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(7)的制备
第一步:(1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)氮杂环丁烷-3-基)氨基甲酸叔丁酯(7a)的制备
将化合物5j(50mg,0.11mmol)溶于1,4-二氧六环(3mL)中,然后依次加入叔丁基氮杂环丁烷-3-基氨基甲酸酯(38mg,0.22mmol)、Cs
第二步:6-(3-氨基氮杂环丁烷-1-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(7)的制备
将化合物7a(22mg,0.04mmol)溶于二氯甲烷(2mL)中,然后加入三氟乙酸(1mL),反应体系置于室温搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物15mg,收率:85%。
LC-MS(ESI)m/z(M+H)
1
实施例8 4-(4-(4-氨基哌啶-1-基)-7-(1-甲基-1H-吲唑-5-基)-3-氧代-2,3-二氢-1H-吡咯[3,4-c]吡啶-6-基)-2-氟苯腈(8)的制备
第一步:(1-(6-(4-氰基-3-氟苯基)-3-氧代-2,3-二氢-1H-吡咯[3,4-c]吡啶-4-基)哌啶-4-基氨基甲酸叔丁酯(8a)的制备
将化合物3f(287mg,0.78mmol)、3-氟-4-氰基苯硼酸(193mg,1.17mmol)、K
第二步:(1-(7-溴-6-(4-氰基-3-氟苯基)-3-氧代-2,3-二氢-1H-吡咯并[3,4-c]吡啶-4-基)哌啶-4-基)氨基甲酸叔丁酯(8b)的制备
将化合物8a(310mg,0.69mmol)溶于乙腈(12mL)中,冰浴条件下向体系中缓慢加入NBS(246mg,1.38mmol),反应体系维持冰浴条件下搅拌反应1小时。反应液倾入水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物364mg,收率:100%。
第三步:(1-(6-(4-氰基-3-氟苯基)-7-(1-甲基-1H-吲唑-5-基)-3-氧代-2,3-二氢-1H-吡咯[3,4-c]吡啶-4-基)哌啶-4-基)氨基甲酸叔丁酯(8c)的制备
将化合物8b(70mg,0.13mmol)、1-甲基吲唑-5-硼酸(35mg,0.20mmol)、K
第四步:4-(4-(4-氨基哌啶-1-基)-7-(1-甲基-1H-吲唑-5-基)-3-氧代-2,3-二氢-1H-吡咯[3,4-c]吡啶-6-基)-2-氟苯腈(8)的制备
将化合物8c(57mg,0.10mmol)溶于二氯甲烷(3mL)中,然后在0℃下加入三氟乙酸(3mL),反应体系维持0℃下搅拌反应2h。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物15mg,收率:32%。
LC-MS(ESI)m/z(M+H)
1
实施例9 4-(4-(4-氨基哌啶-1-基)-7-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)-3-氧代-2,3-二氢-1H-吡咯[3,4-c]吡啶-6-基)-2-氟苯腈(9)的制备
第一步:1-(5-溴-6-氟-1H-吲唑-1-基)-2-甲基丙烷-2-醇(9b)的制备
将化合物9a(430mg,2.00mmol)溶于N,N-二甲基甲酰胺(10mL)中,然后依次加入Cs
第二步:1-(6-氟-5-(4,4,5,5-四甲基-1,3,2-二氧苯甲醛-2-基)-1H-吲唑-1-基)-2-甲基丙烷-2-醇(9c)的制备
将化合物9b(286mg,1.00mmol)、联硼酸频那醇酯(381mg,1.50mmol)、醋酸钾(196mg,0.10mmol)溶于N,N-二甲基甲酰胺(5mL)中,置换氮气,依次加入三苯基膦(52mg,0.20mmol)、Pd(AcO)
第三步:(1-(6-(4-氰基-3-氟苯基)-7-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)-3-氧代)-2,3-二氢-1H-吡咯并[3,4-c]吡啶-4-基)哌啶-4-基)氨基甲酸叔丁酯(9d)的制备
将化合物8b(110mg,0.21mmol)、化合物9c(107mg,0.32mmol)、K
第四步:4-(4-(4-氨基哌啶-1-基)-7-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)-3-氧代-2,3-二氢-1H-吡咯[3,4-c]吡啶-6-基)-2-氟苯腈(9)的制备
将化合物9d(12mg,0.02mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物6mg,收率:59%。
LC-MS(ESI)m/z(M+H)
1
实施例10 6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(1-甲基-1H-吲唑-5-基)异烟酰胺(10)的制备
第一步:(1-(4-氨甲酰-6-氯-5-(1-甲基-1H-吲唑-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(10a)的制备
将化合物4a(190mg,0.41mmol)溶于甲醇溶液(4mL)中,然后依次加入二甲基亚砜(2mL)、NaOH(49mg,1.23mmol)、H
第二步:(1-(4-氨甲酰-6-(4-氰基-3-氟苯基)-5-(1-甲基-1H-吲唑-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(10b)的制备
将化合物10a(100mg,0.21mmol)、3-氟-4-氰基苯硼酸(53mg,0.32mmol)、K
第三步:6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(1-甲基-1H-吲唑-5-基)异烟酰胺(10)的制备
将化合物10b(95mg,0.17mmol)溶于乙酸乙酯(2mL)中,然后加入盐酸-乙酸乙酯溶液(2mL,3M in EA),反应体系置于室温搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物31mg,收率:40%。
LC-MS(ESI)m/z(M+H)
1
实施例11 2-(4-氨基哌啶-1-基)-6-(4-氰基-3-氟苯基)-5-(1-甲基-1H-吲唑-5-基)烟酰胺(11)的制备
第一步:2,6-二氯烟酰胺(11b)的制备
将化合物11a(3.0g,15.63mmol)溶于氯仿(30mL)中,在冰浴下向体系中缓慢加入二氯亚砜(18.6g,156.30mmol),将体系加热至70℃搅拌反应3小时。将反应体系减压浓缩并用四氢呋喃(10mL)溶解,然后滴加到冰浴冷却的氨水(30mL)的四氢呋喃(10mL)溶液中,搅拌反应30分钟。向反应体系加入水,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物2.8g,收率:94%。
第二步:(1-(3-氨基甲酰基-6-氯吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(11c)的制备
将化合物11b(2.8g,14.66mmol)、4-叔丁氧羰基氨基哌啶(4.9g,24.47mmol)、三乙胺(3.0g,29.65mmol)溶于乙腈(30mL)中,将该体系加热至70℃搅拌反应3小时。反应体系冷却至室温,反应液倾入水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物4.4g,收率:88%。
第三步:(1-(3-氨基甲酰基-6-(4-氰基-3-氟苯基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(11d)的制备
将化合物11c(400mg,1.13mmol)、3-氟-4-氰基苯硼酸(280mg,1.70mmol)、K
第四步:(1-(5-溴-3-氨甲酰-6-(4-氰基-3-氟苯基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(11e)的制备
将化合物11d(448mg,1.02mmol)溶于N,N-二甲基甲酰胺(5mL)中,冰浴条件下向体系中缓慢加入NBS(200mg,1.12mmol),反应体系维持0℃下搅拌反应2小时。反应液倾入水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物528mg,收率:100%。
第五步:(1-(3-氨甲酰-6-(4-氰基-3-氟苯基)-5-(1-甲基-1H-吲唑-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(11f)的制备
将化合物11e(518mg,1.00mmol)、1-甲基吲唑-5-硼酸(263mg,1.49mmol)、K
第六步:2-(4-氨基哌啶-1-基)-6-(4-氰基-3-氟苯基)-5-(1-甲基-1H-吲唑-5-基)烟酰胺(11)的制备
将化合物11f(30mg,0.05mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物6mg,收率:24%。
LC-MS(ESI)m/z(M+H)
1
实施例12 2-(4-氨基哌啶-1-基)-6-(4-氰基-3-氟苯基)-5-(1-甲基-1H-吲唑-5-基)烟腈(12)的制备
第一步:(1-(3-氰基-6-(4-氰基-3-氟苯基)-5-(1-甲基-1H-吲唑-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(12a)的制备
将化合物11f(200mg,0.35mmol)溶于四氢呋喃(6mL)中,置换氮气,在冰浴下依次加入吡啶(194mg,2.45mmol)、三氟乙酸酐(147mg,0.70mmol)的二氯甲烷(1mL)溶液,反应体系缓慢恢复至室温搅拌反应过夜。反应液倾入水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物10mg,收率:5%
第二步:2-(4-氨基哌啶-1-基)-6-(4-氰基-3-氟苯基)-5-(1-甲基-1H-吲唑-5-基)烟腈(12)的制备
将化合物12a(10mg,0.02mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物1mg,收率:12%。
LC-MS(ESI)m/z(M+H)
实施例13 6-(4-氨基-3-氟哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(13)的制备
第一步:4-氨基-3-氟哌啶-1-羧酸叔丁酯(13b)的制备
将化合物13-1(1.0g,4.60mmol)溶于甲醇(10mL)中,依次加入乙酸铵(2.5g,32.43mmol)、氰基硼氢化钠(375mg,5.97mmol),反应体系置于室温搅拌反应过夜。向反应体系中加水稀释,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物500mg,收率:50%。
第二步:4-(((苄氧基)羰基)氨基)-3-氟哌啶-1-羧酸叔丁酯(13c)的制备
将化合物13b(500mg,2.29mmol)溶于乙酸乙酯(5mL)中,再加入饱和碳酸氢钠溶液(10mL),冰浴条件下向体系中加入氯甲酸苄酯(391mg,2.29mmol),将反应体系置于室温搅拌反应过夜。反应液用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物420mg,收率:52%。
第三步:(3-氟哌啶-4-基)氨基甲酸苄酯盐酸盐(13d)的制备
将化合物13c(200mg,0.57mmol)溶于乙酸乙酯(10mL)中,然后加入盐酸-乙酸乙酯溶液(4mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,粗品未经纯化,直接用于下一步。
第四步:(1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)-3-氟哌啶-4-基)氨基甲酸苄酯(13e)的制备
将化合物5j(50mg,0.11mmol)溶于1,4-二氧六环(3mL)中,然后依次加入化合物13d(64mg,0.22mmol)、t-BuONa(53mg,0.55mmol),置换氮气,加入RuPhos-Pd-G3(9mg,0.01mmol),再次置换氮气,反应体系置于80℃搅拌反应3小时。反应体系冷却至室温,减压浓缩反应液,向体系中加入水稀释,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物21mg,收率:33%。
第五步:6-(4-氨基-3-氟哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(13)的制备
将化合物13e(21mg,0.04mmol)溶于乙酸乙酯(2mL)中,然后室温下加入Pd/C(2mg,10%),用氢气置换体系中的空气,然后体系在氢气氛围下加热至35℃搅拌反应4小时。将反应体系过滤并减压浓缩,粗品经薄层硅胶板纯化得到标题化合物10mg,收率:61%。
LC-MS(ESI)m/z(M+H)
1
实施例14 6-(4-氨基环己基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(14)的制备
第一步:(4-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)环己-3-烯-1-基)氨基甲酸叔丁酯(14a)的制备
将化合物5j(80mg,0.17mmol)溶于1,4-二氧六环(3mL)和水(0.5mL)中,依次加入(4-(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)环己-3-烯-1-基)氨基甲酸叔丁酯(165mg,0.51mmol)、K
第二步:(4-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)环己基)氨基甲酸叔丁酯(14b)的制备
将化合物14a(40mg,0.07mmol)溶解于乙酸乙酯(2mL)中,然后室温下加入Pd/C(4mg,10%),用氢气置换体系中的空气,然后体系在氢气氛围下室温搅拌反应过夜。将反应体系过滤并减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物23mg,收率:57%。
第三步:6-(4-氨基环己基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(14)的制备
将化合物14b(23mg,0.04mmol)溶于乙酸乙酯(5mL)中,然后加入盐酸-乙酸乙酯溶液(4mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物11mg,收率:59%。
LC-MS(ESI)m/z(M+H)
1
实施例15 2-(4-氰基-3-氟苯基)-6-(1,4-二氮杂环庚烷-1-基)-3-(3-氟-4-甲氧基苯基)异烟腈(15)的制备
第一步:4-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)-1,4-二氮环庚烷-1-羧酸叔丁酯(15a)的制备
将化合物5j(50mg,0.11mmol)溶于1,4-二氧六环(3mL)中,依次加入1,4-二氮杂环庚烷-1-羧酸叔丁酯(44mg,0.22mmol)、Cs
第二步:2-(4-氰基-3-氟苯基)-6-(1,4-二氮杂环庚烷-1-基)-3-(3-氟-4-甲氧基苯基)异烟腈(15)的制备
将化合物15a(24mg,0.04mmol)溶于乙酸乙酯(5mL)中,然后加入盐酸-乙酸乙酯溶液(4mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物10mg,收率:51%。
LC-MS(ESI)m/z(M+H)
1
实施例16 6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)异烟腈(16)的制备
第一步:(1-(4-氰基-6-(4-氰基-3-氟苯基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(16a)的制备
将化合物1b(1.7g,5.05mmol)、3-氟-4-氰基苯硼酸(1.2g,7.28mmol)、K
第二步:(1-(5-溴-4-氰基-6-(4-氰基-3-氟苯基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(16b)的制备
将化合物16a(421mg,1.00mmol)溶于乙腈(15mL)中,在冰浴下向体系中缓慢加入NBS(182mg,1.02mmol),反应体系置于室温下搅拌反应3小时。将反应体系减压浓缩,粗品经硅胶柱层析纯化得到标题化合物450mg,收率:90%。
第三步:叔丁基(1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸酯(16c)的制备
将化合物16b(100mg,0.20mmol)、化合物9c(100mg,0.30mmol)、K
第四步:6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)异烟腈(16)的制备
将化合物16c(30mg,0.05mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物24mg,收率:95%。
LC-MS(ESI)m/z(M+H)
1
实施例17 6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)异烟酰胺(17)的制备
第一步:叔丁基(1-(6-氯-4-氰基-5-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸酯(17a)的制备
将化合物1c(229mg,0.55mmol)、化合物9c(368mg,1.10mmol)、K
第二步:叔丁基(1-(4-氨基甲酰基-6-氯-5-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸酯(17b)的制备
将化合物17a(138mg,0.25mmol)溶于甲醇溶液(1mL)中,随后依次加入二甲基亚砜(1mL)、NaOH(30mg,0.75mmol)、H
第三步:叔丁基(1-(4-氨基甲酰基-6-(4-氰基-3-氟苯基)-5-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸酯(17c)的制备
将化合物17b(126mg,0.22mmol)、3-氟-4-氰基苯硼酸(54mg,0.33mmol)、K
第四步:6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)异烟酰胺(17)的制备
将化合物17c(74mg,0.11mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物24mg,收率:38%。
LC-MS(ESI)m/z(M+H)
1
实施例18 6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(1-甲基-2-氧代吲哚-5-基)异烟腈(18)的制备
第一步:(1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(1-甲基-2-氧代吲哚-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(18a)的制备
将化合物18b(75mg,0.15mmol)、1-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)二氢吲哚-2-酮(63mg,0.23mmol)、K
第二步:6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(1-甲基-2-氧代吲哚-5-基)异烟腈(18)的制备
将化合物18a(36mg,0.06mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物18mg,收率:61%。
LC-MS(ESI)m/z(M+H)
1
实施例19 6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(2-氟-4-(2-羟基-2-甲基丙基)苯基)异烟腈(19)的制备
第一步:2-(4-溴-3-氟苯基)乙酸甲酯(19b)的制备
将化合物19a(1.0g,4.29mmol)溶于乙腈(40mL)中,然后依次加入K
第二步:1-(4-溴-3-氟苯基)-2-甲基丙烷-2-醇(19c)的制备
将化合物19b(815mg,3.30mmol)溶于干燥四氢呋喃(30mL)中,然后在冰水浴下缓慢滴加甲基溴化镁(5.5mL,3M in THF),滴加完毕,反应体系缓慢回温至室温搅拌反应过夜。将反应体系冷却至0℃,缓慢加入适量水淬灭反应,过滤,滤液用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物366mg,收率:45%。
第三步:1-(3-氟-4-(4,4,5,5-四甲基-1,3,2-二氧苯甲醛-2-基)苯基)-2-甲基丙烷-2-醇(19d)的制备
将化合物19c(360mg,1.46mmol)、联硼酸频那醇酯(741mg,2.92mmol)、醋酸钾(429mg,4.37mmol)溶于1,4-二氧六环(15mL)中,置换氮气,加入Pd(dppf)Cl
第四步:叔丁基(1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(2-氟-4-(2-羟基-2-甲基丙基)苯基)吡啶-2-基)哌啶-4-基)氨基甲酸酯(19e)的制备
将化合物16b(100mg,0.20mmol)、化合物19d(118mg,0.40mmol)、K
第五步:6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(2-氟-4-(2-羟基-2-甲基丙基)苯基)异烟腈(19)的制备
将化合物19e(50mg,0.09mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物36mg,黄色固体,收率:87%。
LC-MS(ESI)m/z(M+H)
1
实施例20 2-(4-氰基-3-氟苯基)-6-(1,4-二氮杂苯胺-1-基)-3-(1-甲基-1H-吲唑-5-基)异烟腈(20)的制备
第一步:4-(6-氯-4-氰基吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(20a)的制备
将化合物1a(1.5g,8.67mmol)、1,4-二氮杂环庚烷-1-甲酸叔丁酯(1.7g,8.49mmol)、K
第二步:4-(4-氰基-6-(4-氰基-3-氟苯基)吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(20b)的制备
将化合物20a(1.0g,2.97mmol)、3-氟-4-氰基苯硼酸(735mg,4.46mmol)、Na
第三步:4-(5-溴-4-氰基-6-(4-氰基-3-氟苯基)吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(20c)的制备
将化合物20b(500mg,1.19mmol)溶于乙腈(5mL)中,在冰浴下向体系中缓慢加入NBS(211mg,1.19mmol),反应体系置于室温下搅拌反应3小时。将反应体系减压浓缩,粗品经硅胶柱层析纯化得到标题化合物420mg,收率:71%。
第四步:4-(4-氰基-6-(4-氰基-3-氟苯基)-5-(1-甲基-1H-吲唑-5-基)吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(20d)的制备
将化合物20c(50mg,0.10mmol)、1-甲基吲唑-5-硼酸(53mg,0.30mmol)、K
第五步:2-(4-氰基-3-氟苯基)-6-(1,4-二氮杂苯胺-1-基)-3-(1-甲基-1H-吲唑-5-基)异烟腈(20)的制备
将化合物20d(30mg,0.05mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(2mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物20mg,收率:82%。
LC-MS(ESI)m/z(M+H)
1
实施例21 2-(4-氰基-3-氟苯基)-6-(1,4-二氮杂苯胺-1-基)-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)异烟腈(21)的制备
第一步:叔丁基4-(4-氰基-6-(4-氰基-3-氟苯基)-5-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)吡啶-2-基)-1,4-二氮烷-1-羧酸酯(21a)的制备
将化合物20c(50mg,0.10mmol)、化合物9c(100mg,0.30mmol)、K
第二步:2-(4-氰基-3-氟苯基)-6-(1,4-二氮杂苯胺-1-基)-3-(6-氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)异烟腈(21)的制备
将化合物21a(10mg,0.02mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(2mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物4mg,收率:48%。
LC-MS(ESI)m/z(M+H)
1H NMR(400MHz,DMSO-d6)δ8.11(s,1H),7.79-7.73(m,2H),7.58(d,J=10.4Hz,1H),7.41(s,1H),7.39-7.33(m,1H),7.21-7.16(m,1H),4.67(s,1H),4.32-4.23(m,2H),3.88-3.67(m,4H),2.94-2.85(m,2H),2.76-2.68(m,2H),1.86-1.72(m,2H),1.15(s,3H),1.10(s,3H).
实施例22 2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)-6-(2,7-二氮螺环[3.5]壬-7-基)异烟腈(22)的制备
第一步:7-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)-2,7-二氮螺环[3.5]壬烷-2-羧酸叔丁酯(22a)的制备
将化合物5j(50mg,0.11mmol)溶于1,4-二氧六环(3mL)中,然后依次加入2-叔丁氧羰基-2,7-二氮杂螺[3.5]壬烷(50mg,0.22mmol)、Cs
第二步:2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)-6-(2,7-二氮螺环[3.5]壬-7-基)异烟腈(22)的制备
将化合物22a(30mg,0.05mmol)溶于二氯甲烷(2mL)中,然后加入三氟乙酸(1mL),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物15mg,收率:61%。
LC-MS(ESI)m/z(M+H)
1
实施例23 6-(4-氨基环己基)-2-(4-氰基-3-氟苯基)-3-(1-甲基-1H-吲唑-5-基)异烟腈(23-1,23-2)的制备
第一步:6-氨基-2-氯-3-(1-甲基-1H-吲唑-5-基)异烟腈(23a)的制备
将化合物5g(1.5g,6.45mmol)溶于1,4-二氧六环(20mL)和水(3mL)中,然后依次加入1-甲基吲唑-5-硼酸(1.1g,6.25mmol)、K
第二步:6-氨基-2-(4-氰基-3-氟苯基)-3-(1-甲基-1H-吲唑-5-基)异烟腈(23b)的制备
将化合物23a(1.1g,3.88mmol)溶于1,4-二氧六环(30mL)和水(3mL)中,然后依次加入3-氟-4-氰基苯硼酸(1.9g,11.52mmol)、K
第三步:2-(4-氰基-3-氟苯基)-6-碘-3-(1-甲基-1H-吲唑-5-基)异烟腈(23c)的制备
将化合物23b(500mg,1.36mmol)溶于乙腈(10mL)中,然后依次加入碘化钾(451mg,2.72mmol)、亚硝酸异戊酯(319mg,2.72mmol),将反应体系置于80℃搅拌反应过夜。反应体系冷却至室温,减压浓缩反应液,向体系中加入水稀释,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物90mg,收率:14%。
第四步:(4-(4-氰基-6-(4-氰基-3-氟苯基)-5-(1-甲基-1H-吲唑-5-基)吡啶-2-基)环己-3-烯-1-基)氨基甲酸叔丁酯(23d)的制备
将化合物23c(90mg,0.19mmol)溶于1,4-二氧六环(3mL)和水(0.6mL)中,然后依次加入(4-(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)环己-3-烯-1-基)氨基甲酸叔丁酯(184mg,0.57mmol)、K
第五步:(4-(4-氰基-6-(4-氰基-3-氟苯基)-5-(1-甲基-1H-吲唑-5-基)吡啶-2-基)环己基)氨基甲酸叔丁酯(23e)的制备
将化合物23d(80mg,0.14mmol)溶解于乙酸乙酯(3mL)中,然后室温下加入Pd/C(8mg,10%),用氢气置换体系中的空气,然后体系在氢气氛围下室温搅拌反应过夜。将反应体系过滤并减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物67mg,收率:83%。
第六步:6-(4-氨基环己基)-2-(4-氰基-3-氟苯基)-3-(1-甲基-1H-吲唑-5-基)异烟腈(23-1,23-2)的制备
将化合物23e(67mg,0.12mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(3mL,3M in EA),反应体系置于室温搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化(DCM/MeOH=10:1)得到10mg标题化合物23-1(上点:R
上点:
LC-MS(ESI)m/z(M+H)
1
下点:
LC-MS(ESI)m/z(M+H)
1
实施例24 2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)-6-(7-氧代-1,4-二氮杂-1-基)异烟腈(24)的制备
第一步4-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)-5-氧基-1,4-二氮烷-1-羧酸叔丁酯(24a)的制备
将化合物5j(50mg,0.11mmol)溶于1,4-二氧六环(3mL)中,然后依次加入1-叔丁氧羰基-1,4-二氮杂-5-环庚酮(47mg,0.22mmol)、Cs
第二步:2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)-6-(7-氧代-1,4-二氮杂-1-基)异烟腈(24)的制备
将化合物24a(40mg,0.07mmol)溶于二氯甲烷(2mL)中,然后加入三氟乙酸(1mL),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物16mg,收率:49%。
LC-MS(ESI)m/z(M+H)
1
实施例25 6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(2,2-二氟苯并[1,3-d]二氧杂环-5-基)异烟腈(25)的制备
第一步:叔丁基(1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(2,2-二氟苯并[1,3-d]二氧杂环-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸酯(25a)的制备
将化合物16b(75mg,0.15mmol)、(2,2-二氟苯并[1,3-d]二氧杂环戊烯-5-基)硼酸(46mg,0.23mmol)、K
第二步:6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(2,2-二氟苯并[1,3-d]二氧杂环-5-基)异烟腈(25)的制备
将化合物25a(53mg,0.09mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物34mg,收率:78%。
LC-MS(ESI)m/z(M+H)
1
实施例26 6-(4-氰基-3-氟苯基)-2-(1,4-二氮杂-1-基)-5-(1-甲基-1H-吲唑-5-基)烟酰胺(26)的制备
第一步:4-(3-氨甲酰-6-氯吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(26a)的制备
将化合物11b(550mg,2.88mmol)、1,4-二氮杂环庚烷-1-甲酸叔丁酯(572mg,2.86mmol)和K
第二步:4-(3-氨甲酰-6-(4-氰基-3-氟苯基)吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(26b)的制备
将化合物26a(620mg,1.75mmol)、3-氟-4-氰基苯硼酸(433mg,2.63mmol)、K
第三步:4-(5-溴-3-氨甲酰-6-(4-氰基-3-氟苯基)吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(26c)的制备
将化合物26b(754mg,1.72mmol)溶于N,N-二甲基甲酰胺(9mL)中,冰浴下向体系中缓慢加入NBS (609mg,3.42mmol),反应体系在室温下搅拌反应2小时。反应液倾入水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物315mg,收率:35%。
第四步:4-(3-氨甲酰-6-(4-氰基-3-氟苯基)-5-(1-甲基-1H-吲唑-5-基)吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(26d)的制备
将化合物26c(160mg,0.31mmol)、1-甲基吲唑-5-硼酸(82mg,0.47mmol)、K
第五步:6-(4-氰基-3-氟苯基)-2-(1,4-二氮杂-1-基)-5-(1-甲基-1H-吲唑-5-基)烟酰胺(26)的制备
将化合物26d(72mg,0.13mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应2小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物49mg,收率:83%。
LC-MS(ESI)m/z(M+H)
1
实施例27 6-(4-氰基-3-氟苯基)-2-(1,4-二氮杂苯胺-1-基)-N,N-二甲基-5-(1-甲基-1H-吲唑-5-基)烟酰胺(27)的制备
第一步:2,6-二氯-N,N-二甲基烟酰胺(27a)的制备
将化合物11a(500mg,2.60mmol)溶于四氢呋喃(5mL)中,再加入二甲胺盐酸盐(636mg,7.80mmol)、2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(1.2g,3.16mmol)和N,N-二异丙基乙胺(1.0g,7.74mmol),在室温搅拌反应过夜。反应液倾入水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,得到标题化合物570mg,未经纯化直接用于下一步反应。
第二步:4-(6-氯-3-(二甲基氨甲酰)吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(27b)的制备
将化合物27a(570mg,2.60mmol)、1,4-二氮杂环庚烷-1-甲酸叔丁酯(518mg,2.59mmol)、K
第三步:4-(6-(4-氰基-3-氟苯基)-3-(二甲基氨甲酰)吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(27c)的制备
将化合物27b(520mg,1.36mmol)、3-氟-4-氰基苯硼酸(335mg,2.03mmol)、K
第四步:4-(5-溴-6-(4-氰基-3-氟苯基)-3-(二甲基氨甲酰)吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(27d)的制备
将化合物27c(389mg,0.83mmol)溶于N,N-二甲基甲酰胺(4mL)中,冰浴条件下向体系中缓慢加入NBS(295mg,1.66mmol),反应体系在室温下搅拌反应2小时。反应液倾入水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物311mg,收率:68%。
第五步:叔丁基4-(6-(4-氰基-3-氟苯基)-3-(二甲基氨甲酰)-5-(1-甲基-1H-吲唑-5-基)吡啶-2-基)-1,4-二氮烷-1-羧酸酯(27e)的制备
将化合物27d(160mg,0.29mmol)、1-甲基吲唑-5-硼酸(76mg,0.43mmol)、K
第六步:6-(4-氰基-3-氟苯基)-2-(1,4-二氮杂苯胺-1-基)-N,N-二甲基-5-(1-甲基-1H-吲唑-5-基)烟酰胺(27)的制备
将化合物27e(53mg,0.09mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应3小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物31mg,收率:70%。
LC-MS(ESI)m/z(M+H)
1
实施例28 6-(4-氰基-3-氟苯基)-N-环丙基-2-((1,4-二氮杂苯胺-1-基)-5-(1-甲基-1H-吲唑-5-基)烟酰胺(28)的制备
第一步:2,6-二氯-N-环丙基烟酰胺(28a)的制备
将化合物11a(500mg,2.60mmol)溶于氯仿(10mL)中,置换氮气,在冰浴下向体系中缓慢加入二氯亚砜(3.1g,26.00mmol),再次置换氮气,将体系加热至70℃搅拌反应2小时。将反应体系减压浓缩并用二氯甲烷(1mL)溶解,然后滴加到冰浴冷却的环丙胺(327mg,5.73mmol)的二氯甲烷(1mL)溶液中,搅拌反应1小时。向反应体系加入冷水,析出固体,过滤后得到标题化合物600mg,未经纯化直接用于下一步反应。
第二步:4-(6-氯-3-(环丙基氨甲酰)吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(28b)的制备
将化合物28a(600mg,2.60mmol)、1,4-二氮杂环庚烷-1-甲酸叔丁酯(520mg,2.60mmol)、K
第三步:4-(6-(4-氰基-3-氟苯基)-3-(环丙基氨甲酰)吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(28c)的制备
将化合物28b(520mg,1.32mmol)、3-氟-4-氰基苯硼酸(327mg,1.98mmol)、K
第四步:4-(5-溴-6-(4-氰基-3-氟苯基)-3-(环丙基氨甲酰)吡啶-2-基)-1,4-二氮烷-1-羧酸叔丁酯(28d)的制备
将化合物28c(630mg,1.31mmol)溶于N,N-二甲基甲酰胺(8mL)中,冰浴条件下向体系中缓慢加入NBS(480mg,2.70mmol),反应体系在室温下搅拌反应2小时。反应液倾入水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物315mg,收率:43%。
第五步:叔丁基4-(6-(4-氰基-3-氟苯基)-3-(环丙基氨甲酰)-5-(1-甲基-1H-吲哚唑-5-基)吡啶-2-基)-1,4-二氮烷-1-羧酸酯(28e)的制备
将化合物28d(160mg,0.29mmol)、1-甲基吲唑-5-硼酸(76mg,0.43mmol)、K
第六步:6-(4-氰基-3-氟苯基)-N-环丙基-2-((1,4-二氮杂苯胺-1-基)-5-(1-甲基-1H-吲唑-5-基)烟酰胺(28)的制备
将化合物28e(52mg,0.09mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应5小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物20mg,收率:46%。
LC-MS(ESI)m/z(M+H)
1
实施例29 2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)-6-(2,8-二氮杂螺[4.5]癸-8-基)异烟腈(29)的制备
第一步:8-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)-2,8-二氮螺环[4.5]癸烷-2-羧酸叔丁酯(29a)的制备
将化合物5j(50mg,0.11mmol)溶于1,4-二氧六环(3mL)中,然后依次加入2,8-二氮螺环[4.5]癸烷-2-羧酸叔丁酯(53mg,0.22mmol)、Cs
第二步:2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)-6-(2,8-二氮杂螺[4.5]癸-8-基)异烟腈(29)的制备
将化合物29a(32mg,0.05mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(2mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物16mg,收率:60%。
LC-MS(ESI)m/z(M+H)
1
实施例30 6-(氮杂-4-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(30)的制备
第一步:4-((((三氟甲基)磺酰基)氧基)-2,3,6,7-四氢-1H-氮杂-1-羧酸叔丁酯(30b)的制备
将化合物30a(3.0g,14.07mmol)溶于四氢呋喃(50mL)中,置换氮气,降温至-78℃,滴加双三甲基硅基胺基锂(19mL,1M in THF),该体系在-78℃反应1小时,再加入N-苯基双(三氟甲烷磺酰)亚胺(6.5g,18.19mmol),继续反应0.5小时,该体系恢复至室温反应14小时,再加水稀释,用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物2.0g,收率:41%。
第二步:4-(4,4,5,5-四甲基-1,3,2-二氧苯甲醛-2-基)-2,3,6,7-四氢-1H-氮杂环-1-羧酸叔丁酯(30c)的制备
将化合物30b(500mg,1.45mmol)、联硼酸频那醇酯(1.1g,4.33mmol)、醋酸钾(426mg,4.34mmol)溶于1,4-二氧六环(5mL)中,置换氮气,加入Pd(dppf)Cl
第三步:4-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)-2,3,6,7-四氢-1H-氮杂吡啶-1-羧酸叔丁酯(30d)的制备
将化合物5j(50mg,0.11mmol)、化合物30c(177mg,0.55mmol)、K
第四步:4-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3-氟-4-甲氧基苯基)吡啶-2-基)氮杂环庚烷-1-羧酸叔丁酯(30e)的制备
将化合物30d(20mg,0.04mmol)溶解于乙酸乙酯(2mL)中,然后室温下加入Pd/C(2mg,10%),用氢气置换体系中的空气,然后体系在氢气氛围下室温搅拌反应过夜。将反应体系过滤并减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物10mg,收率:50%。
第五步:6-(氮杂-4-基)-2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)异烟腈(30)的制备
将化合物30e(10mg,0.02mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(2mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物6mg,收率:74%。
LC-MS(ESI)m/z(M+H)
1
实施例31 2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)-6-(6-羟基-1,4-二氮杂-1-基)异烟腈(31)的制备
第一步N,N'-(乙烷-1,2-二基)双(4-甲基苯磺酰胺)(31b)的制备
将化合物31a(10.0g,75.18mmol)溶于吡啶(200mL)中,再加入对甲苯磺酰氯(28.0g,146.87mmol),将该体系加热至100℃搅拌反应16小时。反应体系冷却至室温后倾入水中,析出固体,过滤、真空干燥得到标题化合物18.0g,收率:65%。
第二步:1,4-二对甲苯磺酰-1,4-二氮杂-6-醇(31c)的制备
将化合物31b(5.8g,15.74mmol)溶于乙腈(100mL)中,加入氢氧化钾(2.2g,39.21mmol),反应体系加热至80℃反应0.5小时,体系冷却至室温,再加入1,3-二溴-2-丙醇(4.1g,18.82mmol),反应体系再次加热至80℃反应16小时。将反应体系冷却至室温后减压浓缩,再加水稀释,用二氯甲烷萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物5.3g,收率:79%。
第三步:1,4-二氮杂-6-醇溴化氢盐(31d)的制备
将化合物31c(2.0g,4.71mmol)溶于溴化氢水溶液(20mL,40%)中,反应体系加热至100℃反应22小时。将反应体系冷却至室温后减压浓缩,再用甲基叔丁基醚打浆得粗品600mg,收率:46%。
第四步:2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)-6-(6-羟基-1,4-二氮杂-1-基)异烟腈(31)的制备
将化合物5j(50mg,0.11mmol)溶于1,4-二氧六环(3mL)中,然后依次加入化合物31d(214mg,0.77mmol)、Cs
LC-MS(ESI)m/z(M+H)
实施例32 2-(4-氰基-3-氟苯基)-6-(6-氟-1,4-二氮杂-1-基)-3-(3-氟-4-甲氧基苯基)异烟腈(32-1、32-2)的制备
第一步:6-氟-1,4-二对甲苯磺酰-1,4-二氮烷(32a)的制备
将化合物31c(2.5g,5.89mmol)溶于二氯甲烷(30mL)中,置换氮气,体系降温至0℃,再加入二乙胺基三氟化硫(1.9g,11.79mmol),反应体系加热至室温反应16小时。再加水淬灭反应,用二氯甲烷萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物1.6g,收率:64%。
第二步:6-氟-1,4-二氮杂环庚烷(32b)的制备
将化合物32a(1.6g,3.75mmol)溶于溴化氢水溶液(20mL,40%)中,反应体系加热至120℃反应22小时。将反应体系冷却至室温后减压浓缩,再用甲基叔丁基醚打浆得粗品700mg,收率:67%。
第三步:2-(4-氰基-3-氟苯基)-3-(3-氟-4-甲氧基苯基)-6-(6-羟基-1,4-二氮杂-1-基)异烟腈(32-1、32-2)的制备
将化合物5j(50mg,0.11mmol)溶于1,4-二氧六环(3mL)中,然后依次加入化合物32b(216mg,0.77mmol)、Cs
32-1:LC-MS(ESI)m/z(M+H)
1
32-2:LC-MS(ESI)m/z(M+H)
1
实施例33 6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(5-氟-3-甲苯并[d]异恶唑-6-基)异烟腈(33)的制备
第一步:3-溴-4-氟苯乙酸酯(33b)的制备
将化合物33a(2.0g,10.47mmol)溶于二氯甲烷(20mL)中,冰浴下向体系中缓慢加入三乙胺(1.6g,15.81mmol),然后缓慢加入乙酰氯(1.2g,15.29mmol),将反应体系恢复到室温搅拌反应1小时。将反应液倾入水中,二氯甲烷萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物2.4g,收率:98%。
第二步:1-(4-溴-5-氟-2-羟基苯基)乙烷-1-酮(33c)的制备
将化合物33b(2.4g,10.30mmol)溶于三氟化硼乙酸溶液(26mL,33%BF
第三步:6-溴-5-氟-3-甲苯并[d]异恶唑(33d)的制备
将化合物33c(1.6g,6.87mmol)溶于甲醇(16mL)中,然后加入盐酸羟胺(963mg,13.86mmol)和醋酸钠(853mg,10.40mmol),将反应体系加热至60℃搅拌反应1小时。将反应体系冷却至室温,反应液倾入冷水中,析出固体,过滤,滤饼干燥得棕色固体。将所得固体溶于乙酸酐(6mL)中,将反应体系加热至60℃搅拌反应2小时,将反应体系冷却至室温,反应液倾入冷水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液得到粗品。将得到的粗品溶于N,N-二甲基甲酰胺(10mL)中,然后加入K
第四步:5-氟-3-甲基-6-(4,4,5,5-四甲基-1,3,2-二氧苯甲醛-2-基)苯并[d]异恶唑(33e)的制备
将化合物33d(693mg,3.01mmol)、联硼酸频那醇酯(914mg,3.60mmol)、醋酸钾(588mg,5.99mmol)溶于1,4-二氧六环(7mL)中,置换氮气,加入Pd(dppf)Cl
第五步:(1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(5-氟-3-甲基苯并[d]异恶唑-6-基)吡啶-2-基)哌啶-4-基)氨基甲酸叔丁酯(33f)的制备
将化合物16b(150mg,0.30mmol)、化合物33e(166mg,0.60mmol)、K
第六步:6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(5-氟-3-甲苯并[d]异恶唑-6-基)异烟腈(33)的制备
将化合物33f(41mg,0.07mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物12mg,黄色固体,收率:35%。
LC-MS(ESI)m/z(M+H)
1
实施例34 6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3,3-二氟-1-甲基-2-氧吲哚-5-基)异烟腈(34)的制备
第一步:5-溴-3,3-二氟吲哚-2-酮(34b)的制备
将化合物34a(2.0g,8.85mmol)溶于二氯甲烷(30mL)中,置换氮气,体系降温至0℃,然后加入二乙胺基三氟化硫(4.3g,26.68mmol),反应体系置于室温反应16小时。向体系中加水淬灭反应,用二氯甲烷萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物1.8g,收率:82%。
第二步:5-溴-3,3-二氟-1-甲基吲哚-2-酮(34c)的制备
将化合物34b(1.0g,4.03mmol)溶于乙腈(15mL)中,再加入碘甲烷(1.1g,7.75mmol)、Cs
第三步:3,3-二氟-1-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧苯甲醛-2-基)吲哚-2-酮(34d)的制备
将化合物34c(500mg,1.91mmol)、联硼酸频那醇酯(1.5g,5.91mmol)、醋酸钾(562mg,5.73mmol)溶于1,4-二氧六环(5mL)中,置换氮气,加入Pd(dppf)Cl
第四步:叔丁基(1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3,3-二氟-1-甲基-2-氧代吲哚-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸酯(34e)的制备
将化合物16b(80mg,0.16mmol)、化合物34d(247mg,0.80mmol)、K
第五步:6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3,3-二氟-1-甲基-2-氧吲哚-5-基)异烟腈(34)的制备
将化合物34e(30mg,0.05mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(2mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节 体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物22mg,收率:88%。
LC-MS(ESI)m/z(M+H)
1
实施例35 6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(4-(1,1-二氟-2-羟基-2-甲基丙基)-2-氟苯基)异烟腈(35)的制备
第一步:2-(4-溴-3-氟苯基)-2,2-二氟乙酸乙酯(35b)的制备
将化合物35a(1.3g,4.32mmol)溶于二甲亚砜(20mL)中,置换氮气,加入铜粉(707mg,11.13mmol),室温搅拌1小时,然后加入2-溴-2,2-二氟乙酸乙酯(2.3g,11.33mmol),将反应体系加热至80℃搅拌反应2小时。将反应体系冷却至室温,过滤,将滤液倾入水中,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物719mg,收率:56%。
第二步:1-(4-溴-3-氟苯基)-1,1-二氟-2-甲基丙烷-2-醇(35c)的制备
将化合物35b(719mg,2.42mmol)溶于干燥四氢呋喃(20mL)中,然后在冰水浴下缓慢滴加甲基溴化镁试剂(4.1mL,3M in THF),滴加完毕,反应体系缓慢恢复至室温搅拌反应过夜。将反应体系冷却至0℃,缓慢加入适量水淬灭反应,过滤,滤液用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物577mg,收率:84%。
第三步:1,1-二氟-1-(3-氟-4-(4,4,5,5-四甲基-1,3,2-二氧苯甲醛-2-基)苯基)-2-甲基丙烷-2-醇(35d)的制备
将化合物35c(577mg,2.04mmol)、联硼酸频那醇酯(1.0g,3.94mmol)、醋酸钾(600mg,6.12mmol)溶于1,4-二氧六环(20mL)中,置换氮气,加入Pd(dppf)Cl
第四步:叔丁基(1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(4-(1,1-二氟-2-羟基-2-甲基丙基)-2-氟苯基)吡啶-2-基)哌啶-4-基)氨基甲酸酯(35e)的制备
将化合物16b(100mg,0.20mmol)、化合物35d(132mg,0.40mmol)、K
第五步:6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(4-(1,1-二氟-2-羟基-2-甲基丙基)-2-氟苯基)异烟腈(35)的制备
将化合物35e(57mg,0.09mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(1mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物37mg,黄色固体,收率:77%。
LC-MS(ESI)m/z(M+H)
1
实施例36 6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3,6-二氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)异烟腈(36)的制备
第一步:5-溴-3,6-二氟-1H-吲唑(36a)的制备
将化合物9a(500mg,2.33mmol)溶于乙腈(7.5mL)中,依次加入冰乙酸(0.75mL)、1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟硼酸)盐(1.6g,4.66mmol),将反应体系加热至80℃搅拌反应5小时。将反应体系冷却至室温,向体系中加水淬灭反应,用乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经硅胶柱层析纯化得到标题化合物300mg,收率:55%。
第二步:1-(5-溴-3,6-二氟-1H-吲唑-1-基)-2-甲基丙烷-2-醇(36b)的制备
将化合物36a(300mg,1.29mmol)溶于乙腈(10mL)中,然后依次加入Cs
第三步:1-(3,6-二氟-5-(4,4,5,5-四甲基-1,3,2-二氧苯甲醛-2-基)-1H-吲唑-1-基)-2-甲基丙烷-2-醇(36c)的制备
将化合物36b(150mg,0.49mmol)、联硼酸频那醇酯(373mg,1.47mmol)、醋酸钾(144mg,1.47mmol)溶于1,4-二氧六环(5mL)中,置换氮气,加入Pd(dppf)Cl
第四步:叔丁基(1-(4-氰基-6-(4-氰基-3-氟苯基)-5-(3,6-二氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)吡啶-2-基)哌啶-4-基)氨基甲酸酯(36d)的制备
将化合物16b(107mg,0.21mmol)、化合物36c(74mg,0.21mmol)、K
第五步:6-(4-氨基哌啶-1-基)-2-(4-氰基-3-氟苯基)-3-(3,6-二氟-1-(2-羟基-2-甲基丙基)-1H-吲唑-5-基)异烟腈(36)的制备
将化合物36d(35mg,0.05mmol)溶于乙酸乙酯(1mL)中,然后加入盐酸-乙酸乙酯溶液(2mL,3M in EA),反应体系在室温下搅拌反应1小时。将反应体系减压浓缩,再加水稀释,用饱和碳酸氢钠溶液调节体系至pH值为8左右,乙酸乙酯萃取3次,有机相合并后经饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩滤液,粗品经薄层硅胶板纯化得到标题化合物23mg,收率:78%。
LC-MS(ESI)m/z(M+H)
1
生物学评价
以下试验例中所用的化合物1-36分别由实施例1-36合成方法制备获得。
试验例1:LSD1酶活性测试
1、试验目的
本试验利用时间分辨均相荧光技术(HTRF),采用两步法检测化合物对LSD1酶的抑制活性。第一步为LSD1酶、不同浓度化合物和用生物素标记的甲基化多肽底物(H3(1-21)K4 me1-biotin)室温孵育固定时间,第二步加入去甲基化多肽底物的抗体(Anti-H3K4 me0-Eu(K))和用XL-665标记的链霉素(SA-XL665)的检测试剂,继续室温孵育固定时间。用EnVision读取信号值Intensity(665nM)/Intensity(615nM)测定去甲基化多肽底物的含量,评价测试化合物对LSD1酶活的影响,同时利用抑制率计算测试化合物对LSD1酶的IC
2、试验材料
2.1、待测化合物:
阳性药:CC-90011,成都海博为药业有限公司,批号HBW-013-177-77;
受试药:化合物1-36。
2.2、试验试剂及仪器
LSD1,Active Motif;
DMSO,Sigma;
OptiPlate-384,PerkinElmer;
声波移液器,Labcyte;
酶标仪(EnVision),PerkinElmer;
离心机,Eppendorf。
3、试验方法
3.1、化合物配置
化合物10mM储存液配置:将化合物粉末溶解在100%DMSO中,分别配置成10mM的化合物储存液。
3.2、酶反应过程
(1)按照Active Motif(货号:31432)提供的LSD1酶学试剂盒,配置1×缓冲液。
(2)化合物溶液配置:化合物IC
(3)用1×反应溶液配制2×酶溶液。
(4)用1×反应溶液配制2×底物混合溶液。
(5)在各孔中加5μL的2×酶溶液,Min孔中加入5μL的1×反应溶液,1000rpm离心1min,室温孵育15min。
(6)反应板各孔中加入5μL的2×底物混合溶液,起始反应,1000rpm离心1min,室温孵育60min。
(7)向各孔加入10μL检测液,1000rpm离心1min,室温孵育60min。
(8)用EnVision读取信号值Intensity(665nM)/Intensity(615nM)。
3.3、数据分析
利用最大值、最小值和信号值计算抑制百分率(最大值:5μL的2×酶溶液+5μL的2×底物混合溶液+10nL100%DMSO+10μL检测液的荧光强度;最小值:5μL的1×反应溶液+5μL的2×底物混合溶液+10nL 100%DMSO+10μL检测液的荧光强度;信号值:5μL的2×酶溶液+5μL的2×底物混合溶液+10nL已稀释的待测化合物+10μL检测液的荧光强度),计算公式如下:
以浓度的对数值作为X轴,百分比抑制率为Y轴,采用分析软件Graphpad Prism 5的log(inhibitor)vs.response-Variable slop(Four parameters)公式拟合量效曲线,从而得出各个化合物对酶活性的IC
4、试验结果
本申请受试化合物和阳性对照对LSD1酶抑制活性见表1。
表1本申请受试化合物和阳性对照对LSD1酶抑制活性
从表1中示出的各化合物对LSD1酶抑制活性试验数据可知,本申请的化合物可具有明显的LSD1酶抑制活性。
试验例2:Kasumi-1细胞增殖抑制测试
1、试验原理
研究证实LSD1在包括小细胞肺癌(SCLC)和急性髓系白血病(AML)等多种癌症类型的发生过程中起关键作用。因此采用人急性髓系白血病(Kasumi-1)细胞系,利用Celltiter GLO assay kit测定不同药物浓度下细胞中ATP的含量,通过发光强度反应细胞活性,用抑制率计算不同化合物对Kasumi-1细胞的IC
2、试验材料
2.1、化合物:
阳性药:CC-90011,成都海博为药业有限公司,批号HBW-013-177-77;
受试药:化合物4、16、18和21。
2.2、试验试剂及仪器:
RPMI 1640,Gibico;
青霉素-链霉素:Penicillin-Streptomycin,Gibico;
胎牛血清:Fetal Bovine Serum(FBS),Gibico;
磷酸盐缓冲液:Phosphate Buffered Saline(PBS),Gibico;
二甲基亚砜:DMSO,Sigma;
CelltiterGlo assay Kit(CTG),Promega;
白色384孔细胞培养板,PerkinElmer;
震板器,QILINBE;
离心机,Eppendorf;
CO
显微镜,OLYMMPUS;
全自动细胞计数仪,Gibco;
384孔Source板,Echo Qualified 384-Well Polypropylene,LABCYTE;
Echo,LABCYTE;
多功能酶标仪(Envision),PerkinElmer。
2.3、试验细胞:
Kasumi-1细胞,购自ATCC。
3、试验方法
3.1、试验步骤
Day 1将细胞接种至384孔细胞培养板中,放到37℃、5%CO
Day 0将待测化合物用DMSO配制成10mM母液,以1μM起始,3倍稀释,10个浓度点,双复孔。取稀释化合物加入到384孔细胞培养板,放到37℃、5%CO
Day 7按照培养体系:Celltiter GLO=1:1的体积比,将Celltiter GLO加到384孔细胞培养板,在震板器上避光震荡3min,再室温避光放置30min,用多功能酶标化学发光模块读板。
3.2、数据分析
利用上述DMSO组(加入DMSO)、试验组(加入受试化合物或阳性对照溶液)、空白组(不对细胞进行处理)的发光强度计算抑制率,计算公式如下:
以浓度的对数值作为X轴,百分比抑制率为Y轴,采用分析软件Graphpad Prism 8的log(inhibitor)vs.response-Variable slop(Four parameters)公式拟合量效曲线,从而得出各个化合物对Kasumi-1增殖抑制活性的IC
4、试验结果
本申请受试化合物和阳性对照对Kasumi-1细胞生长抑制活性见表2。
表2本申请受试化合物和阳性对照对Kasumi-1细胞抑制活性
从表2中示出的各化合物对Kasumi-1细胞抑制活性试验数据可知,本申请的化合物可对Kasumi-1细胞的生长具有较强的抑制作用。
试验例3:NCI-H1417细胞增殖抑制测试
1、试验原理
研究证实LSD1在包括小细胞肺癌(SCLC)和急性髓系白血病(AML)等多种癌症类型的发生过程中起关键作用。因此采用人小细胞肺癌细胞(NCI-H1417)细胞系,利用Celltiter GLO assay kit测定不同药物浓度下细胞中ATP的含量,通过发光强度反应细胞活性。用抑制率计算不同化合物对NCI-H1417细胞的IC
2、试验材料
2.1、化合物:
阳性药:CC-90011,成都海博为药业有限公司,批号HBW-013-177-77;
受试药:化合物4、16、18和21。
2.2、试验试剂及仪器:
RPMI 1640,Gibico;
青霉素-链霉素:Penicillin-Streptomycin,Gibico;
胎牛血清:Fetal Bovine Serum(FBS),Gibico;
磷酸盐缓冲液:Phosphate Buffered Saline(PBS),Gibico;
二甲基亚砜:DMSO,Sigma;
CelltiterGlo assay Kit(CTG),Promega;
白色384孔细胞培养板,PerkinElmer;
震板器,QILINBE;
离心机,Eppendorf;
CO
显微镜,OLYMMPUS;
全自动细胞计数仪,Gibco;
384孔Source板,Echo Qualified 384-Well Polypropylene,LABCYTE;
Echo,LABCYTE;
多功能酶标仪(Envision),PerkinElmer。
2.3、试验细胞:
NCI-H1417细胞,购自ATCC。
3、试验方法
3.1、试验步骤
Day 1将细胞接种至384孔细胞培养板中,放到37℃、5%CO
Day 0将待测化合物用DMSO配制成10mM母液,以1μM起始,3倍稀释,10个浓度点,双复孔。取稀释化合物加入到384孔细胞培养板,放到37℃、5%CO
Day 7按照培养体系:Celltiter GLO=1:1的体积比,将Celltiter GLO加到384孔细胞培养板,在震板器上避光震荡3min,再室温避光放置30min,用多功能酶标化学发光模块读板。
3.2、数据分析
利用上述DMSO组(加入DMSO)、试验组(加入测试化合物和阳性对照溶液)、空白组(不对细胞进行处理)的发光强度计算抑制百分率,计算公式
以浓度的对数值作为X轴,百分比抑制率为Y轴,采用分析软件Graphpad Prism 8的log(inhibitor)vs.response-Variable slop(Four parameters)公式拟合量效曲线,从而得出各个化合物对NCI-H1417增殖抑制活性的IC
4、试验结果
本申请受试化合物和阳性对照对NCI-H1417细胞生长抑制活性见表3。
表3本申请受试化合物和阳性对照对NCI-H1417细胞抑制活性
从表3示出的各化合物对NCI-H1417细胞抑制活性试验数据可知,本申请的化合物可对NCI-H1417 细胞的生长具有较强的抑制作用。
试验例4:大鼠口服给药药代动力学的研究
1、试验原理
以SD大鼠为受试动物,应用LC-MS/MS法测定了大鼠澄清盒式口服本申请化合物后不同时刻血浆中的血药浓度。获取本申请的化合物在大鼠体内的药代动力学参数,研究其药代动力学特征。
2、试验材料
2.1、化合物:
阳性药:CC-90011,成都海博为药业有限公司,批号HBW-013-177-77;
受试药:化合物1、4、14-16、18、20、21和36。
2.2、试验仪器:
岛津LC-30A AB API4500串联质谱仪、真空采血管、采血针、滤纸、注射器等。
2.3、试验动物
SD大鼠,雄性,体重180-220g,每组3只。动物购回后饲养于动物房,适应期至少3天,检疫合格后用于试验。
3、试验方法
3.1、分组:根据表4随机分组,分组后,SD大鼠体重组间无统计学差异。
表4试验分组及给药方案
3.2、血样采集测定:
按照表4分别对各组大鼠灌胃给予相应的受试药或阳性药,给药前、给药后15min、30min、1h、2h、4h、6h、8h、24h,经眼眶采血取固定体积的血量,置于EDTA-K2抗凝管中,8000rpm,离心1min,分离血浆于离心管中,-20℃冰箱冷冻。
3.3、分析方法
取出-20℃保存的各时间点血浆,再加入乙腈,涡旋1500转2min后,离心15min(3500r/min),取固定体积的溶液上清液进行LC-MS/MS分析。
4、药代动力学参数计算
对受试化合物的药代动力学行为进行非房室模型拟合,并采用DAS3.31软件计算主要药代动力学参数(T
5、试验结果
表5实施例化合物药代动力学参数
从表5中的试验结果可以看出,与阳性组(CC-90011)比较,化合物1、化合物4、化合物14-16、化合物18、化合物20-21及化合物36给予组在血浆暴露量等方面优于CC-90011,说明本申请实施例1、4、14、15、16、18、20、21、36制备的化合物与CC-90011相比,药代动力学性质明显改善。因此,本申请的化合物可表现出良好的药代动力学性质。
试验例5:大鼠口服和静脉给药药代动力学研究
1、试验原理
以SD大鼠为受试动物,应用LC-MS/MS法测定大鼠口服和静脉给药后,本申请化合物在不同时刻血浆中的血药浓度,获取本申请的化合物在大鼠体内的药代动力学参数和生物利用度,研究其药代动力学特征。
2、试验材料
2.1、化合物:
阳性药:CC-90011,成都海博为药业有限公司,批号HBW-013-177-77;
受试药:化合物16;
2.2、试验仪器:
岛津LC-30A AB API4500串联质谱仪、真空采血管、采血针、滤纸、注射器等。
2.3、试验动物
SD大鼠,雄性,体重180-220g,每组3只。动物购回后饲养于动物房,适应期至少3天,检疫合格后用于试验。
3、试验方法
3.1、分组:根据表6随机分组,分组后,SD大鼠体重组间无统计学差异。
3.2、溶媒:p.o.:0.5%甲基纤维素钠水溶液
i.v.:5%DMSO+10%Solutol+85%Saline
表6试验分组及给药方案
3.2、血样采集测定:
按照表6分别对各组大鼠灌胃给予相应的受试药或阳性药,给药前、给药后5min、15min、30min、1h、2h、4h、6h、8h、10h、24h,经眼眶采血取固定体积的血量,置于EDTA-K2抗凝管中,8000rpm,离心1min,分离血浆于离心管中,-20℃冰箱冷冻。
3.3、分析方法
取出-20℃保存的各时间点血浆,再加入乙腈,涡旋1500转2min后,离心15min(3500r/min),取固定体积的溶液上清液进行LC-MS/MS分析。
4、药代动力学参数计算:
对受试化合物的药代动力学行为进行非房室模型拟合,并采用DAS3.31软件计算主要药代动力学参数(T
5、试验结果:
表7实施例化合物药代动力学参数
从表7中的试验结果可以看出,同等剂量下,化合物16口服和静脉给药的血浆暴露量、最大血药浓度、生物利用度等方面优于CC-90011,说明本申请实施例16制备的化合物与CC-90011相比,药代动力学性质明显改善。
试验例6:小鼠口服给药药代动力学研究
1、试验原理
以Balb/c小鼠为受试动物,应用LC-MS/MS法测定本申请化合物在不同溶媒条件下,小鼠给药后不同时刻血浆中的血药浓度。获取本申请的化合物在不同溶媒条件下的小鼠体内的药代动力学参数,研究其药代动力学特征。
2、试验材料
2.1、化合物:
阳性药:CC-90011,成都海博为药业有限公司,批号HBW-013-177-77;
受试药:化合物16。
2.2、试验仪器:
岛津LC-30A AB API4500串联质谱仪、真空采血管、采血针、滤纸、注射器等。
2.3、试验动物
Balb/c小鼠,雄性,体重18-22g,每组6只。动物购回后饲养于动物房,适应期至少3天,检疫合格后用于试验。
3、试验方法
3.1、分组:根据表8随机分组,分组后,Balb/c小鼠体重组间无统计学差异。
表8试验分组及给药方案
3.2、血样采集测定:
按照表8分别对各组小鼠灌胃给予相应受试药或阳性药,给药前、给药后15min、30min、1h、2h、4h、6h、8h、10h、24h,经眼眶采血取固定体积的血量,置于EDTA-K2抗凝管中,8000rpm,离心1min,分 离血浆于离心管中,-20℃冰箱冷冻。
3.3、分析方法
取出-20℃保存的各时间点血浆,再加入乙腈,涡旋1500转2min后,离心15min(3500r/min),取固定体积的溶液上清液进行LC-MS/MS分析。
4、药代动力学参数计算:
对受试化合物的药代动力学行为进行非房室模型拟合,并采用DAS3.31软件计算主要药代动力学参数(T
5、试验结果:
表9实施例化合物药代动力学参数
从表9中的试验结果可以看出,与阳性组(CC-90011)比较,化合物16在不同溶媒条件下的血浆暴露量、最大血药浓度等方面优于CC-90011,说明本申请实施例16制备的化合物与CC-90011相比,药代动力学性质明显改善,且在不同溶媒条件下均具有药物代谢动力学优势。
实验例7:肝微粒体稳定性测试实验
1、试验原理
肝微粒体体外温孵法是采用肝微粒体,辅以NADPH再生系统,在体外模拟生理环境条件进行代谢反应,经过一定时间的反应后,采用LC-MS/MS测定温孵液中原型药物和代谢产物,并对含量进行初步的分析的方法。研究药物在不同种属中肝微粒体稳定性对于了解药物在体内的变化过程至关重要。
2、试验材料
2.1、大鼠、小鼠、人、犬和猴不同种属肝微粒体
2.2、化合物:
阳性药:CC-90011,成都海博为药业有限公司,批号HBW-013-177-77;
受试药:化合物16和36。
试验阳性参照化合物:睾酮、双氯芬酸、普罗帕酮
2.3、试验仪器:
岛津LC-30A AB API4500串联质谱仪、电热恒温摇床、多管涡旋振荡器、各量程移液器。
3、试验方法
按照Ⅰ相代谢稳定性试剂盒说明书要求,配制肝微粒体孵育体系根据先后顺序,分别依次加入PBS缓冲液、肝微粒体、NADPH再生系统A液、NADPH再生系统B液的混合体系和待测物,在37℃孵育。每个样品平行3次,以不包含NADPH发生系统的样品为阴性对照,分别在0、5、15、30、45、60min后加入等体积预冷的乙腈终止反应,在采用LC-MS/MS测定温孵育液中原型药含量。
4、试验结果
表10化合物在不同种属中肝微粒体稳定性测试结果
上述实验结果表明,化合物16和36在人、大鼠、小鼠、犬和猴肝微粒体中具有较高的代谢稳定性。因此,本申请的化合物可表现出良好的成药性。
试验例8:体外hERG抑制活性测定实验
1、试验原理
快速激活的人延迟整流外向钾电流主要由hERG离子通道介导,参与人类心肌细胞复极化。药物阻断这一电流是导致临床上出现QT间期延长综合症,甚至急性心律紊乱乃至猝死的主要原因。利用全细胞膜片技术,在稳定表达hERG通道的HEK293细胞上检测化合物对hERG通道的阻断作用并且测定该化合物的半数抑制浓度IC
2、试验材料
2.1、试验细胞系:
hERG离子通道稳态表达的HEK293细胞
2.2、化合物:
阳性药:CC-90011,成都海博为药业有限公司,批号HBW-013-177-77;
受试药:化合物16。
2.3、试验仪器:
膜片钳仪器:PC-505B
微操控仪器:MP-225
拉制电极仪器:PC-10(Narishige,Japan)
3、试验方法
将细胞转移到灌流槽中,采用137mM NaCl、4mM KCl、1.8mM CaCl
4、试验结果
表11.化合物对hERG电流的IC
上述实验结果表明,与阳性相(CC-90011)比较,化合物16对hERG抑制IC
本申请的化合物比阳性对照具有更低的心脏毒性风险。
试验例9:Kasumi-1细胞皮下异种移植肿瘤模型中的体内药效试验
1、试验方法
1.1细胞培养
含20%胎牛血清(FBS)的RPMI 1640培养基,37℃、5%CO
1.2化合物:
溶媒:0.5%甲基纤维素水溶液;
受试药:化合物16和36。
1.3实验过程
CB17 SCID鼠,雌性,6-8周,体重约为18-22克,每只小鼠在右侧皮下接种0.1mL(1×10
TGI(%)的计算:TGI(%)=[1-(化合物当次平均瘤体积-化合物开始时平均瘤体积)/(溶媒组当次平均瘤体积-溶媒组开始时平均瘤体积)]×100%
各组小鼠的肿瘤体积和瘤重的变化情况在图1和图2中示出。
2、试验结果
表12.体内抑瘤实验数据
3、试验结论
上述试验结果表明,本申请化合物16和36在人急性原粒细胞白血病Kasumi-1细胞皮下异种移植肿瘤模型中展示出良好的体内药效,具有显著的抑瘤作用(TGI>60%),且受试动物对化合物16和36耐受性良好,给药后下小鼠体重未下降(如图3所示)。因此,本申请的化合物可表现出良好的体内抑瘤效果。
试验例10:HL-60细胞皮下异种移植肿瘤模型中的体内药效试验
1、试验方法
1.1细胞培养
含10%胎牛血清(FBS)的DMEM培养基,37℃、5%CO
1.2化合物:
阳性药:CC-90011,成都海博为药业有限公司,批号HBW-013-203-33;
溶媒:0.5%甲基纤维素水溶液;
受试药:化合物16。
1.3实验过程
Balb/C nu鼠,雌性,6-8周,体重约为18-22克,每只小鼠在右侧皮下接种0.1mL(1×10
TGI(%)的计算:TGI(%)=[1-(化合物当次平均瘤体积-化合物开始时平均瘤体积)/(溶媒组当次平均瘤体积-溶媒组开始时平均瘤体积)]×100%
各组小鼠的肿瘤体积的变化情况在图4中示出。
2、试验结果
表12.体内抑瘤实验数据
3、试验结论
上述试验结果表明,比起阳性化合物CC-90011,本申请的化合物在人原髓细胞白血病HL-60细胞皮下异种移植肿瘤模型中展示出良好的体内药效,具有显著的抑瘤作用(TGI>60%),且受试动物对本申请的化合物耐受性良好,给药后下小鼠体重未下降(如图5所示)。
要理解的是,上文的详述和附随实施例仅是示例性的,且不应被视为限制本申请的范围,该范围仅由所附权利要求及其对等物规定。本领域技术人员容易看出对所公开的实施方案的各种变动和修改。可以在不背离其精神和范围的情况下作出这样的变动和修改,包括但不限于与本申请的化学结构、取代基、衍生物、中间体、合成法、制剂和/或使用方法相关的那些。本文中引用的所有出版物、专利和专利申请出于各种目的全文经此引用并入本文。
- 一种四氢异喹啉类衍生物的制备方法及用途
- 一种氟硼二吡咯类衍生物及其制备方法和用途
- 一种吡唑并嘧啶衍生物及其制备方法和在药物制备中的用途
- 一类2位苯基取代噻唑衍生物及其制备方法和用途
- 叔胺类衍生物或其盐及其制备方法和用途
- 一类新的咪唑并1,2-a吡啶类衍生物、其制备方法及医药用途
- 一类含呋喃的吡啶联吡唑甲酰胺类衍生物及其制备方法和用途