一种环烯烃动态交联共聚物及其制备方法和用途
文献发布时间:2024-04-18 19:58:26
技术领域
本发明涉及材料领域,特别是涉及一种环烯烃动态交联共聚物及其制备方法和用途。
背景技术
开环易位聚合(ROMP)是利用环烯烃合成具有特殊结构和功能的聚合物的一种有效方法,由于Ru类催化剂引发ROMP反应具有很高的活性,分子量及其分布可以很好地控制,所以ROMP反应在制备性能可调控的新型功能性材料中具有重要作用。
环烯烃动态交联共聚物是通过引入动态共价键获得的材料,动态共价键的引入使得环烯烃线形共聚物形成动态交联网络,这一方面保留了环烯烃共聚物所具有的在包装、医用和光学领域的应用价值,另一方面赋予其耐化学性、结构稳定性和动态性。
而现存动态交联材料中大多通过小分子缩聚反应合成,以环烯烃开环聚合形成的功能性聚合物为基体的动态交联材料鲜有报道。
发明内容
鉴于以上所述现有技术的缺点,本发明的目的在于提供一种环烯烃共聚物及由其形成的具有动态功能的新材料。
为实现上述目的及其他相关目的,本发明是通过以下技术方案获得的。
本发明第一方面提供一种中间体化合物A,其结构式如式Ⅰ所示:
本发明第二方面提供如上所述中间体化合物A的制备方法,采用化合物a和化合物b接触发生开环反应,合成路线如下所示:
优选地,化合物b和化合物a的摩尔比至少为1。更优选地,所述化合物b是过量的,化合物b和化合物a的摩尔比为(1.05~1.5):1,例如可以为1.05:1、1.1:1、1.2:1、1.3:1、1.4:1或1.5:1。
优选地,反应中还采用有缚酸剂。优选地,所述缚酸剂选自三乙胺、三异丁基胺、吡啶中的一种或多种。这些化学物质具有缚酸的作用,可以促进反应的进行。
优选地,反应中还采用有反应介质,所述反应介质选自二氯甲烷、四氢呋喃中的一种或多种。
优选地,在室温下进行反应时,反应时间至少为10h。室温为20~25℃。反应时间的增加有利于转化率的提高。室温下,当反应时间小于10h,获得中间体化合物A的效率极低。具体地,申请人发现:当在室温条件下,8h是不能够完全转化为中间体化合物A的。
优选地,所述反应结束后还包括后处理步骤,所述后处理包括加热和提纯。更具体地,所述后处理包括第一次加热、提纯和第二次加热。更优选地,所述第一次加热为旋蒸,通过加热旋蒸去除反应介质,得到粗制中间体化合物A样品。更优选地,所述提纯为通过硅胶柱进行提纯,淋洗剂为正己烷和乙酸乙酯的混合物,使用淋洗剂的目的是除去多余的化合物b,三乙胺等杂质。更优选地,正己烷和乙酸乙酯体积比为(4.5~5.5):1。更优选地,第二次加热为旋蒸,通过加热旋蒸去除淋洗剂,得到纯中间体化合物A。
本发明第三方面公开一种环烯烃线形无规共聚物,所述环烯烃线形无规共聚物包含结构单元
优选地,结构单元
本发明第四方面提供如上所述环烯烃线形无规共聚物的制备方法,采用如上述所述的中间体化合物A和中间体化合物B进行无规共聚反应,合成路线图如下所示:
优选地,中间体化合物A和中间体化合物B的摩尔比(1~15):(5~10)。此处范围的选择考虑到了交联共聚物的性能,如果化合物A和B摩尔比高于3会导致材料又硬又脆,化合物A和B的摩尔比低于0.1会导致材料力学性能极弱,通过改变化合物B与A摩尔比从而调控材料的性能。更优选地,中间体化合物A和中间体化合物B的摩尔比2:(5~10)、3:(5~10)、4:(5~10)、5:(5~10);更优选地,中间体化合物A和中间体化合物B的摩尔比(2~5):(5~10);进一步优选为(2~5):(5~8)。
优选地,所述无规共聚反应中还采用催化剂,催化剂选自格拉布Grubbs催化剂第三代(G3)、格拉布Grubbs催化剂第二代(G2)中的一种或多种。使用催化剂可以使降冰片烯改性单体的双键断裂并以头尾相连的方式形成大分子
优选地,采用终止剂或封端剂终止所述无规共聚反应。优选地,所述终止剂或封端剂为乙烯基乙醚。乙烯基乙醚将通过与G3催化剂的[Ru]卡宾物质反应从而从聚合物主链上分离G3催化剂。
优选地,所述无规共聚反应中还采用有反应溶剂,所述反应溶剂选自四氢呋喃、二氯甲烷中的一种或多种。
优选地,无规共聚反应结束后还包括后处理步骤,所述后处理包括沉淀和干燥。更优选地,所述沉淀过程采用有沉淀剂。特别优选地,所述沉淀剂为无水冷甲醇、无水冰乙醚中的一种或多种,用于除去部分溶剂、反应单体和催化剂。更优选地,所述干燥的温度为40~70℃。
本发明第五方面公开了一种环烯烃动态交联共聚物,所述环烯烃动态交联共聚物采用如上所述的环烯烃线形无规共聚物与伯胺交联剂进行交联反应形成。
优选地,所述伯胺交联剂选自1,2-双(2-氨基乙氧基)乙烷、1,3-苯二甲胺、三(2-氨基乙基)胺中的一种或多种。
优选地,交联反应中还采用有有机溶剂。更优选地,所述有机溶剂为四氢呋喃、二氯甲烷、乙腈中的一种或多种。
优选地,伯胺交联剂与环烯烃线形无规共聚物所含结构单元
优选地,所述交联反应的温度为80~150℃。如交联反应的温度可以为80~90℃、90~100℃、100~110℃、110~120℃、120~130℃、130~140℃、140~150℃,在某个具体的实施例中为120℃。该温度范围用于挥发溶剂及环烯烃动态交联共聚物材料的固化。温度过低或时间短则会导致没有反应完成,温度高或者反应时间过长则会导致游离伯胺氧化,材料动态性变差。
优选地,所述交联反应的反应时间为6~8小时。如可以为6小时,7小时,8小时。
优选地,所述环烯烃动态交联共聚物在150℃的松弛温度下,松弛时间小于1050s;更优选地,所述环烯烃动态交联共聚物在150℃的松弛温度下,松弛时间小于1050s且大于150s。如可以为150~180s、160~190s、190~300s、300~500s、500~600s、600~700s、700~800s、800~900s、900~1000s、1000~1050s,在某个具体的实施例中为174s、565s、740s、956s、1042s。
优选地,所述环烯烃动态交联共聚物在160℃的松弛温度下,松弛时间小于850s;更优选地,所述环烯烃动态交联共聚物在160℃的松弛温度下,松弛时间小于850s且大于120s。如可以为120~130s、130~140s、140~200s、200~300s、300~400s、400~600s、600~700s、700~800s、800~850s,在某个具体的实施例中为134s、373s、628s、738s、824s。
优选地,所述环烯烃动态交联共聚物在170℃的松弛温度下,松弛时间小于650s。更优选地,所述环烯烃动态交联共聚物在170℃的松弛温度下,松弛时间小于650s且大于100s。如可以为100~110s、110~120s、120~200s、200~300s、250~350s、350~450s、450~550s、500~600s、600~650s,在某个具体的实施例中为113s、297s、558s、506s、630s。
本发明第七方面公开了所述环烯烃动态交联共聚物用于可回收加工材料的用途。
本申请提供的制备方法,通过改变两个降冰片烯改性单体的比例可以很容易地调控环烯烃动态交联共聚物的热性能,力学性能和动态性能。由此形成的环烯烃动态交联共聚物不仅可以很好保持环烯烃共聚物所具有的优异特性,同时表现出的可调节的力学及热机械性能、优异的热稳定性及耐化学性有助于推动环烯烃共聚物材料在更宽广领域的应用。此外乙烯胺酯动态键的存在使环烯烃动态交联共聚物材料网络拓扑可以在高温下进行重排,具有重新回收再加工的性质,延长了环烯烃动态交联共聚物的使用寿命。
附图说明
图1显示为实施例1产物5-降冰片烯-2-乙酰乙酸甲酯的核磁氢谱图。
图2显示为实施例1产物5-降冰片烯-2-乙酰乙酸甲酯的质谱图。
图3显示为实施例2获得的环烯烃线形无规共聚物的核磁氢谱图。
图4显示为实施例2获得的环烯烃线形无规共聚物的GPC流出曲线图。
图5显示为实施例4获得的环烯烃线形无规共聚物的核磁氢谱图。
图6显示为实施例4获得的环烯烃线形无规共聚物的GPC流出曲线图。
图7显示为实施例5获得的环烯烃线形无规共聚物的核磁氢谱图。
图8显示为实施例5获得的环烯烃线形无规共聚物的GPC流出曲线图。
图9显示为对比例2获得的环烯烃线形无规均聚物的核磁氢谱图。
图10显示为对比例2获得的环烯烃线形无规均聚物的GPC流出曲线图。
图11显示为实施例3~7及对比例1~2获得的环烯烃动态交联共聚物的红外光谱图。
图12显示为实施例3~7及对比例1~2获得的环烯烃动态交联共聚物的扫描量热图。
图13显示为实施例3~7及对比例1~2获得的环烯烃动态交联共聚物的热机械分析温度扫描图。
图14显示为实施例3~7获得的环烯烃动态交联共聚物的应力松弛图。
图15显示为实施例3~7获得的环烯烃动态交联共聚物的拉伸性能效果图。
具体实施方式
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。下列实施例中未注明具体条件的试验方法,通常按照常规条件,或者按照各制造商所建议的条件。
当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本技术领域技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本技术领域的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实现本发明。
在本申请中,申请人提供一种新型的具有商业利用价值的动态交联共聚物,其技术构思是:利用降冰片烯改性单体,通过开环易位聚合反应构建了官能化的环烯烃共聚物,并通过伯胺交联剂引入乙烯胺酯动态键形成了的环烯烃动态交联共聚物。
具体地,所述环烯烃共聚物为无规共聚物,环烯烃线形无规共聚物结构示意图如下所示:
其可以通过中间体化合物A和中间体化合物B通过聚合反应获得。且通过原料投料就可以调控最终环烯烃无规共聚物的数均分子量。优选地,所述环烯烃无规共聚物的数均分子量为1w~20w,如可以为1w~1.5w,1.5w~2w,1.6w~1.8w,2w~3w,3w~4w,4w~5w,5w~10w,11w~13w,13w~15w,15w~18w,18w~20w。分子量分布可以为1.1~1.5。如可以为1.1~1.2,1.2~1.3,1.3~1.4,1.4~1.5,1.17~1.29,1.1~1.3。
具体地,所述交联剂选自1,2-双(2-氨基乙氧基)乙烷、1,3-苯二甲胺、三(2-氨基乙基)胺中的一种或多种。
由于构建的官能化的环烯烃共聚物上形成有乙酰乙酸基团,能够与交联剂中的伯胺基团发生反应,形成具有动态性能的乙烯胺酯动态键的材料。
具体来说,在下述实施例和对比例中,环烯烃动态交联共聚物采用环烯烃线形无规共聚物与交联剂1,2-双(2-氨基乙氧基)乙烷进行交联反应形成的合成路线如下:
本发明中当中所采用中间体化合物B是通过商业购买获得。
具体地,在下述实施例和对比例中,中间体化合物B是采用化合物a和化合物c接触发生反应,合成路线如下所示:
实施例1
本实施例为中间体化合物A的合成例。
中间体化合物A的合成路线如下:
具体步骤如下:
1)取3g(1eq.)化合物a(97%)溶解在25ml无水二氯甲烷中;
2)向上述溶液中缓慢加入2.24g化合物b(1.1eq.,97%);
3)在室温条件下磁力搅拌上述溶液,并滴入100ul三乙胺(99.0%),室温条件下继续搅拌上述溶液12小时;
后处理步骤:
a)将反应后的溶液旋蒸除去二氯甲烷溶剂,得到粗制5-降冰片烯-2-乙酰乙酸甲酯样品;
b)将粗制5-降冰片烯-2-乙酰乙酸甲酯样品通过硅胶柱进行提纯,淋洗剂为正己烷:乙酸乙酯的体积比为5:1;
c)旋蒸除去淋洗剂得到纯5-降冰片烯-2-乙酰乙酸甲酯。
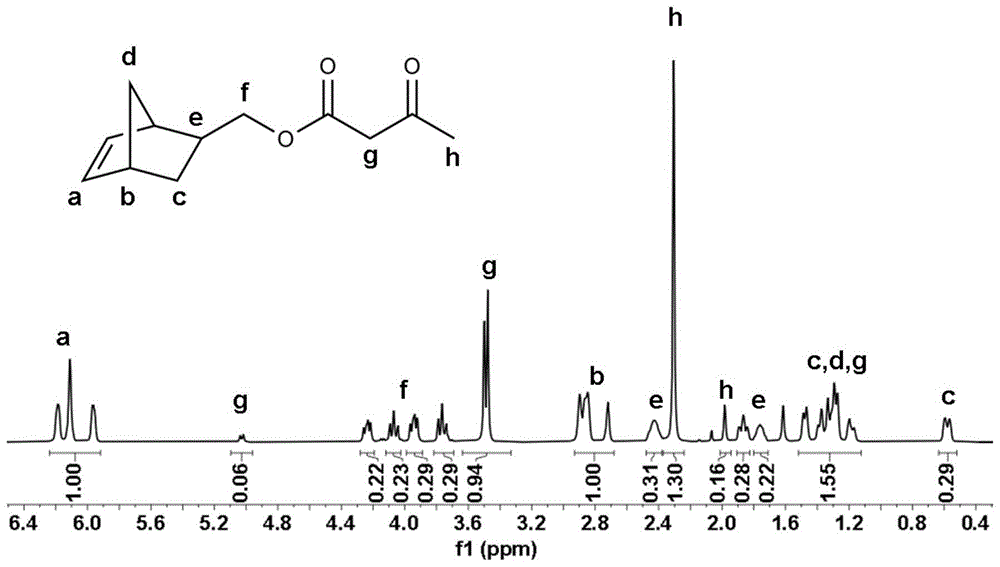
图1为本实施例中产物5-降冰片烯-2-乙酰乙酸甲酯的核磁氢谱图。由图1可以看出核磁氢谱峰位置及积分比率均与5-降冰片烯-2-乙酰乙酸甲酯理论结构相吻合,证明产物为5-降冰片烯-2-乙酰乙酸甲酯且没有杂质。
图2为本实施例中产物5-降冰片烯-2-乙酰乙酸甲酯的质谱图。由图2可以看出,质谱结果为208.1与理论分子量相吻合,为目标化合物。
经计算收率为91.2%。
实施例2
本实施例为中间体化合物A占比35%的环烯烃线形无规共聚物的制备。
具体的合成路线如下:
具体步骤如下:
1)在手套箱中取实施例1制备的单体中间体化合物A(262.4mg,1.26mmol),以及中间体化合物B(585.9mg,2.34mmol),加入到装有16ml无水四氢呋喃的反应瓶中;
2)另在手套箱中取Grubbs三代催化剂(63.7mg,0.072mmol)加入到4ml无水四氢呋喃中,使催化剂完全溶解在溶剂中;
3)然后在磁力搅拌下将催化剂溶液快速加入含有反应单体的溶液中,在室温条件下继续搅拌上述溶液1小时;
4)在上述溶液中加入2.4ml乙烯基乙醚进行封端反应半小时;
后处理步骤阶段:
a)待反应结束后,从手套箱中取出生成物溶液,将其逐滴滴入大量的无水冷甲醇(生成物溶液的20倍体积)中沉淀三次;
b)将获得的沉淀物在40℃真空干燥箱中干燥16小时,得到环烯烃线形无规共聚物。
图3显示为上述制备获得的环烯烃线形无规共聚物的核磁氢谱图。由图3中环烯烃线形无规共聚物中化合物A的乙酰乙酸甲酯基团所对应的化学位移3.4和4.9处特征峰积分面积之和为0.35可以看出,共聚物中化合物A占比为35%,表明成功合成了结构单元
图4显示为上述制备获得的环烯烃线形无规共聚物的GPC流出曲线图。由图4中可以看出上述制备获得的环烯烃线形无规共聚物的相对分子质量约为16621g/mol,分散度为1.235。
产品收率76.5%。
实施例3
本实施例为中间体化合物A占比35%,且交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比为1.8的环烯烃动态交联共聚物的制备。
具体步骤如下:
a)取500mg实施例2制备的环烯烃线形无规共聚物溶解在装有20ml无水四氢呋喃的聚四氟乙烯烧杯中;在上述溶液加入101.1mg1,2-双(2-氨基乙氧基)乙烷(98%)。
b)滴加完成后室温搅拌上述溶液30分钟;
c)将搅拌均匀后的混合溶液放入120℃烘箱6小时挥发溶剂及固化,继续将样品放入70℃真空烘箱过夜继续固化,并除去小分子水等,最终得到环烯烃动态交联共聚物。
将固化好的样品取出,切成小块,放置在平板热压机用18MPa的压力在180℃下热压30min,得到厚度约为1mm的矩形样片用于后续性能表征。
实施例4
本实施例为中间体化合物A占比20%,且交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比为1.8的环烯烃动态交联共聚物的制备。
具体步骤如下:
1)环烯烃线形无规共聚物的制备。
与实施例2步骤完全相同,除了采用实施例1制备的单体中间体化合物A的量为149.9mg,0.72mmol;中间化合物B的量为721.1mg,2.88mmol之外,其他步骤完全与实施例2相同。
图5显示为本实施例中制备获得的环烯烃线形无规共聚物的核磁氢谱图,由图5中环烯烃线形无规共聚物中化合物A的乙酰乙酸甲酯基团所对应的化学位移3.4和4.9处特征峰积分面积之和为0.20可以看出,共聚物中化合物A占比为20%,表明成功合成了结构单元
图6显示为本实施例中制备获得的环烯烃线形无规共聚物的GPC流出曲线图,由图6中可以看出本实施例中制备获得的环烯烃线形无规共聚物的相对分子质量为16742g/mol,分散度为1.175。
2)环烯烃动态交联共聚物的制备。
与实施例3步骤完全相同,除了采用的环烯烃线形无规共聚物为本实施例中1)制备得的环烯烃线形无规共聚物,且1,2-双(2-氨基乙氧基)乙烷(98%)的加入量为56.3mg(1.8eq.)之外,其他步骤完全与实施例3相同。
实施例5
本实施例为中间体化合物A占比50%,且交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比为1.8的环烯烃动态交联共聚物的制备。
具体步骤如下:
1)环烯烃线形无规共聚物的制备。
与实施例2步骤完全相同,除了采用实施例1制备的单体中间体化合物A的量为374.9mg,1.8mmol;中间化合物B的量为450.7mg,1.8mmol之外,其他步骤完全与实施例2相同。
产品收率82.9%。
图7显示为上述制备获得的环烯烃线形无规共聚物的核磁氢谱图,由图7中环烯烃线形无规共聚物中化合物A的乙酰乙酸甲酯基团所对应的化学位移3.4和4.9处特征峰积分面积之和为0.50可以看出,共聚物中化合物A占比为50%,表明成功合成了结构单元
图8显示为上述制备获得的环烯烃线形无规共聚物的GPC流出曲线图,由图8中可以看出本实施例中制备获得的环烯烃线形无规共聚物的相对分子质量为18183g/mol,分散度为1.220。
2)环烯烃动态交联共聚物的制备。
与实施例3步骤完全相同,除了采用的环烯烃线形无规共聚物为本实施例中1)制备得的环烯烃线形无规共聚物,且1,2-双(2-氨基乙氧基)乙烷(98%)的加入量为148.4mg(1.8eq.)之外,其他步骤完全与实施例3相同。
实施例6
本实施例为中间体化合物A占比35%,且交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比为1.6的环烯烃动态交联共聚物的制备。
具体步骤如下:
与实施例3步骤完全相同,除了步骤a)中加入1,2-双(2-氨基乙氧基)乙烷(98%)的量为89.9mg之外,其他步骤完全与实施例3相同。
实施例7
本实施例为中间体化合物A占比35%,且交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比为2.0的环烯烃动态交联共聚物的制备。
具体步骤如下:
与实施例3步骤完全相同,除了加入步骤a)中1,2-双(2-氨基乙氧基)乙烷(98%)的量为112.3mg之外,其他步骤完全与实施例3相同。
对比例1
本实施例为中间体化合物A占比35%,且交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比为1.4的环烯烃动态交联共聚物的制备。
具体步骤如下:
与实施例3步骤完全相同,除了加入步骤a)中1,2-双(2-氨基乙氧基)乙烷(98%)的量为78.6mg之外,其他步骤完全与实施例3相同。
对比例2
本实施例为中间体化合物A占比100%,且交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比为1.8的环烯烃动态交联均聚物的制备。
具体步骤如下:
1)环烯烃线形无规均聚物的制备。
与实施例2步骤完全相同,除了采用实施例1制备的单体中间体化合物A的量为749.7mg,3.6mmol;且不加入中间化合物B之外,其他步骤完全与实施例2相同。
图9显示为本实施例中制备获得的环烯烃线形无规均聚物的核磁氢谱图,由图9中环烯烃线形无规均聚物中化合物A的乙酰乙酸甲酯基团所对应的化学位移3.4和4.9处特征峰积分面积之和为1.00可以看出,均聚物中化合物A占比为100%,表明成功合成了结构单元
图10显示为本实施例中制备获得的环烯烃线形无规均聚物的GPC流出曲线图,由图10中可以看出本实施例中制备获得的环烯烃线形无规均聚物的相对分子质量为16765g/mol,分散度为1.282。
2)环烯烃动态交联均聚物的制备。
与实施例3步骤完全相同,除了采用的环烯烃线形无规均聚物为本对比例中1)制备得的环烯烃线形无规均聚物,且1,2-双(2-氨基乙氧基)乙烷(98%)的加入量为326.8mg之外,其他步骤完全与实施例3相同。
实施例3~7及对比例1~2获得的环烯烃动态交联共聚物的性能测试结果如下:
红外光谱:红外光谱使用FTIR(Spectrum 100)在室温条件下测试,波数范围为650~4000cm
具体的测试结果如图11所示。由图11左图中的实施例3-5及对比例2的红外测试效果图可以看出,当交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比一定,都为1.8时,环烯烃动态交联共聚物在波数为1730cm
由图11右图中对应实施例3、实施例6-7及对比例1的红外测试效果图可以看出,当中间体化合物A占比一定为35%时,环烯烃动态交联共聚物在波数为3200~3500cm
扫描量热:扫描量热实验在DSC(TA Q2000)上测试,在氮气气氛下以10℃/min的加热/冷却速率进行了从-60℃到120℃加热/冷却/加热三个循环,使用第三个加热循环曲线获得玻璃化转变温度。
测试结果如图12所示,由图12左图中对应的实施例3(化合物A含量35%)、实施例4(化合物A含量20%)、实施例5(化合物A含量50%)及对比例2(化合物A含量100%)的DSC曲线可以看出,当交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比一定,都为1.8时,环烯烃动态交联共聚物的玻璃化转变温度随着化合物A和B摩尔比的增加而升高。由图12右图对应的实施例3、实施例6-7及对比例1的DSC曲线可以看出,当中间体化合物A占比一定为35%时,环烯烃动态交联共聚物的玻璃化转变温度随着交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比的增加而降低。
温度扫描:温度扫描实验在DMA(TA Q850)上测试,裁取尺寸为1.0mm(T)×5.5mm(W)×15mm(L)矩形样条,温度扫描范围从-60℃至120℃,升温速率为5℃/min,振荡应变为1%。
测试结果如图13所示,由图13左图中对应的DMA曲线可以看出,实施例3(化合物A含量35%)、实施例4(化合物A含量20%)、实施例5(化合物A含量50%)及对比例2(化合物A含量100%)获得的环烯烃动态交联共聚物的平台模量分别是实施例3获得的环烯烃动态交联共聚物的平台模量的0.48倍、1.41倍、2.92倍;图13右图中对应的实施例6、实施例7、对比例1获得的环烯烃动态交联共聚物的DMA曲线可以看出,平台模量分别是实施例3获得的环烯烃动态交联共聚物的平台模量的1.09倍、0.82倍、1.12倍。这表明环烯烃动态交联共聚物的平台模量随着交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比的增加而降低。
应力松弛:应力松弛实验在旋转流变仪(TA ARES-G2)上使用8mm平行板测试,分别在150℃至170℃每间隔10℃测试。测试得到的松弛模量G(t)用初始松弛模量G
具体结果如图14所示,图14为归一化后的应力松弛图,由图上可以看出在相同温度下实施例3获得的环烯烃动态交联共聚物的动态交换速度远大于实施例4和实施例5的环烯烃动态交联共聚物,这表明,在交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比一定时,中间体化合物A占比为35%时,获得的环烯烃动态交联共聚物的动态交换速度最快,动态性最佳。实施例3、实施例6和实施例7可以看出,当中间体化合物A占比一定为35%时,环烯烃动态交联共聚物的动态交换速度随着交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比的增加而加快。具体对应温度的松弛时间如表1所示。
表1
拉伸性能:在室温下(25℃)在万能试验机(Instron 3343)上对1.0mm(T)×2mm(W)×35mm(L)的哑铃型样条进行拉伸应力-应变曲线测试,单轴拉伸速率为5mm/min。
测试结果如图15所示,曲线结果表明实施例3(化合物A含量35%)获得的环烯烃动态交联共聚物的延展性高于实施例4(化合物A含量20%)和实施例5(化合物A含量50%)的环烯烃动态交联共聚物,实施例3(摩尔比1.8)、实施例6(摩尔比1.6)和实施例7(摩尔比2.0)获得的环烯烃动态交联共聚物的延展性随着交联剂中伯胺基团与中间体化合物A中乙酰乙酸基团摩尔比的增加而增加。由于对比例1中获得的最终产物动态性差热压后愈合效果不理想、对比例2中获得的最终产物材料又硬又脆,导致两对比例获得的环烯烃动态交联共聚物机械性能极差且难以表征。
在本申请中,申请人利用如本申请所述的结构式为式Ⅰ、式Ⅱ的降冰片烯改性单体通过开环易位聚合反应构建了官能化的环烯烃共聚物,并通过伯胺交联剂引入乙烯胺酯动态键形成了的环烯烃动态交联共聚物。通过改变两个降冰片烯改性单体的比例可以很容易地调控环烯烃动态交联共聚物的热性能,力学性能和动态性能,并且其热稳定性、耐化学性相比传统环烯烃共聚物大大提升,此外乙烯胺酯动态键的存在使环烯烃动态交联共聚物可进行回收重加工,延长了环烯烃动态交联共聚物的使用寿命。
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
- 一种环烯烃共聚物的制备方法
- 由再循环交联的聚烯烃泡沫材料制备共挤出交联的多层聚烯烃泡沫结构体的方法
- 一种阻燃交联聚烯烃绝缘电缆及其制备方法
- 一种自交联型氯乙烯共聚物乳液及其制备方法
- 一种可交联环烯烃共聚物及其制备方法和应用
- 一种可交联热塑性环烯烃共聚物及其制备方法和应用