一种Fe基MOFs玻璃的制备方法及其在高级氧化中的应用
文献发布时间:2024-04-18 19:58:26
技术领域
本发明属于功能化催化剂用于高级氧化技术处理废水中难降解有机污染物的研究领域,具体涉及一种Fe基MOFs玻璃(FeMOFs glass)及其制备方法与应用。
背景技术
在环境催化等领域中催化剂通过复杂的活性位点来催化氧化剂去除有机污染物。在这个过程中催化剂通过质子耦合电子转移(Proton Coupled Electron Transfer,PCET)反应可实现对污染物去除过程巨大速率加速和无与伦比的选择性,被视为发展更绿色、更高效清洁生产的关键技术。
在污染物的转化去除过程中质子和电子转移的相互依赖带来了丰富的反应机理。通过PCET,电子和质子可以同时发生转移且使反应活性中间体只经历单一的反应过渡态,避免了分步降解途径中所经历的高能量中间体的形成,从而实现污染物去除过程反应热力学的最优化。因此,适当的电子转移和质子转移协同对于PCET发挥着核心作用。关于单独的电子转移过程的调控已有许多深入研究,例如,轨道杂化增强金属性来促进电子转移效率,内建电场促进界面电子定向转移等。然而,关于质子转移过程的研究尚处于起步阶段。有研究人员发现,(100)取向双钙钛矿相氧化物的去质子化过程最容易发生,从而使该取向的电化学测试和计算结果中观察到更好的质子电子耦合转移过程。还有学者构建了垂直异质结构来协同氢溢流和去质子化过程。然而,短寿命活性氢的传质效率差固有缺点尚无法克服。
已有研究报道设计了高比表面积的MOFs材料用于催化降解有机污染物,可见光激发产生的光电子在多孔MOFs结构中快速迁移,高比表面积的多空结构使吸附和降解交替进行,从而实现高催化性能。但是,这种明确的晶体构型使活性位点和吸附位点等规律性排列和交替组合,无法形成不平衡配位和电荷分布。这种结构在催化反应界面上无法同步调控电子转移和质子转移协同,从而导致电子和质子转移无法协同反应的缺陷(Li P,Kim S,Jin J,et al.Efficient photodegradation of volatile organic compounds by iron-based metal-organic frameworks with high adsorption capacity[J].AppliedCatalysis B:Environmental,2020,263:118284.)。
发明内容
金属有机骨架(MOFs)是结构有序的热门晶体材料,不过随着研究的深入,越来越多的人认识到这种“软晶体”材料家族其他不寻常的物理性质也值得重视,如缺陷、框架柔性和无序性。在较高的温度和压力下,沸石咪唑酯框架(ZIF)能与结构类似的沸石一样保持微孔结构。如果温度和压力进一步升高,ZIF最终会发生相变并熔化,再经淬火会形成一类新的玻璃材料,包含非晶态、类似SiO
本发明的目的在于提供一种Fe基MOFs玻璃(FeMOFs glass)的制备方法及其在高级氧化中的应用,通过将ZIF-62与Fe-N配位前驱体混合后急速升温至熔融态,再快速冷却淬火得到具有大孔-中孔-微孔的多孔FeMOFs glass,用于高级氧化技术来去除水中有机污染物。本发明特征是采用熔融玻璃化法,将混合物急速加热至熔融态,而没有达到分解态,从而使材料的晶体结构无序化,得到长程无序、短程有序的,并具有多级孔的MOFs玻璃材料。其具有以下方面的特点和作用:(1)由于长程无序、短程有序的微结构,使得依托于MOFs玻璃的Fe-N配位作为活性中心可以发生电子转移行为,并且得到Fe-N-C极化电场促进的电子传输并诱导界面带电的质子迁移,从而调控质子耦合电子转移反应;(2)由于FeMOFsglass整体熔融淬火后会形成一种宏观形貌结构,具有较好的机械强度,因此在废水中可以便于回收再利用、易于工程化;(3)由于玻璃化MOFs孔道的限域效应,使得污染物、氧化剂、活性物种的传质速率加快,使得污染物去除效率和氧化剂利用率提高,降低了成本。最重要是MOFs玻璃中1.9nm以下的纳米孔道限域作用,可以使得质子约束在双电层的亥姆霍兹层内加速转移,从而极大地促进质子耦合电子转移反应,对有机污染物表现出超高的降解效率。
为了达到上述目的,本发明采用如下技术方案。
一种Fe基MOFs玻璃的制备方法,包括如下步骤:
(1)ZIF-62的制备:将金属盐Zn(NO
(2)ZIF-62与Fe-N配位前驱体混合物的制备:将ZIF-62和Fe-N配位前驱体在研钵中混合均匀,得到预混合前体;
(3)FeMOFs glass的制备:在步骤(2)得到的预混合前体置于惰性气体氛围下保持20-60min排除氧气,然后以10-20℃/min升温速率急速升温至430-500℃,使其熔融,保持20-60min后,以150-250℃/h的降温速率迅速冷却使其玻璃化,得到以Fe-N为配位中心的类玻璃态FeMOFs glass催化剂。
进一步地,步骤(1)中,所述咪唑的投加量与苯丙咪唑的投加量摩尔比为4:1-1:2;Zn(NO
进一步地,步骤(2)中,所述Fe-N配位前驱体为ZIF-8(Zn,Fe)、酞菁铁、Fe掺杂g-C
进一步地,步骤(2)中,所述ZIF-62与Fe-N配位前驱体质量比为20:1-1:2。
进一步地,本发明提供所述合成方法制备的FeMOFs glass,具有长程无序和短程有序的配位结构、纳米孔道限域效应以及宏观形貌结构。
本发明还提供所述FeMOFs glass应用于高级氧化技术中去除废水中有机污染物,将FeMOFs glass加入含有有机污染物的废水中,再加入氧化剂,置于恒温振荡培养箱中振荡反应,反应温度为20-40℃。
进一步地,所述有机污染物为内分泌干扰物类的新污染物,比如给电子酚类污染物,包括双酚A(BPA)、罗丹明B、甲基橙等。
进一步地,所述氧化剂为过二硫酸盐、过一硫酸盐或H
进一步地,所述有机污染物和氧化剂的摩尔比为10:1-1:200,FeMOFs glass在废水中的浓度为为0.1g/L-5.0g/L,有机污染物在废水中的浓度为0.5mg/L-50mg/L。
本发明针对废水深度处理过程中新污染物难降解去除的问题,公开了一种Fe基MOFs玻璃的制备方法及其应用于高级氧化技术处理废水中的有机污染物,本发明提供的一种Fe基MOFs玻璃的制备方法具有以下优点和增益效果:
(1)本发明首次将ZIF-62与Fe-N配位的前驱体结合来构建一种长程无序、短程有序结构的类玻璃MOFs催化剂。所述催化剂的长程无序状态使其拓扑结构非常态,具有独特的非晶态结构性质。在此基础上的短程有序状态使其局部配位结构之间可以形成极化电场,能够促进局部电子定向传输,从而加速活性位点的Fe循环,提高催化性能与选择性。由于长程无序、短程有序的微结构,使得依托于MOFs玻璃的Fe-N配位作为活性中心可以发生电子转移行为,并且得到Fe-N-C极化电场促进的电子传输并诱导界面带电的质子迁移,从而调控质子耦合电子转移反应。
(2)本发明提供的FeMOFs glass由于其玻璃化具有小于1.9nm的孔道结构,因此可以将有机污染物、氧化剂、活性物种约束在孔道内,加速传质、增大反应碰撞几率,从而加速反应过程。同时,纳米孔道可以防止废水中大分子杂质干扰催化降解有机污染物,避免氧化剂和活性物种的浪费,从而降低成本。此外,小于1.9nm的孔道在局部会使界面双电层结构重叠,从而使双电层内氢离子富集,构建一种酸性微环境,可以在中性和碱性溶液中保持和酸性溶液一样好的反应效果。最重要的是FeMOFs glass中1.9nm以下的纳米孔道限域作用,可以使得质子约束在双电层的亥姆霍兹层内加速转移,从而极大促进质子耦合电子转移反应。
(3)本发明提供的一种FeMOFs glass的制备方法可以直接得到一种具有宏观形貌结构的催化剂,具有较好的机械强度和大孔-中孔-微孔的多级孔结构,不需要再进行催化剂负载、成形等处理,可以直接用于废水处理工程中,便于回收再利用和工程化。
(4)本发明提出的一种FeMOFs glass的制备方法设备简单,反应条件温和,过程易于控制,无需添加有毒有害试剂,便于催化剂的产业化和工程应用。
附图说明
图1为本发明提供的FeMOFs glass制备原理图。
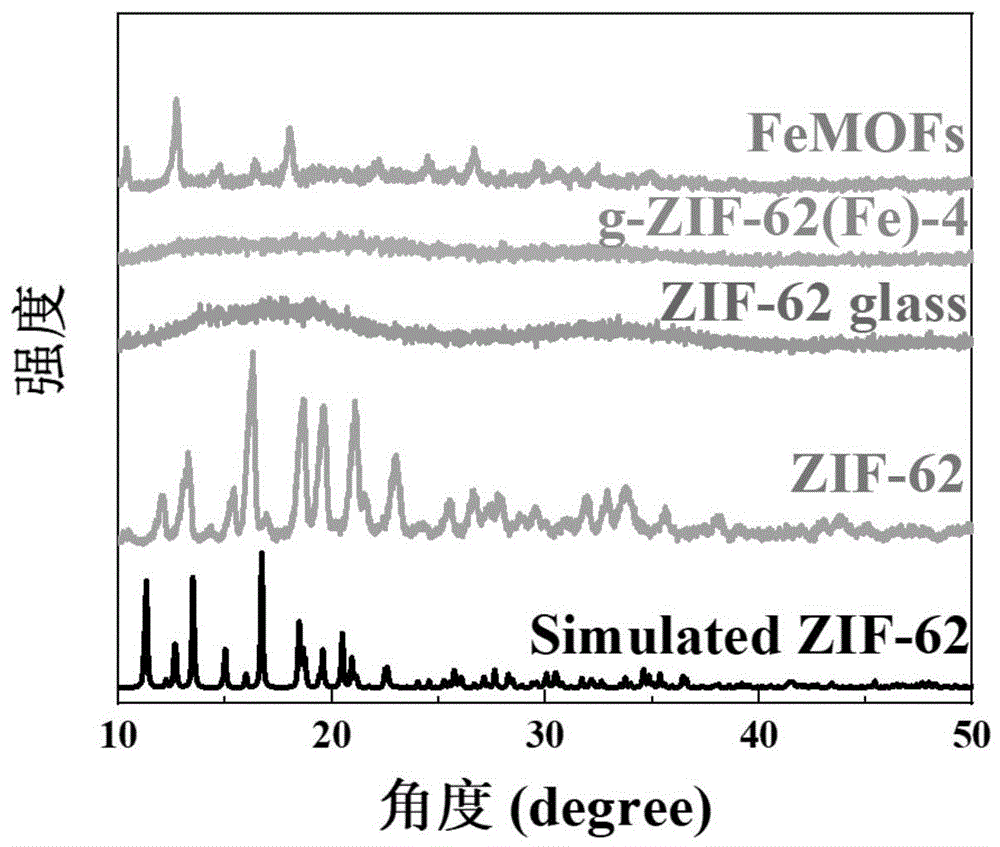
图2为FeMOFs glass的X射线晶体衍射图。
图3为实施例1中FeMOFs glass催化剂ZIF-62(Fe)-4的扫描电子显微镜图。
图4为本发明提供的FeMOFs glass催化剂的傅里叶变换红外光谱图。
图5为FeMOFs glass催化剂ZIF-62(Fe)-4的长程无序、短程有序的结构图。
图6为FeMOFs glass催化剂ZIF-62(Fe)-4的Fe-N配位中心电势分布图。
图7为FeMOFs glass催化剂ZIF-62(Fe)-4电子转移性能的电化学阻抗图。
图8为FeMOFs glass催化剂ZIF-62(Fe)-4的孔径分布图。
图9为FeMOFs glass催化剂ZIF-62(Fe)-4的紫外-可见-近红外光吸收谱图。
图10为利用不同Fe-N配位前体制备FeMOFs glass催化剂用于降解污染物效率图。
图11为FeMOFs glass催化剂ZIF-62(Fe)和对比例1产物的外层电子轨道分布图。
图12为FeMOFs glass和几种对比例产物的X射线晶体衍射图。
图13为FeMOFs glass催化剂ZIF-62(Fe)的催化降解污染物效率图。
图14为FeMOFs glass催化剂ZIF-62(Fe)-4的活性物种掩蔽实验图。
图15为FeMOFs glass催化剂ZIF-62(Fe)-4的活性物种定量检测图。
图16为FeMOFs glass催化剂ZIF-62(Fe)-4的在太阳光照下催化降解污染物的增益效果图。
图17为FeMOFs glass催化剂ZIF-62(Fe)-4的催化降解污染物的热力学能垒图。
图18为FeMOFs glass催化剂ZIF-62(Fe)-4的宏观形貌图。
图19为FeMOFs glass催化剂ZIF-62(Fe)-4的吸附量和比表面积。
图20为FeMOFs glass催化剂ZIF-62(Fe)催化降解不同污染物的效果图。
具体实施方式
本发明通过提供一种Fe基MOFs玻璃(FeMOFs glass)的制备方法及其在高级氧化中的应用,解决了现有催化剂催化效率差、活性物种选择性差、电子转移效率低、无稳定的宏观形貌结构、在废水中难以回收、无小于2nm的孔道、在中性和碱性溶液中反应效果差等技术缺陷。本发明制备的FeMOFs glass,具有长程无序、短程有序结构,还具有小于1.9nm的孔道和稳定的宏观形貌。
为解决上述技术缺陷,本发明实施例的主要思路是:
本发明实施例提供一种FeMOFs glass的制备方法,包括以下步骤:
提供一Fe前驱体,所述Fe前驱体包括Fe-N配位的材料;
将所述Fe-N配位的前驱体与ZIF-62按20:1-1:2的质量比混合,获得混合物;将所得混合物在惰性气氛中以10-20℃/min的升温速率急速加热至430-500℃,保持20-60min,然后以150-250℃/min的降温速率快速冷却,获得FeMOFs glass。这种制备方法的原理如图1所示,将ZIF-62与Fe-N配位前驱体结合,经急速高温熔融后冷却淬火,得到类玻璃态的FeMOFs glass。
本发明实施例将ZIF-62和Fe-N配位前驱体混合后急速升温至熔融态,再经快速冷却后得到类玻璃态的FeMOFs。结合可以获得惰性玻璃类的多孔ZIF-62材料和高催化活性Fe-N配位的材料,最终获得具有宏观形貌结构、整体惰性稳定的玻璃态、高催化活性Fe-N中心的催化剂。对制备的催化剂进行结构、形貌和性质表征,并以双酚A为模型污染物评估其催化去除水中有机污染物的性能。本发明实施例的增益效果归因于FeMOFs glass中长程无序、短程有序的玻璃化结构,以及小于1.9nm的纳米孔道限域效应和稳定的宏观形貌结构。
本发明实施例提供的FeMOFs glass可催化氧化剂降解水中有机污染物。该催化剂对双酚A的降解速率达到了96%,对其他污染物的降解效果也均可达到预期。
为了让本发明上述和其他目的、特征、优势和技术可行性能更明显易懂,下文结合附图对本发明的实施例进行详细说明,但本发明的实施和保护不限于此。
实施例1
本实施例提供了一种FeMOFs glass的制备方法,主要步骤如下:
(1)ZIF-62的制备:将3mM Zn(NO
(2)ZIF-8(Zn,Fe)的制备:将4g 2-甲基咪唑溶于60mL甲醇中,记为第一溶液,将1.6g Zn(NO
(3)预混合前体的制备:将ZIF-62和ZIF-8(Zn,Fe)混合物在研钵中混合均匀,质量比1:1,得到预混合前体。
(4)FeMOFs glass的制备:将步骤(3)得到的预混合前体置于惰性气体氛围下保持30min排出氧气,然后以10℃/min的速率急速升温至500℃,使其熔融,保持30min后,以200℃/h的降温速率迅速冷却,得到玻璃类FeMOFs材料,记为g-ZIF-62(Fe)-4。其晶体结构如图2所示,g-ZIF-62(Fe)-4呈现非晶态,表明g-ZIF-62(Fe)-4是一种长程无序的类玻璃态结构。g-ZIF-62(Fe)-4的扫描电镜如图3所示,表明其是一种无规则的非晶体结构。
实施例2
(1)ZIF-62的制备:将5mM Zn(NO
(2)ZIF-8(Zn,Fe)的制备:将4g 2-甲基咪唑溶于60mL甲醇中,记为第一溶液,将1.6g Zn(NO
(3)预混合前体的制备:将ZIF-62和ZIF-8(Zn,Fe)混合物在研钵中混合均匀,质量比1:2,得到预混合前体。
(4)FeMOFs glass的制备:在步骤(3)得到的预混合前体置于惰性气体氛围下保持50min排出氧气,然后以10℃/min的速率急速升温至500℃,使其熔融。保持30min后,以200℃/h的降温速率迅速冷却,得到玻璃类FeMOFs glass材料。记为g-ZIF-62(Fe)-5。其配体结构如图4所示,由Fe-N、C=N、甲基咪唑、咪唑、苯丙咪唑配位键共同构成FeMOFs glass的配位结构,表明FeMOFs glass仍是一种短程有序的配位结构。其微观结构如图5所示,以Fe-N
实施例3
(1)ZIF-62的制备:将5mM Zn(NO
(2)ZIF-62与含Fe-N配位混合物的制备:将ZIF-62分别与酞箐铁在研钵中混合均匀,质量比5:1,得到预混合前体。
(3)FeMOFs glass的制备:在步骤(2)得到的预混合前体置于惰性气体氛围下保持50min排出氧气,然后以10℃/min的速率急速升温至500℃,使其熔融。保持30min后,以180℃/h的降温速率迅速冷却,得到FeMOFs glass材料,记为g-ZIF-62(Fe)(酞菁铁)。
实施例4
(1)ZIF-62的制备:将5mM Zn(NO
(2)Fe掺杂g-C
(3)ZIF-62与含Fe-N配位混合物的制备:将ZIF-62分别与Fe掺杂g-C
(4)FeMOFs glass的制备:在步骤(3)得到的预混合前体置于惰性气体氛围下保持50min排出氧气,然后以10℃/min的速率急速升温至500℃,使其熔融。保持30min后,以180℃/h的降温速率迅速冷却,得到FeMOFs glass材料,记为g-ZIF-62(Fe)(Fe掺杂g-C
实施例5
(1)ZIF-62的制备:将5mM Zn(NO
(2)Fe/N掺杂石墨烯、Fe/N掺杂碳纳米管的制备:将Fe盐、氮源和石墨烯或碳纳米管在研钵中均匀混合,置于马弗炉以2-10℃/min的升温速率到450-550℃加热1-3h,得到Fe/N掺杂石墨烯或Fe/N掺杂碳纳米管。其中Fe盐可以是FeCl
(3)ZIF-62与含Fe-N配位混合物的制备:将ZIF-62分别与Fe/N掺杂石墨烯、Fe/N掺杂碳纳米管在研钵中混合均匀,质量比5:1,得到预混合前体。
(4)FeMOFs glass的制备:在步骤(3)得到的预混合前体置于惰性气体氛围下保持50min排出氧气,然后以10℃/min的速率急速升温至500℃,使其熔融。保持30min后,以180℃/h的降温速率迅速冷却,得到FeMOFs glass材料。依次分别记为g-ZIF-62(Fe)(Fe/N掺杂石墨烯)、g-ZIF-62(Fe)(Fe/N掺杂碳纳米管)。
实施例6
(1)ZIF-62的制备:将5mM Zn(NO
(2)Fe/N掺杂生物质碳的制备:将Fe盐、氮源和生物质在研钵中均匀混合,置于马弗炉以2-10℃/min的升温速率到450-550℃加热1-3h,得到Fe/N掺杂生物质碳。其中Fe盐可以是FeCl
(3)ZIF-62与含Fe-N配位混合物的制备:将ZIF-62分别与Fe/N掺杂生物质、Fe/N掺杂生物质碳在研钵中混合均匀,质量比5:1,得到预混合前体。
(4)FeMOFs glass的制备:在步骤(3)得到的预混合前体置于惰性气体氛围下保持50min排出氧气,然后以10℃/min的速率急速升温至500℃,使其熔融。保持30min后,以180℃/h的降温速率迅速冷却,得到FeMOFs glass材料,记为g-ZIF-62(Fe)(Fe/N掺杂生物质碳)。
实施例1~6所制备产物催化去除水中有机污染物的效果如图10所示,在30min内都可以去除80%以上的双酚A,表明ZIF-8(Zn,Fe)、酞箐铁、Fe掺杂g-C
实施例7
本实施例提供了一种FeMOFs glass的制备方法,主要步骤如下:
(1)ZIF-62的制备:将1mM Zn(NO
(2)ZIF-8(Zn,Fe)的制备:将4g 2-甲基咪唑溶于60mL甲醇中,记为第一溶液,将1.6g Zn(NO
(3)预混合前体的制备:将ZIF-62和ZIF-8(Zn,Fe)混合物在研钵中混合均匀,质量比20:1,得到预混合前体。
(4)FeMOFs glass的制备:将步骤(3)得到的预混合前体置于惰性气体氛围下保持50min排出氧气,然后以20℃/min的速率急速升温至500℃,使其熔融,保持30min后,以150℃/h的降温速率迅速冷却,得到玻璃类FeMOFs材料。
实施例8
本实施例提供了一种FeMOFs glass的制备方法,主要步骤如下:
(1)ZIF-62的制备:将3mM Zn(NO
(2)ZIF-8(Zn,Fe)的制备:将4g 2-甲基咪唑溶于60mL甲醇中,记为第一溶液,将1.6g Zn(NO
(3)预混合前体的制备:将ZIF-62和ZIF-8(Zn,Fe)混合物在研钵中混合均匀,质量比1:2,得到预混合前体。
(4)FeMOFs glass的制备:将步骤(3)得到的预混合前体置于惰性气体氛围下保持30min排出氧气,然后以20℃/min的速率急速升温至430℃,使其熔融,保持60min后,以250℃/h的降温速率迅速冷却,得到玻璃类FeMOFs材料。
实施例9
本实施例提供了一种FeMOFs glass的制备方法,主要步骤如下:
(1)ZIF-62的制备:将3mM Zn(NO
(2)ZIF-8(Zn,Fe)的制备:将4g 2-甲基咪唑溶于60mL甲醇中,记为第一溶液,将1.6g Zn(NO
(3)预混合前体的制备:将ZIF-62和ZIF-8(Zn,Fe)混合物在研钵中混合均匀,质量比20:1,得到预混合前体。
(4)FeMOFs glass的制备:将步骤(3)得到的预混合前体置于惰性气体氛围下保持30min排出氧气,然后以15℃/min的速率急速升温至480℃,使其熔融,保持30min后,以150℃/h的降温速率迅速冷却,得到玻璃类FeMOFs材料。
对比例1
(1)ZIF-62的制备:将4mM Zn(NO
(2)MOFs glass的制备:将ZIF-62置于惰性气体氛围下保持50min排出氧气,然后以15℃/min的速率急速升温至500℃,使其熔融。保持30min后,以200℃/h的降温速率迅速冷却,得到玻璃类MOFs glass(g-ZIF-62glass)材料。其最外层电子轨道分布如图11所示,与本发明实施例制备的g-ZIF-62(Fe)相比,g-ZIF-62glass由于没有Fe-N配位中心,其外层电子轨道在费米能级附近的分布更少,这表明其金属性较弱,电子跃迁能力弱,不能有效激发电子跃迁来催化氧化剂进而降解有机污染物。
对比例2
(1)ZIF-62的制备:将4mM Zn(NO
(2)ZIF-8(Zn,Fe)的制备:将4g 2-甲基咪唑溶于60mL甲醇中,记为第一溶液,将1.6g Zn(NO
(3)预混合前体的制备:将ZIF-62和ZIF-8(Zn,Fe)混合物在研钵中混合均匀,质量比1:5,得到预混合前体。
(4)FeMOFs的制备:在步骤(3)得到的预混合前体置于惰性气体氛围下保持30min排出氧气,然后以10℃/min的速率急速升温至500℃,使其熔融。保持30min后,以200℃/h的降温速率迅速冷却,得到晶体态、非玻璃态的FeMOFs材料。记为ZIF-62:ZIF-8=1:5。图12显示以ZIF-8分解的特征峰为主,而不是FeMOFs glass的峰型,意味着ZIF-62含量过少无法形成FeMOFs glass。说明控制ZIF-62与Fe-N配位前驱体的混合比例在合适范围内才能够制备出FeMOFs glass。
对比例3
(1)ZIF-62的制备:将4mM Zn(NO
(2)ZIF-8(Zn,Fe)的制备:将4g 2-甲基咪唑溶于60mL甲醇中,记为第一溶液,将1.6g Zn(NO
(3)预混合前体的制备:将ZIF-62和ZIF-8(Zn,Fe)混合物在研钵中混合均匀,质量比1:1,得到预混合前体。
(4)FeMOFs的制备:在步骤(3)得到的预混合前体置于惰性气体氛围下保持30min排出氧气,然后以10℃/min的速率急速升温至400℃或者600℃。保持30min后,以200℃/h的降温速率迅速冷却,得到晶体态、非玻璃态的FeMOFs材料。分别记为ZIF-62(Fe)400℃和ZIF-62(Fe)600℃。图12显示400℃的材料还保留了ZIF-62的特征峰,没有实现玻璃化;600℃的材料则显示了Fe化合物的峰,说明温度过高导致FeMOFs glass分解。这些说明低于430℃的温度导致FeMOFs无法熔融进而玻璃化,无法形成玻璃态的FeMOFs glass;高于500℃的温度将导致FeMOFs分解。结果表明合适的加热温度430-500℃是成功制备FeMOFs glass的关键。
对比例4
(1)ZIF-62的制备:将4mM Zn(NO
(2)ZIF-8(Zn,Fe)的制备:将4g 2-甲基咪唑溶于60mL甲醇中,记为第一溶液,将1.6g Zn(NO
(3)预混合前体的制备:将ZIF-62和ZIF-8(Zn,Fe)混合物在研钵中混合均匀,质量比1:1,得到预混合前体。
(4)FeMOFs的制备:在步骤(3)得到的预混合前体置于惰性气体氛围下保持30min排出氧气,然后以5℃/min的速率升温至500℃。保持30min后,以200℃/h的降温速率迅速冷却,得到晶体态、非玻璃态的FeMOFs材料。记为ZIF-62(Fe)5℃/min。图12显示材料还存在ZIF-62晶体的衍射峰,说明低于10℃/min的升温速率将导致FeMOFs无法玻璃化,无法形成玻璃态的FeMOFs glass,说明保持升温速率10℃/min以上的快速淬火是成功制备FeMOFsglass的关键。
应用例1
应用例1比较ZIF-62与FeMOFs glass的催化降解有机污染物性能效果。
根据本发明的制备方法,选取ZIF-62与ZIF-8(Zn,Fe)质量比分别为10:0、10:1、10:2、10:5、1:1、1:2制备得到的FeMOFs glass即ZIF-62glass(对比例1)、g-ZIF-62(Fe)-1、g-ZIF-62(Fe)-2、g-ZIF-62(Fe)-3、g-ZIF-62(Fe)-4、g-ZIF-62(Fe)-5,探索实施例1制备的ZIF-62(Fe)-4催化降解有机污染物性能。催化剂浓度为0.5g/L、污染物(双酚A)浓度为5g/L、氧化剂浓度为200mg/L,在50mL的上述混合溶液中分别加入不同催化剂,将反应溶液置于搅拌混合中,转速控制在500rpm,温度为25℃。在反应不同时间节点进行取样检测。
上述几种催化剂对污染物的降解性能如图13所示,g-ZIF-62(Fe)-4对目标污染物双酚A的降解率可达86%,而ZIF-62glass、g-ZIF-62(Fe)-1、g-ZIF-62(Fe)-2、g-ZIF-62(Fe)-3、g-ZIF-62(Fe)-5也分别有2%、8%、23%、47%、77%。说明本发明制备的FeMOFsglass具有良好的催化性能;ZIF-62(Fe)glass对污染物的降解率很低,60min内污染物几乎都没有降解,说明没有Fe-N配位中心的MOFs glass无法有效去除水中有机污染物,体现了本发明将ZIF-62与Fe-N配位前驱体混合的增益效果。
应用例2
应用例2研究了在实施例1制备的g-ZIF-62(Fe)-4催化剂催化降解有机污染物的主要活性物种。
通过添加不同活性物种的淬灭剂来探索实施例1制备的g-ZIF-62(Fe)-4催化剂在降解污染物的主要活性物种。所施加模拟太阳光波长范围为400~2200nm,催化剂浓度为0.5g/L、污染物浓度为5g/L、氧化剂浓度为200mg/L,淬灭剂为100mM,在50mL的上述混合溶液中加入g-ZIF-62(Fe)-4催化剂,将反应溶液置于磁力搅拌混合的烧杯容器中,转速控制在500rpm,温度为25℃,在反应不同时间节点进行取样检测。乙醇(EtOH)可以淬灭体系中的羟基自由基和硫酸根自由基,N
应用例3
应用例3比较太阳光照对实施例1制备的g-ZIF-62(Fe)-4催化剂在太阳光照下对污染物降解的增益效果
通过辐照模拟太阳光,探索实施例1制备的g-ZIF-62(Fe)-4催化剂在光照下降解污染物的增益效果。所施加模拟太阳光波长范围为400~2200nm,催化剂浓度为0.5g/L、污染物浓度为5g/L、氧化剂浓度为200mg/L,在50mL的上述混合溶液中加入g-ZIF-62(Fe)-4催化剂,将反应溶液置于磁力搅拌混合的烧杯容器中,转速控制在500rpm,温度为25℃,在反应不同时间节点进行取样检测。结果如图16所示,太阳光照条件下污染物降解速率显著提升至0.0332min
应用例4
应用例4证实本发明实施例1制备的g-ZIF-62(Fe)-4催化剂的孔道限域效应和极化电场促进电子/质子转移对催化降解污染物能垒的影响。
将Fe-N
应用例5
应用例5证实本发明实施例1制备的g-ZIF-62(Fe)-4催化剂经制备出后即具有较高机械强度且多孔的宏观形貌的类玻璃态。
将实施例1制备的g-ZIF-62(Fe)-4催化剂取出后直接得到具有宏观形貌结构的催化剂,如图18所示,该催化剂具有宏观形状且肉眼可见毫米级大孔,结合孔径分布结果可发现其具有大孔-中孔-微孔多级孔道结构(微孔孔径<2nm;中孔孔径2~50nm;大孔孔径>50nm)。图19显示了FeMOFs glass这种多级孔结构的大吸附量和高比表面积(13.5474m
应用例6
应用例6证实本发明实施例1制备的g-ZIF-62(Fe)-4催化剂对水种多种有机污染物具有优异的去除效果。
将实施例1制备的g-ZIF-62(Fe)-4催化剂在光照下催化过二硫酸盐降解有机污染物。结果如图19所示,双酚A、罗丹明B、甲基橙都被快速降解,降解速率分别为79%、92%、97%。因此,本发明提供的的FeMOFs glass可以直接用于去除水中多种有机污染物,无需特殊处理,处理效果好且易于回收利用。
尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例作出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
- 一种高强度氧化物弥散强化Fe基合金的制备方法
- 一种高电导玻璃粉及其制备方法,及基于其的钛酸钡基玻璃陶瓷及其制备方法
- 一种由MOFs衍生中空Fe/N/C燃料电池氧还原催化剂及其制备方法
- 一种MOF衍生的二氧化锰/四氧化三锰的层状复合材料的制备方法
- 一种利用MOF衍生双金属氧化物模板制备金属氧化物多级结构的方法
- 一种耐高温氧化的钴基Co-Fe-Ni-Al共晶中熵合金及其制备方法和应用
- 一种石墨烯基三维多孔基团改性Fe-MOFs气凝胶催化剂及其制备方法和应用