一种基于Cre重组酶的人腺病毒HAd5C改造方法
文献发布时间:2024-04-18 19:58:30
技术领域
本发明属于生物医药技术领域,具体涉及一种基于Cre重组酶的人腺病毒HAd5C改造方法。
背景技术
腺病毒是从呼吸道感染的婴儿扁桃体腺中分离培养得到,可分为A-G七个种,超过100个血清型。人腺病毒(Human adenovirus,HAdV)属于腺病毒科(Adenoviridae)哺乳动物腺病毒属(Mastadenovirus)。腺病毒具有外源基因较大的装载容量,因此腺病毒可作为基因表达载体,被广泛应用于基因治疗和病毒溶瘤等方面。腺病毒也是疫苗开发中常采用的载体,以5亚型腺病毒载体居多。
腺病毒属线性双链DNA病毒,病毒颗粒直径65-90nm,为无包膜二十面体结构,由240个六邻体组成,每个顶点处有12个五邻体蛋白,并有从每个五邻体突出的12个纤维蛋白三聚体。人腺病毒基因组全长约36kb,包括早期转录基因(E1A、E1B、E2、E3、E4)和晚期转录基因(L1-L5)。E1A是基因组中最早被转录的基因,其编码产物与宿主细胞pRb蛋白结合,导致E2F转录因子与pRb蛋白分离,进而激活E2F介导其他早期转录基因(E1B、E2、E3、E4)的转录,启动病毒DNA的复制。随后,E1B编码的E1B-55K蛋白与细胞的p53蛋白结合并使之失活,同时E1B-19K蛋白作为细胞凋亡因子阻止细胞凋亡。E2则编码病毒基因组复制所必需的3种蛋白:前体末端蛋白(pTP)、聚合酶(Pol)和单链DNA结合蛋白(DBP)。E3基因编码可破坏宿主免疫反应的蛋白(10.4K、14.5K、14.7K和19K),其中19K糖蛋白(gp19)下调MHCⅠ介导的抗原呈递给细胞毒性T淋巴细胞,从而阻止细胞毒性T淋巴细胞对病毒的杀伤作用;在病毒复制晚期,E3基因编码的蛋白可促进病毒粒子的释放和加快被感染细胞的死亡。E4的基因产物主要参与DNA的复制、转录、宿主细胞蛋白质合成的终止和细胞周期的调节。晚期基因的转录起始于主要晚期启动子(MLP),转录产物参与腺病毒基因组的调控,并编码成熟病毒颗粒装配所必需的衣壳结构蛋白。
腺病毒能在人体细胞中高效复制,被感染宿主细胞48h内能产生约10
专利CN202010055543.9公开了一种人3型腺病毒复制缺陷型重组病毒,其以野生型人3型腺病毒衣壳蛋白六邻体基因为保护性抗原基因,以E1、E3区基因缺失的人5型复制缺陷型腺病毒为载体,以整合E1区基因的AD293细胞为包装细胞系,制备得到的疫苗免疫小鼠后,可以诱导机体产生更高的体液免疫。专利CN202110193614.6则公开了一种人工改造的重组腺病毒载体,其基于黑猩猩型腺病毒AdC68基因组序列,完全删除其E1序列,改造其E3序列和E4序列,所述改造包括:(1)改造其E3序列,保留orf9和orf1;(2)改造其E4序列,以C亚型人腺病毒基因组E4序列中orf6和/或orf6/7替换其E4序列中orf6和/或orf6/7;其所述的腺病毒表达载体能够高效地包装,病毒产量高,且外源基因装载量高。
人腺病毒HAd5C是目前临床研究中最常用的溶瘤病毒载体,而要实现其溶瘤病毒的安全性及特异性则需要对HAd5C基因骨架进行改造,如fiber,hexon,penton等。目前常用的腺病毒骨架改造的方法有galK正负筛选,Kana/SacB正负筛选等,其阴性筛选普遍存在假阴性(阴性筛选压力下筛选基因galK或SacB基因发生突变)比例较高且阴性筛选效率较低耗时较长的缺点,基于此,开发一种避免进行阴性筛选的构建策略尤为关键。
发明内容
针对以上问题,本发明提供了一种基于Cre重组酶的人腺病毒HAd5C改造方法。本发明利用腺病毒AdMax系统中的载体pBHGlox(delta)E13Cre,该载体敲除了腺病毒复制关键基因E1和E3,其余骨架基因完整,因此可以用于HAd5C骨架基因的改造,通过将阳性筛选的抗性基因构建至该载体上,避免了阴性筛选效率低耗时长的缺点,筛选得到阳性载体后,进一步将抗性基因敲除,不影响外源基因的插入与表达。
本发明中,“Δ”表示缺失,如“ΔE3”表示E3基因缺失。
为了实现上述发明目的,本发明的技术方案如下:
一方面,本发明提供了一种改造人腺病毒HAd5C的方法,所述的方法包括以下步骤:
(1)对于敲除腺病毒复制关键基因的Admax系统腺病毒骨架载体,构建含抗性筛选标记1的表达框,表达框包含目的基因,同时在抗性筛选标记1两端设置重组酶切位点,得到抗性标记载体1;
(2)对抗性标记载体1进行抗性筛选,筛选后的载体利用重组酶删除抗性筛选标记;
所述的步骤(1)中所述的腺病毒复制关键基因为E3,目的基因的插入位点在ΔE3处。
优选地,所述的步骤(1)中的Admax系统腺病毒骨架载体为pBHGlox(delta)E13Cre载体。
优选地,步骤(1)中所述的抗性为非氨苄抗性,进一步优选为Kana抗性。本发明中,Kana抗性指卡那霉素抗性。
所述的重组酶包括但不限于Cre重组酶、Flpe重组酶或Dre重组酶。
所述的重组酶识别位点优选为loxs位点、FRT或roxP。具体地,所述的Cre重组酶识别loxs位点,所述的Flpe重组酶识别FRT,所述的Dre重组酶识别roxP。
优选地,所述的重组酶选自Cre重组酶,识别位点可以是loxs位点,能被重组酶识别并重组,所述的loxs位点包括但不限于lox2272、loxN或lox511。
步骤(3)中所述的利用重组酶删除抗性筛选标记的方法包括但不限于体外Cre同源重组和大肠杆菌SW106 Cre同源重组。
优选地,所述的方法中包括:
S1、敲除腺病毒复制关键基因的Admax系统腺病毒骨架载体,插入与目的基因具有同源臂的含抗性筛选标记2的表达框,抗性筛选得到空载体;
S2、构建载体插入片段:重组酶识别位点-抗性筛选标记1-重组酶识别位点-目的基因;所述的片段两端包含目的基因同源臂;
S3、步骤S2构建的载体插入片段插入步骤S1构建的空载体,替换含抗性筛选标记2的表达框,然后进行抗性条件筛选得到抗性标记载体;
S4、重组酶删除抗性标记载体的抗性筛选标记1。
根据以上优选方案,在一些具体实施例中,本发明所述的人腺病毒HAd5C改造方法包括以下步骤:
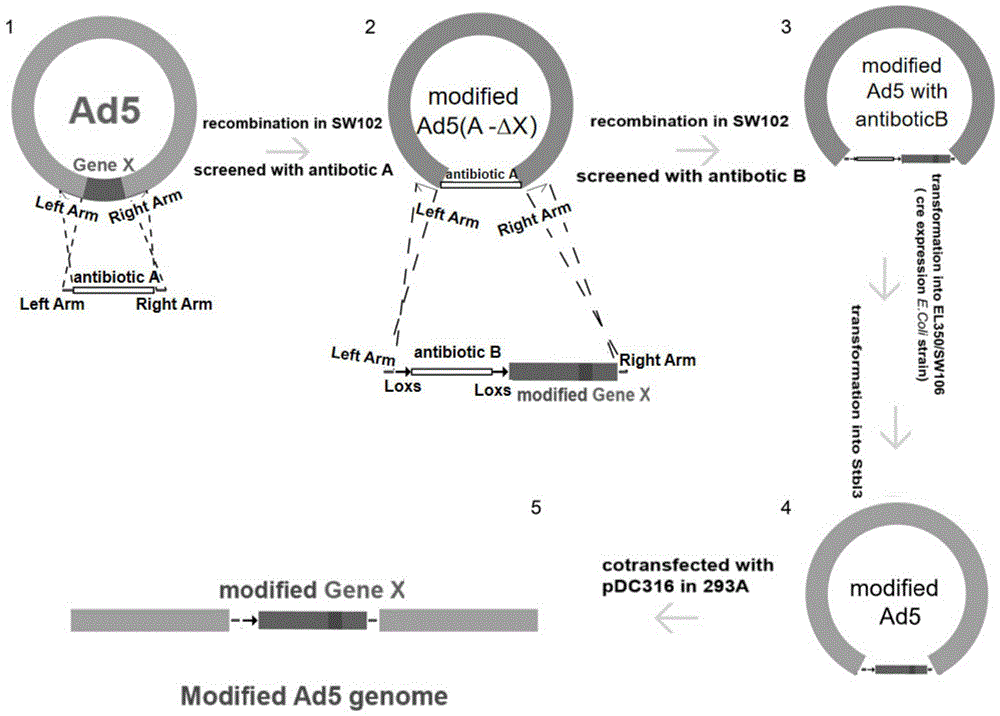
首先对于HAd5C的Gene X,先构建用于阳性筛选的anbiotic A(非氨苄抗性)的表达框,在LB平板上验证其功能正常后,设计引物primer1-F,primer1-R使其分别携带50nt左右(40-60nt)同源臂,与Gene X上下游序列同源,这样通过将pBHGlox(delta)E13Cre与含有同源臂的anbiotic A表达框共转SW102感受态细胞,并涂布anbiotic A平板对其进行筛选,仅成功将anbiotic A插入pBHGlox(delta)E13Cre中替换Gene X的克隆能够生长,进一步通过测序证明序列的完整性后可得到pBHGlox(delta)E13Cre-A-ΔX载体。
由于pBHGlox(delta)E13Cre自身携带一个loxp位点,该位点不能被Cre酶识别,而其他位点如lox2272、loxN、lox511等,能够被Cre酶所识别并重组。利用其他载体如pCDNA3.1+构建,将基因loxs-antibiotic B-loxs-modified Gene X(合成或构建)插入pCDNA3.1+中,并利用antibiotic B进行筛选验证其功能正常,然后设计引物primer2-F,primer2-R使其携带50nt左右(40-60nt)同源臂,与Gene X上下游序列同源,扩增出loxs-antibiotic B-loxs-modified Gene X插入序列,并与pBHGlox(delta)E13Cre-A-ΔX载体共转SW102感受态,涂布antibiotic B抗性平板进行筛选并测序,阳性克隆即为Gene X改造后的腺病毒载体pBHGlox(delta)E13Cre-loxs-B-loxs-modified GeneX。
此时获得的改造后Gene X腺病毒骨架中携带有antibiotic B表达框(~1Kbp),一方面减少了后续多基因改造的筛选备选抗性基因,另一方面pBHGlox(delta)E13Cre载体的容量有限,经过多轮筛选后无效的抗性基因序列在腺病毒骨架中大小不可忽略,会影响后续外源基因的插入,因此需要将抗性基因进行敲除,这时就需要利用Cre重组酶将两个loxs之间的抗性基因进行删除,主要有两种方式,体外Cre同源重组和大肠杆菌SW106 Cre同源重组。
体外Cre同源重组:直接将pBHGlox(delta)E13Cre-loxs-B-loxs-modified GeneX载体取2.5μg(50μL体系)加入1unit Cre重组酶(NEB)37℃酶切30min,70℃失活10min,然后取5ng转化Stbl3感受态细胞,通过菌落PCR鉴定抗性基因B的删除情况,并对删除的克隆进行测序,即可得到删除antibiotic B的Gene X改造后的腺病毒载体pBHGlox(delta)E13Cre-loxs-modified Gene X。
大肠杆菌SW106 Cre同源重组:直接转化SW106感受态细胞,将SW106感受态细胞在阿拉伯糖诱导后可表达Cre重组酶对两个loxs位点间的抗性基因B进行删除,通过菌落PCR筛选antibiotic B删除的克隆并测序,同样可得到删除antibiotic B的Gene X改造后的腺病毒载体pBHGlox(delta)E13Cre-loxs-modified Gene X。
最后将得到的Gene X改造后的腺病毒载体pBHGlox(delta)E13Cre-loxs-modified Gene X与AdMax系统的穿梭载体pDC316共转到293A细胞中即可获得Gene X完成改造后的腺病毒骨架。
又一方面,本发明公开了上述方法在制备人腺病毒HAd5C重组病毒中的应用。
又一方面,本发明公开了一种通过上述方法制备的人腺病毒HAd5C重组载体,所述的载体包括CD19t基因,所述的CD19t基因为SEQ ID NO.9所示的核苷酸序列。
又一方面,本发明公开了上述载体的制备方法,所述的方法包括以下步骤:
(1)构建载体pLQ-35(pDC316-lox2272-Kana);
(2)扩增pLEO-CD19t基因,与步骤(1)制备的pLQ-35载体进行同源重组,制备得到pLQ-36(pDC316-lox2272-Kana-lox2272-pLEO-CD19t)载体;
(3)利用重组酶删除步骤(2)构建的载体中的Kana抗性基因,得到pLQ-44((pBHGlox(delta)E13Cre)-lox2272pLEO-CD19t-bGH)载体。
又一方面,本发明提供了上述人腺病毒HAd5C重组载体在制备抗肿瘤药物中的应用。
所述的应用包括但不限于与CAR T细胞联用,优选为CD19-CAR T细胞。
又一方面,本发明提供了一种抗肿瘤药物,所述的药物包括上述人腺病毒HAd5C重组载体。
具体地,所述的药物还包括药学上可接受的辅料。
所述的药学上可接受的辅料包括但不限于溶剂、乳化剂、崩解剂、增溶剂、抗氧剂、pH调节剂、渗透压调节剂、抑菌剂、稀释剂、润湿剂、粘合剂、成膜剂等。
与现有技术相比,本发明的积极和有益效果在于:
(1)本发明提供了一种人腺病毒HAd5C改造方法,避免了阴性筛选效率低耗时长的缺点,筛选得到阳性载体后,进一步将抗性基因敲除,不影响外源基因的插入与表达。
(2)本发明将pLEO-CD19t基因插入到pBHGlox(delta)E13CreΔE3处获得的pLQ-44载体,包装腺病毒后与CD19-CAR T联用治疗肿瘤具有更好的效果,为肿瘤的治疗提供了新的思路与方法。
附图说明
图1为人腺病毒HAd5C基因组改造流程图。
图2为pLQ-37测序结果比对图。
图3为pLQ-44测序结果比对图。
图4为筛选试验结果,其中,PC表示positive control阳性对照。
图5为对比例1阴性筛选测序结果。
图6为对比例1阴性筛选阳性结果统计。
具体实施方式
下面结合具体实施例,对本发明作进一步详细的阐述,下述实施例不用于限制本发明,仅用于说明本发明。以下实施例中所使用的实验方法如无特殊说明,实施例中未注明具体条件的实验方法,通常按照常规条件,下述实施例中所使用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例中未注明具体技术或条件者,均按照本领域内的文献所描述的技术或条件(如参考J.萨姆布鲁克等著,黄培堂等译的《分子克隆实验指南》,第三版,科学出版社)或者按照产品说明书进行。
实施例1一种包含pLEO-CD19t基因的HAd5C载体的制备
1、目的基因
由人工合成的早期启动子pLEO启动的CD19t(truncated CD19)基因删除了CD19基因胞内信号转导功能,仅保留其胞外识别区域,通过与CD19-CAR T联用,可增强其对肿瘤的靶向性。具体的增强作用表现在:CAR T能够识别肿瘤表面特定抗原(靶标)以发挥肿瘤杀伤作用,但并非所有目的肿瘤都具有合适的靶标,因此CAR T可能会造成健康细胞的误伤或者肿瘤细胞的逃避攻击。通过构建表达CD19t的溶瘤病毒,使用溶瘤病毒感染肿瘤后,可以使肿瘤细胞表达CD19抗原,这些CD19抗原即可以作为靶标使CD19-CAR T精准识别肿瘤,起到靶向性增强作用。
本实施例中目的基因CD19t基因的插入载体的位置为pBHGlox(delta)E13Cre骨架中在ΔE3(29495nt)处,其上游同源臂UHA序列为SEQ ID NO.1,下游同源臂序列DHA为SEQID NO.2。
2、重组酶识别位点和抗性标记
构建载体pLQ-35(pDC316-lox2272-Kana),对载体pDC316(Microbix Biosystems,PD-01-64)用XbaI/EcoRI线性化,合成引物F1(SEQ ID NO.3)及引物R1(SEQ ID NO.4)扩增由amp promoter(SEQ ID NO.5)启动的Kana基因,扩增出来的基因序列为SEQ ID NO.6所示的序列。并将扩增出的序列与pDC316线性化载体进行同源重组,转化Stbl3感受态细胞,涂布kana抗性平板进行筛选测序,得到pLQ-35(pDC316-lox2272-Kana)载体,其序列为SEQ IDNO.7所示的序列。
3、目的基因连接
构建pLQ-36(pDC316-lox2272-Kana-lox2272-pLEO-CD19t):
pLQ-35(pDC316-lox2272-Kana)利用EcoRI/SalI线性化得pLQ-35(pDC316-lox2272-Kana)线性化载体。
合成引物F2(SEQ ID NO.8)及引物R2(SEQ ID NO.9)扩增pLEO-CD19t基因,其序列为SEQ ID NO.10所示的序列。
pLEO-CD19t基因序列与pLQ-35(pDC316-lox2272-Kana)线性化载体进行同源重组,转化Stbl3涂Kana平板筛选测序,得到pLQ-36(pDC316-lox2272-Kana-lox2272-pLEO-CD19t),其序列为SEQ ID NO.11所示的序列。
4、目的基因重组片段获得
利用含有上游同源臂UHA引物F3(SEQ ID NO.12)及下游同源臂DHA引物R3(SEQ IDNO.13)对载体pLQ-36进行扩增得到插入片段。
5、热激诱导RecA基因表达,SW102(明舟生物,货号B98002)genotype(F-mcrAΔ(mrr-hsdRMS-mcrBC)Φ80dlacZ M15ΔlacX74 deoR recA1 endA1araD139Δ(ara,leu)7649galU galK rspL nupGλcI857(cro-bioA)<>tetΔgalK);通过42℃热激诱导其表达同源重组酶表达的SW102电转感受态细胞制备:
1)挑选单克隆于含有四环素的LB培养基中32℃过夜震荡培养;
2)第二天以1:500接种到50mL无抗LB培养基中,32℃震荡培养5h左右至OD6000.4-0.6之间;
3)立即转移至42℃恒温水浴摇床中,200rpm震荡培养,热激15min后立即至于冰上冰浴10min,期间对离心机进行提前预冷至4℃,并将10%甘油提前置于冰上预冷;
4)将菌转移至50mL离心管中4℃、4000g离心10min,尽量去除上清;
5)加入1mL 10%甘油温和重悬菌体,然后加入10%甘油补齐体积至50mL,4℃、4000g离心10min,尽量去除上清;
6)重复步骤5)两次;
7)尽量去除上清后,加入500μL 10%甘油重选菌体,并每管50μL分装至提前预冷的1.5mL离心管中;
8)在液氮中快速冻存制备的热激电转SW102感受态细胞,然后转移至-80℃冰箱中保存。
6、抗性标记载体筛选
将步骤4扩增得到的插入片段100ng与pBHGlox(delta)E13Cre载体(HonorGene,货号HG-VXM0739)20ng共电转SW102感受态细胞,然后加入1mL无抗LB培养基恢复2h,涂Kana抗性平板筛选并测序,得到pLQ-37((pBHGlox(delta)E13Cre)-lox2272-kana-lox2272-pLEO-CD19t-bGH);
测序引物F4:SEQ ID NO.14;
测序引物F5:SEQ ID NO.15;
测序引物BGH:SEQ ID NO.16。
测序结果比对如图2所示。
7、制备SW106电转感受态细胞
1)SW106大肠杆菌(宝赛生物,货号pL042)在阿拉伯糖诱导下可表达Cre重组酶,首先挑选单克隆至无抗LB培养基中32℃震荡培养过夜;
2)第二天以1:500接种到50mL无抗、0.2%阿拉伯糖LB培养基中,32℃震荡培养5h左右至OD600 0.4-0.6之间;
3)立即转移至冰上冰浴10min,期间对离心机进行提前预冷至4℃,并将10%甘油提前至于冰上预冷;
4)将菌转移至50mL离心管中4℃、4000g离心10min,尽量去除上清;
5)加入1mL 10%甘油温和重悬菌体,然后加入10%甘油补齐体积至50mL,4℃、4000g离心10min,尽量去除上清;
6)重复步骤5)两次;
7)尽量去除上清后,加入500μL 10%甘油重选菌体,并每管50μL分装至提前预冷的1.5mL离心管中;
8)在液氮中快速冻存制备的热激电转SW106感受态细胞,然后转移至-80℃冰箱中保存。
8、电转50ng pLQ-37至SW106感受态细胞中,涂氨苄平板进行筛选并测序,得到pLQ-44((pBHGlox(delta)E13Cre)-lox2272pLEO-CD19t-bGH);测序引物为F4和BGH。
测序比对结果如图3所示。
由此利用本改造策略,成功将由pLEO启动的CD19t基因插入到腺病毒载体pBHGlox(delta)E13CreΔE3处,并不携带额外的抗性筛选基因序列。
对比例1阴性筛选效果对照
阴性筛选(Kana-SacB)的具体方法为:
1、合成基因pSacB-Kana-SacB(北京擎科生物),序列为SEQ ID NO.17所示的序列;
2、利用含有UHA的上游引物SacB-f(序列为SEQ ID NO.18所示)及含有DHA的下游引物Kana-r(序列为SEQ ID NO.19所示)扩增基因pSacB-Kana-SacB;
3、取200ng扩增的片段与200ng载体ad5(pBHGlox(delta)E13Cre)充分混匀后加入到SW102热激电转感受态细胞中进行电转,构建载体pLQ32(ad5(pBHGlox(delta)E13Cre)-SacB-Kana);
4、电击后加入1mL无抗LB培养基32℃恢复1hr后,5000rpm离心1min,去上清仅保留100μL左右,涂Kana抗性平板(阳性筛选平板),置于32℃培养箱培养过夜;
5、第二天挑选32个克隆鉴定,鉴定引物为:SacB-f及Kana-r,大小约3k bp,鉴定结果如图4,阳性率20/32,挑选其中1、4、17、18、25、32测序;
6、测序引物F6:SEQ ID NO.20;
7、测序引物F7:SEQ ID NO.21;
8、测序引物F8:SEQ ID NO.22;
测序结果如图5,插入序列完成正确,选择其中18号克隆进行下一步SacB阴性筛选;
9、按照实施例1中制备SW102热激电转感受态细胞的方法,制备pLQ32/SW102热激电转感受态细胞;
10、由于SacB基因代谢sucrose后产物polymeri clevans对于革兰氏阴性菌(如SW102)为致死的,所以用只有当SacB基因被同源重组去除后才能够在含有sucrose的平板上生长,配置含7%sucrose LB-NaCl Amp+阴性筛选平板;
11、利用含有UHA的上游引物F9(SEQ ID NO.23)及含有DHA的下游引物R9(SEQ IDNO.24)扩增基因pLEO-CD19t(SEQ ID NO.10),并电转片段100ng至pLQ32/SW102热激电转感受态细胞中,并加入1mL LB培养基32℃恢复培养1hr后,涂阴性筛选平板培养过夜;
12、第二天挑选64个克隆用引物F9/R9鉴定,结果见图6。
结果对比:
(1)基于Kana-SacB筛选策略,阳性筛选插入Kana-SacB基因时效率是很高的(20/32,图4),但进行阴性筛选插入目的基因时,可以看到大量克隆生长,挑选了64个克隆进行鉴定,其中仅一个克隆(图6)有疑似插入条带,测序后无信号,这也说明了该方法假阳性率太高,不适合进行腺病毒骨架的改构;另外galK阴性筛选策略则少有克隆生长,同样还存在假阳性高的问题;
(2)而本发明的基于Cre酶的阳性筛选策略,则其阳性率可达12/32,效率足够,可在腺病毒骨架中进行应用。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- DNA重组酶Cre基因的改造、重组蛋白表达方法及应用
- 一种改造重组酶Cre的方法及其在植物中的应用