两种基因编辑工具酶鉴定及其在核酸检测中的应用
文献发布时间:2024-04-18 19:58:30
技术领域
本发明属于生物技术领域,具体地涉及两种新鉴定的RNA引导的核酸内切酶Gs12-2和Gs12-14及其在核酸检测中的应用。
背景技术
传统的基因编辑工具主要包括锌指核酸酶(ZFNs)、转录激活效应核酸酶(TALENs)和成簇规则间隔短回文重复序列(CRISPR)。ZFN和TALEN是第一代和第二代基因编辑技术,两者都属于包含DNA识别结合域和DNA切割域的核酸酶。在这些技术中,DNA结合域是经过专门设计,可以靶向被DNA核酸酶域切割的特定DNA序列。然而,这些第一代和第二代基因编辑技术的广泛应用存在靶标识别率低、成本高、脱靶概率高和结构复杂等问题。这些缺点推动了第三代基因编辑技术的发展,即CRISPR/Cas9系统,它源自细菌中的适应性免疫系统。CRISPR基因编辑依赖于两个关键成分:CRISPR相关(Cas)蛋白和反式活性单向导RNA(sgRNA)。
随着对CRISPR相关蛋白的组成和发挥功能的形式进行更深入的研究,可将CRISPR/Cas系统分为两类:第1类Cas蛋白是多个亚基组成的多蛋白效应复合物,包括Ⅰ、Ⅲ和Ⅳ型;第2类Cas蛋白是单个多结构域的效应蛋白,包括Ⅱ型的Cas9蛋白、Ⅴ型的Cas12蛋白和Ⅵ型的Cas13蛋白。相对而言,第二类系统相对更为简单、高效,并且操作方便。在2015年被发现的2类Ⅴ型效应蛋白Cas12a,其长度为1200-1500个氨基酸。Cas12a蛋白可以在crRNA(CRISPR RNA)的引导下与含有PAM序列(Protospacer-Adjacent Motif,前间隔区序列邻近基序,一段富含碱基T的序列)的靶标双链DNA结合并切割基因组DNA,该crRNA的长度仅为42-44个碱基。并且,CRISPR/Cas12a系统具有RNase和DNase活性,不需要额外的反式激活crRNA(tracrRNA)。2018年,Cas12a被发现能在crRNA的引导下特异性结合和切割目标双链DNA(dsDNA),并在切割完成后仍保持活性,继续切割其他的非靶标单链DNA(ssDNA),这种非特异性的切割活性被称为“附带切割”能力。科学家利用Cas12a的“附带切割”能力,与核酸扩增技术相结合,同时在系统中引入带有荧光基团和淬灭基团修饰的ssDNA报告基因,开发出了许多快速、简便、低成本的核酸检测平台。
CRISPR/Cas12a系统具有操作简单、价格低廉等优点,在核酸检测方面具有巨大的应用潜力,但特异性、PAM限制等缺陷使其应用受到限制。而这些均源自于Cas12a核酸酶的自身特性。因此,本领域仍然亟需寻找编辑活性高、PAM序列简单且基因组覆盖范围广、特异性高的新型CRISPR/Cas12a基因编辑系统。
发明内容
本发明首次挖掘并鉴定出两种Cas12a家族同源核酸内切酶Gs12-2和Gs12-14,还建立了基于Gs12-2和Gs12-14介导的核酸可视化检测方法。
为了实现上述目的,本发明采用以下技术方案:
CRISPR/Cas12a系统中的同源核酸内切酶,包括以下蛋白:
I、SEQ ID NO.1所示氨基酸序列的Gs12-2蛋白和SEQ ID NO.2所示氨基酸序列的Gs12-14蛋白;
II、与SEQ ID NO.1和SEQ ID NO.2所示的氨基酸序列相比,具有80%以上的序列相似性的蛋白,并且基本保留了其源自序列的生物学功能;
III、与SEQ ID NO.1和SEQ ID NO.2所示的氨基酸序列相比,具有一个或多个氨基酸的置换、缺失或添加的蛋白,并且基本保留了其源自序列的生物学功能。
融合蛋白,包含上述核酸内切酶,以及与所述核酸内切酶的N端或C端连接的多肽。
多核苷酸,所述多核苷酸为编码上述核酸内切酶的多核苷酸,或编码上述融合蛋白的多核苷酸。含有所述多核苷酸的载体或宿主细胞。
一种可视化核酸检测试剂盒,包括上述核酸内切酶,单链DNA荧光-淬灭报告基因,以及与靶标核酸配对的向导RNA。
一种可视化检测伪狂犬病毒(PRV)核酸的试剂盒,包括上述Gs12-2蛋白或Gs12-14蛋白,单链DNA荧光-淬灭报告基因,以及与PRV核酸靶标配对的Gs12-2或Gs12-14蛋白向导RNA,所述的Gs12-2向导RNA序列为5’-AAUUUCUACUAUUGUAGAUU
本发明的技术方案具有如下主要的有益效果:
1.本发明首次提供了一种结合宏基因组学与实验手段挖掘和鉴定到的新型Cas12a家族同源核酸内切酶Gs12-2和Gs12-14。
2.本发明发现PAM识别序列范围更广的核酸内切酶Gs12-2和Gs12-14,其优点在于可以在基因组中识别PAM为“HVH”或“TTYV”(V=A/C/G、Y=C/T、H=A/T/C)的靶标DNA位点,极大拓展了基因编辑靶标覆盖范围。
3.本发明首次提供了CRISPR/Gs12-2和Gs12-14系统介导的核酸可视化检测技术。
附图说明
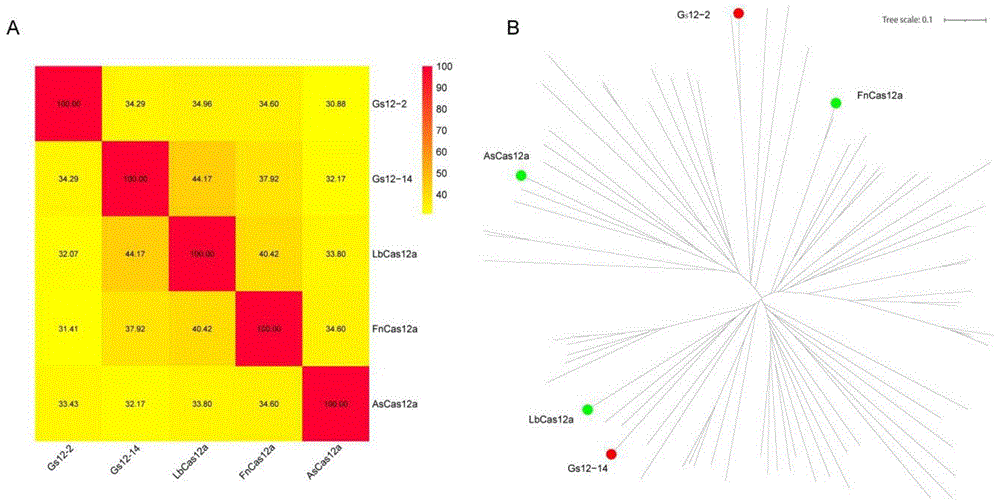
图1.利用宏基因组学方法预测的Cas12a家族同源核酸内切酶Gs12-2和Gs12-14系统进化树分析。A.Gs12-2和Gs12-14与已知Cas12a蛋白(AsCas12a、LbCas12a和FnCas12a)同源性分析;B.Gs12-2和Gs12-14系统进化树分析。
图2.核酸内切酶Gs12-2和Gs12-14基因座、结构域及向导RNA的DR序列模式图。A.Gs12-2和Gs12-14基因座示意图;B.向导RNA的DR序列二级结构折叠与多序列比对。
图3.预测的Gs12-2和Gs12-14蛋白氨基酸序列与已知Cas12a蛋白(AsCas12a、LbCas12a和FnCas12a)氨基酸序列保守性分析。
图4.凝胶电泳检测Gs12-2和Gs12-14切割双链DNA靶标活性。靶标为非洲猪瘟病毒ASFV p72基因扩增片段,识别的靶标位点PAM为“TTTV”。
图5.在细菌中利用PAM文库消减实验鉴定Gs12-2和Gs12-14识别PAM的特征。A.PAM文库消减实验示意图;B.Gs12-2和Gs12-14可识别的PAM。
图6.鉴定Gs12-2和Gs12-14切割双链DNA靶标的温度范围。靶标为猪伪狂犬病毒PRV gB基因扩增片段。A.Gs12-2体外切割双链靶标1h后凝胶电泳图;B.Gs12-14体外切割双链靶标1h后凝胶电泳图。
图7.鉴定Gs12-2和Gs12-14反式切割活性的酶切温度范围。靶标为ASFV p72基因。A.Gs12-2反式切割活性的酶切温度范围;B.Gs12-14反式切割活性的酶切温度范围。
图8.评估靶标中单个碱基错配对Gs12-2和Gs12-14反式切割活性的位置效应。靶标为猪PRV gB基因扩增片段,PC为阳性对照,PAM位点为GTTG,NC为阴性对照。A.靶点单碱基错配模式图;B、D.通过蓝光和荧光强度检测Gs12-2在单碱基错配时的不同反式切割活性。C、E.通过蓝光和荧光强度检测Gs12-14在单碱基错配时的不同反式切割活性。
图9.基于RPA-CRISPR/Gs12-2系统介导的猪伪狂犬病毒核酸可视化检测技术。A.利用qPCR明确PRV阳性或阴性样品;B.通过蓝光检测RPA-CRISPR/Gs12-2结果。
具体实施方式
术语说明
除非另外定义,否则本文中所用的全部技术与科学术语均具有如本发明所属领域的普通技术人员通常理解的相同含义。
Genie scissor(灵剪)核酸内切酶家族,其中Genie是精灵的意思,代表为细菌来源,scissor代表基因剪刀,表明其可能发挥的基因编辑功能。Genie scissor核酸内切酶对应的中文名称为“灵剪”核酸内切酶,Genie scissor基因编辑系统代表“灵剪”核酸内切酶介导的基因编辑系统,简称为“灵剪基因编辑”。
前间隔序列邻近基序(protospacer adjacent motif,简称PAM)是一个短的DNA序列(通常为2-6碱基对长度)。传统观点认为,PAM是Cas核酸酶切割所必需的,通常在切割位点下游3-4个核苷酸。有许多不同Cas内切酶可以从不同的细菌中纯化,并且每种酶可能识别不同的PAM序列。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,例如《分子克隆:实验室手册》(New York:Cold Spring Harbor Laboratory Press,1989)中所述的条件,或按照制造厂商所建议的条件。
实施例1.基于宏基因组学方法挖掘新型Cas12家族同源核酸内切酶
基于发明人搭建的Cas12a家族同源核酸内切酶的生物信息学鉴定流程,对NCBInr(Non-Redundant Protein Sequence Database)非冗余蛋白库、全球微生物基因目录数据库(GMGC)等公共数据库中的海量宏基因组测序数据进行了细菌编码蛋白深度挖掘。大致分析流程为:针对目标数据库中所有的contig序列,使用minced软件搜寻与定位CRISPRarray,接着使用prodigal软件预测CRISPR array邻近表达的蛋白质,通过CD-hit软件对预测到的所有蛋白去冗余、并利用mega软件进行蛋白质聚类分析、利用hmmer软件进行CRISPR-Cas相似性蛋白鉴定与分类,最终获得两种新的未知细菌蛋白。
通过系统发育进化树分析,发现这两种新的细菌蛋白位于CRISPR-Cas12a系统进化不同分支上(图1),推测它们可能为新的RNA引导型核酸内切酶。本发明对这类来自不同细菌中新发现的蛋白命名为Genie scissor(灵剪,Gs)核酸内切酶。为了方便后续研究,进一步基于细菌种属来源,发明人将这种新的未知细菌蛋白分别命名为Gs12-2和Gs12-14,其命名规则为:“核酸内切酶+数字编号”,Gs12-2蛋白的氨基酸序列如SEQ ID NO.1所示,Gs12-14蛋白的氨基酸序列如SEQ ID NO.2所示。
接着,发明人利用本地化blast程序,对这种新发现的细菌蛋白与NCBI nr数据库进行序列相似性比对。结果发现,Gs12-2蛋白与已知核酸内切酶LbCas12a、FnCas12a和AsCas12a的氨基酸序列保守性分别为34.96%、34.60%和30.88%,新的Gs12-14蛋白与已知核酸内切酶LbCas12a、FnCas12a和AsCas12a的氨基酸序列保守性分别为44.17%、37.92%和32.17%(图1)。
进一步,发明人通过对这两种蛋白的基因座用CRISPRCasFinder软件进行分析。结果发现,Gs12-2和Gs12-14具有CRISPR array序列,包含多个重复和间隔序列,但是Gs12-14没有Cas4、Cas1和Cas2。通过使用hmmer软件与Pfam数据库中的结构域序列进行隐马尔可夫模型比对分析,分析得到了REC1domain(Alpha helical recognition lobe domain),RuvCnuclease domain和NUC domain(Nuclease domain),推测这两个新的细菌蛋白可能具有核酸切割活性;接着发明人对Gs12-2和Gs12-14的DR序列二级结构通过RNAfold web server在线网站进行预测与多序列比对,结果发现这两个新预测的细菌蛋白与已知Cas12a蛋白的DR二级结构类似,Gs12-2仅有一个碱基存在差异;Gs12-14 DR序列多一个碱基,并且有四个碱基存在差异(图2)。
最后发明人对Gs12-2和Gs12-14蛋白的RuvC和REC结构域分别与已知的LbCas12a、FnCas12a和AsCas12a蛋白进行氨基酸多序列比对。如图3所示,发现Gs12-2和Gs12-14蛋白结构域与已知Cas12a蛋白的氨基酸序列相似性存在较大差别,因此亟需通过进一步实验确定它是否具有核酸定向切割活性。
实施例2.新型核酸酶Gs12-2和Gs12-14具有体外双链DNA切割活性
本实施例通过体外实验测试Gs12-2和Gs12-14蛋白对双链DNA的切割活性。利用与靶核酸配对的向导RNA引导Gs12-2和Gs12-14蛋白识别并结合在靶核酸上,从而激发Gs12-2和Gs12-14蛋白对靶核酸的切割活性,切割体系里的双链靶核酸。接着进行琼脂糖凝胶电泳观察目标条带大小变化来鉴定它的酶切活性。
本实施例中选择的靶标双链DNA(dsDNA)为非洲猪瘟P72基因,PAM为TTTA,其序列:
加粗标记为PAM,下划线为靶向序列。Gs12-2蛋白的向导RNA序列为:AAUUUCUACUAUUGUAGAUU
结果如图4所示,与不加向导RNA的对照组相比,实验组中的Gs12-2和Gs12-14蛋白在30min内就能够切割反应溶液里的双链DNA,存在1条或2条明显的切割条带,切割效率分别为Gs12-2:4.7%,Gs12-14:87.2%,enAsCas12a:47.9%,LbCas12a:11.2%由此可见,通过宏基因组学策略预测的细菌蛋白跟预期推测一样具有核酸靶向切割活性。
实施例3.鉴定Gs12-2和Gs12-14识别PAM的特征
通过在细菌上进行PAM文库消减实验,对同源性低且具有体外目标核酸切割活性的Gs12-2和Gs12-14蛋白所识别的PAM序列进行了鉴定。其中随机混合PAM载体库构建流程为:合成DNA oligo序列GGCCAGTGAATTCGAGCTCGGTACCCGGGNNNNNNN
细菌PAM文库消减实验:将构建好的Gs12-2和Gs12-14蛋白分别与crRNA共表达的载体pACYC-Duet-1-Gs12-2-crRNA和pACYC-Duet-1-Gs12-14-crRNA转化至DE3(BL21)感受态中,制备稳定表达的细菌株。不含Gs12-2/14蛋白表达载体pACYC-Duet-1构建的稳转细菌株作为阴性对照。将100ng的PAM文库质粒分别电转至稳定表达的细菌株中,通过氨苄霉素和氯霉素双抗性的板子进行筛选,16h后将板子上的菌落刮下进行质粒提取。分别以100ng提取的质粒为模板,用文库测序引物Seq-F:5’-GGCCAGTGAATTCGAGCTCGG-3’和PAM-Seq-R:5’-CAATTTCACACAGGAAACAGCTATGACC-3’进行PCR扩增,产物回收后分别将实验组和对照组进行二代高通量测序,对测序结果通过Weblogo3.0分析展示。
鉴定Gs12-2和Gs12-14蛋白识别的PAM序列特征:对起始载体库中含有的16,384种不同类型的PAM序列,分别统计它们在高通量测序中实验组和对照组中出现的次数高低,并用各自组所有PAM序列总数进行标准化。针对每条PAM消耗变化的计算方式为log
结果如图5所示,发现两种松散型PAM序列的CRISPR/Cas12a基因编辑系统的Gs12-2和Gs12-14,分别可以在基因组中PAM为“HVH”和“TTYV”(V=A/C/G、Y=C/T、H=A/T/C)的序列中切割靶标DNA。
实施例4.鉴定Gs12-2和Gs12-14体外切割双链DNA靶标的温度作用范围
本实施例通过体外实验测试Gs12-2和Gs12-14蛋白在不同温度梯度下对双链DNA的切割活性。利用与靶核酸配对的向导RNA引导Gs12-2和Gs12-14蛋白识别并结合在靶核酸上,从而激发Gs12-2和Gs12-14蛋白对靶核酸的切割活性,切割体系里的双链靶核酸。接着进行琼脂糖凝胶电泳观察目标条带大小变化来鉴定它的酶切活性。
本实施例中选择靶标双链DNA(dsDNA)为伪狂犬病毒gB基因,PAM为TTTG,其序列:
加粗标记为PAM,下划线为靶向序列。Gs12-2蛋白的向导RNA序列为:AAUUUCUACUAUUGUAGAUU
实施例5.CRISPR/Gs12-2和CRISPR/Gs12-14系统可介导核酸现场可视化快速检测
进一步评估Gs12-2和Gs12-14蛋白是否具有反式切割(trans cleavage)活性。利用可以与靶核酸配对的向导RNA引导核酸内切酶Gs12-2和Gs12-14识别并结合在靶核酸上;随之激发其对任意单链核酸的“反式切割”活性,从而切割反应体系里的单链DNA荧光-淬灭报告基因(ssDNA-FQ);进一步可以通过激发的荧光强度、背景噪音和肉眼颜色变化来判断候选细菌蛋白的反式切割功能。本实施例评估Gs12-2和Gs12-14反式切割活性作用温度范围中采用的靶标双链DNA(dsDNA)为伪狂犬病毒PRV gB基因扩增片段,序列如下:
加粗标记为PAM,下划线为靶向序列。Gs12-2蛋白向导RNA序列为:AAUUUCUACUAUUGUAGAUU
实施例6.评估CRISPR/Gs12-2和CRISPR/Gs12-14系统的特异性
进一步鉴定CRISPR/Gs12-2和CRISPR/Gs12-14系统对靶标区域单碱基错配识别能力。本实施例中采用的靶标双链DNA(dsDNA)为猪伪狂犬病毒PRV的gB部分保守基因,序列如下:
加粗标记为PAM,下划线为靶向序列。首先PCR扩增出含有从1-20位连续靶标位点突变的双链DNA模板,分别以Target-F至Target-p72-F-20C为上游引物,以Target-p72-R为下游引物进行扩增得到靶标双链基因。本发明使用的引物序列表如下:
Gs12-2蛋白向导RNA序列为:AAUUUCUACUAUUGUAGAUU
实施例7.利用RPA-CRISPR/Gs12-2技术可视化检测猪伪狂犬病毒
评估CRISPR/Gs12-2系统介导的核酸现场可视化检测技术的可靠性。针对PRV,建立RPA-CRISPR/Gs12-2检测技术。先收集PRV感染和未感染细胞的培养液作为模拟临床病毒感染样本,与定量PCR相比较,以此来评估新开发技术的检测准确性。其中采用伪狂犬病毒PRV gB基因扩增片段为阳性对照组,仅加灭菌水作为阴性对照。序列如下:
/>
加粗标记为PAM,下划线为靶向序列。Gs12-2蛋白向导RNA序列为:AAUUUCUACUAUUGUAGAUU
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本领域的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
- 两种新型核酸内切酶及其在核酸检测中的应用
- 新的组件式Ⅱ型限制性内切核酸酶CspCI和组件式内切核酸酶在产生具有新的特异性的内切核酸酶中的应用