一种细胞表面通用DNA传感工具箱及其制备方法和应用
文献发布时间:2024-04-18 19:58:53
技术领域
本发明涉及DNA纳米技术领域,特别是涉及一种细胞表面通用DNA传感工具箱及其制备方法和应用。
背景技术
细胞作为生物体的关键组成部分,经过数十亿年的生物进化,通过自然信号感知/响应途径感知环境信号,通过细胞状态的变化(生长、分泌、迁移、粘附、分化、繁殖或死亡)做出反应,进而执行复杂的功能。通过将这些信号感应与不同的细胞状态反应重新组合,各种基因工程细胞,包括免疫细胞、干细胞和细菌,已经被开发出来,并应用于疾病的治疗,如肿瘤的破坏、炎症的控制和组织的修复。然而,看似庞大的信号传感/响应途径库很少用于基因工程。例如,工程免疫细胞通过抗原受体基因修饰实现特异性抗原识别和免疫激活。然而,抗原受体独特的结构和性质极大地限制了工程修饰的多样性。因为天然抗原受体的整个结合-识别-触发机制是为单个配体-抗原蛋白量身定制的,从而阻碍了人工修饰抗原受体对其他类型配体(氢离子、活性氧或肿瘤微环境中的特征性疾病标志物)的反应的扩展。同样,工程细菌可用的信号传感/反应途径也有局限性。在工程细菌中常用的方法之一是双组分信号转导系统,在该系统中细菌通过控制基因表达来感知和响应环境变化。虽然这对大多数小分子信号传感器是可行的,但通过基因改造来感知蛋白质或核酸等生物大分子是困难的。
独特的自我识别能力和高度可预测的自组装特性赋予了DNA设计和构建任意二维和三维结构的能力。近期,DNA纳米装置已被用于设计和功能化细胞膜,以控制细胞状态和编程细胞功能。具有可预测性和可设计性的DNA纳米装置被用于控制细胞粘附和相互作用,杀死肿瘤细胞,在空间中排列多个细胞,甚至合成三维组织。然而,虽然一系列细胞表面DNA纳米装置可以为单细胞状态控制和细胞功能定制,但缺乏从信号传感到细胞状态控制的集成和通用系统。
发明内容
本发明的目的是提供一种细胞表面通用DNA传感工具箱及其制备方法和应用,以解决上述现有技术存在的问题,通过利用DNA纳米技术,构建细胞表面通用的传感工具箱,赋予细胞非常规信号的感知能力来调控细胞多种功能。
为实现上述目的,本发明提供了如下方案:
本发明提供一种DNA传感工具箱,包括DNA折纸框架,还包括分别组装在所述DNA折纸框架内表面和外表面的传感核心和锚定组块;
其中,所述传感核心包块传感开关和功能组件,所述传感开关为响应H
优选的是,所述I-motif传感开关包括第一链和第二链,其中,所述第一链通过碱基互补配对连接于所述DNA折纸框架,所述第二链与所述功能组件偶联,所述第一链修饰荧光基团,所述第二链修饰淬灭基团,根据荧光的淬灭和恢复表征所述I-motif传感开关的打开和关闭状态。
优选的是,当pH>响应阈值时,所述第一链和所述第二链结合并保持稳定状态,荧光淬灭,开关关闭;当pH<响应阈值时,所述第一链折叠形成四链结构,所述第二链释放,荧光强度增强,开关打开。
优选的是,所述第一链和所述第二链的摩尔比为1:2,所述第一链的核苷酸序列如SEQ ID NO:3所示,所述第二链的核苷酸序列如SEQ ID NO:4所示。
优选的是,所述核酸-蛋白偶联物包括CV1-PE38和CD47-PD1。
优选的是,所述锚定组块包括MUC1适配体和胆固醇标记的核苷酸链。
优选的是,所述细胞包括肿瘤细胞和免疫细胞。
优选的是,所述DNA折纸框架是利用核酸自组装技术用staple链将M13 ssDNA固定而成。
优选的是,将传感开关、功能组件、锚定组块和三棱柱折纸框架混合孵育进行组装,组装完成后,经超滤,得到所述DNA传感工具箱;其中所述传感开关、功能组件、锚定组块和三棱柱折纸框架的摩尔配比为5:5:5:1。
本发明还提供所述的DNA传感工具箱的应用,所述应用包括:
(1)在制备调控肿瘤细胞凋亡的药物或者递药载体中的应用;
(2)在制备调控免疫细胞和肿瘤细胞互作控制方面的药物或者药物载体中的应用。
本发明公开了以下技术效果:
本发明通过细胞表面工程改造重组细胞的天然信号感知/响应通路进而调控细胞功能,构建工程细胞用于重大疾病的诊疗是极具创新的发展方向。但天然细胞信号感知-响应通路数量种类和自身结构属性的局限性、基因工程技术操作的复杂性、耗时以及存在干扰细胞天然状态的风险等问题极大地限制了基因工程策略对细胞表面工程化改造的普适性和灵活性。本发明利用DNA纳米技术,构建细胞表面通用的传感工具箱,赋予细胞非常规信号的感知能力来调控细胞多种功能。
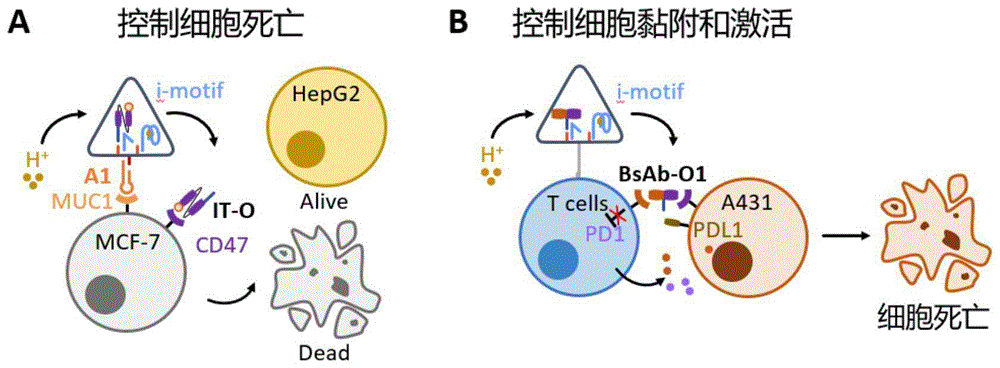
传感工具箱整体结构包括三棱柱折纸框架和传感核心。折纸框架外表面可以锚定化学修饰核酸链,以疏水作用或共价修饰固定工具箱于细胞膜及细菌细胞壁表面。折纸内部组装传感核心,内部限域反应空间既能提高核心在复杂生理环境中的长效稳定性,也可避免功能元器件非特异性结合造成脱靶毒性、信号泄露等副作用,从而实现精准控制。传感核心由传感开关和功能组件两部分构成,包括:1)传感开关包括:响应低pH环境(肿瘤微环境)的I-motif开关及响应核酸的链置换反应开关;2)功能组件包括:细胞功能蛋白-核酸偶联物用于控制细胞凋亡、附着及去抑制等多种功能。通过选配不同的传感开关和功能组件,实现多种细胞功能的重编程,进一步应用到免疫调节及肿瘤杀伤实用场景中。更具体的为:(1)肿瘤细胞凋亡控制:DNA传感工具箱通过粘蛋白1(MUC1)适配体组成的锚定模块组装至肿瘤细胞表面,内部传感核心为pH响应性I-motif开关,功能基团为靶向跨膜蛋白CD47的免疫毒素,酸性环境下,工具箱特异性释放免疫毒素杀伤肿瘤细胞。(2)免疫细胞和肿瘤细胞互作控制:DNA传感工具箱利用胆固醇基团的疏水性组装在免疫细胞膜表面,内部传感核心为pH响应性I-motif开关,功能基团为靶向CD47和PD-1(程序性死亡受体1)的双功能融合纳米抗体,当出现酸性环境时,工具箱释放出双功能纳米抗体,介导免疫细胞和肿瘤细胞的相互作用,阻断PD-1/PD-L1通路,解除抑制,同时增强免疫细胞杀伤活性。本发明通过构建细胞表面通用DNA传感工具箱,嫁接新型细胞信号传感通路,重编程细胞功能,构建纳米机械-天然杂合细胞,拓展工程细胞及工程菌响应信号和状态变化的多样性,为工程细胞及工程菌的诊疗应用提供新策略。
附图说明
图1为DNA传感工具箱重编程免疫细胞和肿瘤细胞;A:肿瘤细胞;B:免疫细胞和肿瘤细胞;
图2为使用CADnano设计的三棱柱折纸结构;
图3为三棱柱折纸的结构预测;
图4为三棱柱折纸的结构表征;A:三棱柱折纸结构的琼脂糖凝胶电泳图像;B:三棱柱折纸结构的透射电子显微镜图像;C:三棱柱折纸结构图;
图5为I-motif三链开关的优化;A:三种不同I链核心序列;B:对I链核心序列的优化;C:两种O链的序列及结构;D:对O链的优化;E:不同序列组合的三链开关的稳定性;
图6为酶标仪测试折纸内部I-motif开关的可行性;
图7为用于将核酸锚定在细胞表面的胆固醇修饰策略;A:胆固醇功能化折纸的共聚焦图像;B:流式验证胆固醇修饰DNA的细胞锚定;C:流式验证胆固醇功能化折纸的细胞锚定;
图8为用于将核酸锚定在细胞表面的MUC1适配体靶向策略;A:流式验证MUC1适配体的细胞锚定;B:流式验证MUC1适配体功能化折纸的细胞锚定;C:MUC1适配体功能化折纸的共聚焦图像;
图9为CV1-PE38、CD47-PD1的质粒构建图谱;
图10为SDS-PAGE图显示大肠杆菌表达系统成功表达IT;
图11为CV1-PE38(IT)对MCF-7和HepG2细胞的毒性。
图12为IT-O的功能表征;A、B分别为流式细胞仪验证IT-O(Ni-NTA策略)对MCF-7和HepG2的靶向性;C:IT-O对MCF-7和HepG2细胞的毒性;
图13为共聚焦成像表征传感工具箱的靶向及内部免疫毒素的动态释放;
图14为共聚焦成像表征传感工具箱的靶向及内部免疫毒素的动态释放;
图15为纳米工具箱在DMEM+10%FBS条件下的不同pH的细胞毒性实验;
图16为CD47-PD1的表达纯化以及靶向性测试;A:CD47-PD1的SDS-PAGE图;B、C分别为流式分析CD47-PD1对A431和T细胞的靶向性;
图17为共聚焦表征CD47-PD1促进A431和T细胞的细胞黏附;
图18为CD47-PD1对T细胞的激活;A:CD47-PD1对被抑制T细胞IFN-γ的影响;B:CD47-PD1对被抑制T细胞杀伤肿瘤细胞的影响;
图19为纳米工具箱响应不同的pH控制细胞黏附;
图20为工具箱响应pH控制T细胞激活和对A431的杀伤;A:T细胞杀伤能力控制;B:T细胞激活控制。
具体实施方式
现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为还具体公开了该范围的上限和下限之间的每个中间值。在任何陈述值或陈述范围内的中间值,以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。
在不背离本发明的范围或精神的情况下,可对本发明说明书的具体实施方式做多种改进和变化,这对本领域技术人员而言是显而易见的。由本发明的说明书得到的其他实施方式对技术人员而言是显而易见得的。本发明说明书和实施例仅是示例性的。
关于本文中所使用的“包含”、“包括”、“具有”、“含有”等等,均为开放性的用语,即意指包含但不限于。
本发明以下实施例中所使用的修饰核酸链序列及折纸订书钉链序列均由生工生物工程(上海)股份有限公司合成,未修饰核酸序列由上海擎科生物科技有限公司合成(见表1)。实验中所用到的细胞、菌种及质粒信息见表2,所用生物试剂见表3。
表1实验中所涉及的核酸序列信息
表2细胞、菌种及质粒信息
表3实验材料
表4M13 Staple链序列
*带下划线未斜体的序列为I链锚定位点,其与I1(Tri-prism)链序列的阴影部分互补配对;带下划线斜体的序列为A链锚定位点。
培养基及溶液配制:
(1)LB培养基:每25g LB培养基粉末溶于1000mL水中,溶解后分装至离心管或锥形瓶中,在121℃条件下高压灭菌20min。
(2)PBS缓冲液:商用化20×PBS加水稀释到1×PBS。
(3)抗生素:称取卡那霉素粉末、氨苄青霉素粉末溶于无菌水中,浓度为50mg/mL。于超净台中使用无菌0.22μm的滤头过滤,分装至无菌的EP管中,置于-20℃保存。
(4)IPTG母液:异丙基-β-D-硫代半乳糖苷(IPTG)粉末溶于无菌水中,终浓度为96mg/mL,于超净台中使用无菌0.22μm的滤头过滤,分装至无菌的EP管中,置于-20℃保存。
(8)10×SDS-PAGE电泳缓冲液:称取144g Gly,30.3g Tris于烧杯中,加入去离子水定容至1000mL。电泳液:10×SDS-PAGE电泳缓冲液100mL,10mL 10% SDS水溶液并加入去离子水定容至1000mL。
(9)考马斯亮蓝染色液:称取0.25g考马斯亮蓝G250于烧杯中,加入45mL蒸馏水及10mL冰乙酸充分溶解。
(11)10%过硫酸铵(APS):称量0.1g过硫酸铵加入含有1mL水的棕色EP管中,充分溶解。
(12)咪唑平衡缓冲液:3g NaH
(13)DNA变性PAGE胶:胶板0.75mm、梳子10孔、上样10-20μL(12μL,500nM-1μM浓度的DNA)。浓缩胶:0.84g尿素、1M,pH 6.8的Tris 250μL、330μL丙烯酰胺/双丙烯酰胺(29:1,30%),用去离子水定容到2mL。分离胶:2.1g尿素,650μL的Tris(3M,pH 8.8),2.75mL丙烯酰胺/双丙烯酰胺(19:1,40%),去离子水定容到5mL。上样缓冲液:0.48g尿素,93.75μL的Tris(1M,pH 6.8),62.5μL 1%溴酚蓝染料,用去离子水定容至1mL。电泳缓冲液:2.5mM Tris,0.19M甘氨酸。
M13 ssDNA,7249nt,购自New England Biolabs,货号与规格:B3003-50pmol。
实施例1
1、DNA三棱柱折纸载体的设计、构建、表征
DNA三棱柱折纸结构由一条长链的M13噬菌体基因组DNA以及178条短的订书钉链混合后梯度退火构建而成。以上DNA折纸结构的设计和序列均是在CADnano上面完成并导出的。
折纸合成的具体步骤如下:
(1)将7249nt的M13 ssDNA与混合后的178条staple链按照1:5的摩尔比例混合在1×Tris-EDTA-Mg
(2)混合物按每管50μL分装至PCR管中,退火程序:75℃维持5min,然后从65℃梯度退火到5℃,每1℃维持15min,程序总耗时是15h左右。
(3)取5管得到的混合物各加入5μL的10×DNA Loading buffer跑琼脂糖凝胶电泳(TAE)。
(4)刀片切除目的条带并切碎以最大限度地回收和纯度。
(5)放入Quantum Prep Freeze'N Squeeze DNA凝胶提取旋转柱的过滤柱中。柱子放入套管中。
(6)在室温下以13000g旋转样品3min。
(7)从收集管中收集纯化的DNA。
(8)纯化后的折纸进行TEM电镜表征:在铜网上面制样,首先使用2M醋酸镁加入到铜网上预处理2min,并使用滤纸去除多余液体。
(9)纯化后的折纸框架加入铜网中,静置30min。
(10)将1%的醋酸双氧铀加入到铜网中并使用滤纸快速吸去。
(11)将1%的醋酸双氧铀加入铜网上染色2min。在室温完全干燥后使用透射电子显微镜对三棱柱折纸进行表征。
(12)对折纸进行琼脂糖凝胶电泳表征:配制1%的琼脂糖凝胶,使用1×TAE微波炉加热溶解,并加入核酸染料,倒板后等待其凝固。
(13)在点样孔中加入混合了10×DNA loading buffer的M13 ssDNA及DNA折纸框架后在160V条件下跑电泳。
(14)电泳完成后,在凝胶成像仪中使用UV通道拍照。
2、开关的设计和优化
(1)为了优化方便,将I-motif开关设计成3链结合的形式(L+O+I),分别在O链和L链上修饰淬灭基团BHQ1和荧光基团FAM,互补序列位于I-motif的富含C的核心序列,因此当pH降低时,I-motif就会在核心序列处折叠,从而使功能基团释放出来并发挥功能。L链修饰有FAM荧光基团,O链带有BHQ1淬灭基团,通过荧光的淬灭与恢复来反映I-motif开关的两种状态。
(2)将L链、O链、I链按照1:4:2的摩尔比混合在1×PBS中制备L终浓度为1μM的母液(同时保留一组不加O链的阳性对照组)并退火。95℃加热5min,然后在冰上放置20min。
(3)配制2×PBS,并且使用HCl滴定到1×PBS后pH为7.37、7.11、6.49、6.01、5.63和5.09。
(4)将30μL母液,150μL的2×PBS,7.5μL的200mM的Mg
(5)在37℃的条件下反应2h,并且在反应终点的时候将反应液按照每孔100μL,每组3个平行加入到黑底的384孔板,并测量初始和终点的各个孔在激发波长485nM,以及发射波长为520nM的荧光强度。
(6)继续在37℃的条件下反应24h,测量此时的荧光强度用于表征开关结合的稳定性及泄漏率。
(7)按照同样的方法继续优化I1、I2和O1、O2的序列组合,主要区别是I和O结合在核心序列上的互补碱基的数量。
(8)测量A液在1×PBS+5mM Mg
(9)制作F1链的荧光强度随浓度的标准曲线,将F1在1×PBS+5mM Mg
3、质粒构建
(1)构建CV1-PE38和CD47-PD1表达质粒,利用PCR技术将目的基因融合、连接,骨架选用pET28a质粒来构建表达质粒,所涉及的引物序列如下:
表4引物序列
(2)反应体系为50μL,其中2×Taq Master Mix 25μL,水21μL,F引物1μL,R引物1μL,模板1μL,Taq DNA聚合酶1μL,dNTP 1μL。PCR反应条件设置为第一步95℃,5min;第二步95℃,20s;第三步58℃,30s;第四步72℃,1kb/1min,其中2-4步共34个循环;第五步72℃,10min。
(3)上述样品加入5μL的10×DNA上样缓冲液并进行琼脂糖凝胶电泳。
(4)在紫外灯下切除目的条带,切碎后放置于离心管中,根据胶回收试剂盒说明书进行胶回收,测量回收的DNA浓度。
(5)将回收目的片段,骨架片段和2×Mix同源重组酶混合。体系为4μL目的片段,2μL骨架,6μL酶,浓度低的组分适当多加。置于50℃水浴30min。
(6)将DH5α感受态置于冰上融化,将同源重组产物置于冰上,于超净台中将两者混合,冰上静置30min。
(7)42℃热激90s后立即冰浴2min。
(8)加入500μL的LB培养基,在37℃摇床,220rpm培养45min。
(9)融化固体培养基(150mL),流动水冷却至可用手握住10s,加入卡那霉素晃动20次左右倒平板。
(10)8000rpm离心1min,吸去500μL上清,重悬剩余的菌液涂板,过夜培养。
(11)超净台内挑单菌落,每个PCR管加10μL无菌水,再挑单菌落,混匀,取1μL当模板。
(12)菌液PCR体系为Mix 12.5μL,水10.5μL,F引物0.5μL,R引物0.5μL,模板1μL。程序为第一步95℃,5min;第二步95℃,20s;第三步58℃,30s;第四步72℃,1kb/15s,其中2-4步共34个循环;第五步72℃,10min。
(13)产物取5μL跑琼脂糖凝胶电泳,剩余PCR原液DNA测序。
(14)测序成功后的单菌落接种到5mL的LB中过夜培养。
(15)根据质粒抽提试剂盒说明书收集菌体并且提取质粒,使用酶标仪测量质粒浓度。
(16)按照上述转化步骤将质粒转化到大肠杆菌感受态BL21(DE3)中。
(17)挑取单菌落并且活化、保种,保种体系:500μL菌液加500μL 50%甘油。
4、功能蛋白的表达与纯化
(1)保种的菌液按百分之一加入到含有千分之一的卡那霉素的5mL LB中过夜培养,相同步骤二次活化。
(2)二次活化后的菌液全部接到250mL锥形瓶中(含有100mL的LB),培养3-4h后,等到OD
(3)转移菌液到50mL离心管,4℃8000rpm离心10min。
(4)使用1×PBS洗两次菌体后在PBS中超声破碎。(程序设置45%功率,超声3s,关闭5s)。
(5)破碎后的样品8000rpm离心10min,分离沉淀和上清。
(6)上清进行镍柱纯化,首先10mL咪唑平衡缓冲液平衡镍柱,随后加入上清,使用3倍柱体积的咪唑洗涤缓冲液洗脱杂蛋白,最后咪唑洗脱缓冲液洗脱目的蛋白。
(7)镍柱用10mL 0.5M NaOH清洗,再加入10mL咪唑平衡缓冲液置于4℃保存。
(8)纯化后的蛋白使用合适的超滤管浓缩并将蛋白洗脱缓冲液换成PBS,浓缩后的蛋白短期保存置于4℃,长期保存加入等量50%甘油置于-80℃保存。
(9)蛋白浓度使用BCA蛋白定量,工作液A:B=50:1,96孔板每孔加200μL工作液及20μL蛋白样品,37℃孵育30min。使用酶标仪在562nm处测量吸光度。
(10)配制SDS-PAGE胶:分离胶:2.5mL 10%下层胶、2.5mL下层胶缓冲液和65μL促凝剂,使用异丙醇压平界面。浓缩胶:将1mL上层胶、1mL彩色上层胶缓冲液、35μL促凝剂插入对应的梳子,待其凝固后使用。
(11)电泳体系:15μL蛋白样品加3μL 6×Protein buffer,浓缩胶电压80V,分离胶电压120V。
(12)小心的分离出蛋白胶并将其浸泡在考马斯亮蓝中染色约30min。加入适量的水放入微波炉中煮沸脱色,最后将其置于凝胶成像仪中观察结果并拍照。
5、蛋白质-DNA偶联策略
5.1荧光链标准曲线的绘制
将DNA溶解在PBS中依次稀释成200nM、100nM、50nM、25nM、12.5nM、6.25nM、0nM。测量485nm激发、520nm发射处的荧光强度。
5.2Ni-NTA策略:
(1)巯基修饰的DNA用水溶解,对于氨基修饰的DNA,首先加入10mM SPDP在室温孵育1h。
(2)(初始浓度为100mM,终浓度为10mM)的TCEP和(初始浓度为100μM,终浓度为20μM)的巯基修饰的DNA,室温孵育15-30min去除二硫键。
(3)加入70倍过量的Maleimido-C3-NTA,室温1h孵育,除去过量交联剂,保存到PBS中4℃保存。
(4)NTA-DNA(20μM)和0.5mM的NiCl
(5)3k超滤管除去过量的Ni
(6)纯化后蛋白(初始浓度为8μM)和DNA(初始浓度为20μM)按摩尔比2:3混合,室温孵育1h。
(7)PAGE胶观察DNA是否修饰成功,样品与上样缓冲液按1:1的比例混匀,95℃加热5min。
(8)上样,浓缩胶电压150V,分离胶电压300V。
(9)使用SYBR Gold染色。2.5μL+25mL 1×TBE染色20-30min。
(10)紫外观察条带并拍照。
(11)使用AKTA pure蛋白纯化仪分析制备的核酸-蛋白质偶联物并纯化。
6、传感工具箱的组装
将过量的开关链、功能核酸链和DNA-蛋白偶联物一起与纯化的折纸框架混合,并在25℃下在补充有10mM镁离子的1×PBS缓冲液中孵育过夜。为了确保锚定在折纸上的所有接头链与DNA-蛋白质偶联物的正确连接,在孵育过程中折纸结构与DNA-蛋白质偶联物的摩尔比例1:200。组装后,使用100k的超滤管纯化纳米工具箱,以去除多余的链和功能基团。所得产物储存在4℃以保持其完整性以备将来使用。
7、细胞培养
MCF-7、HEK-293T、HepG2和A431四种细胞系的培养使用90% DMEM培养基、10%的胎牛血清(FBS)和青霉素(100U/mL)/链霉素(0.1mg/mL)双抗的培养基培养,置于37℃、含有5% CO
细胞传代:将所需的细胞培养基以及PBS预热,吸去细胞培养瓶中的培养液。加入PBS清洗一遍,加入适量的EDTA-胰蛋白酶(T25方瓶加1mL,T75方瓶加2mL),在37℃放置2-3min,等到细胞变圆脱落后加入等体积的细胞培养基,并转移到离心管中,800g离心1min,吸去培养液,加入新的培养基重悬并取适量的液体接种到细胞培养瓶中(T25方瓶培养体积为5mL,T75方瓶培养体积为15mL)。
T细胞扩增:在离心管中储存并重悬磁珠,然后转移至新的离心管中。使用等体积或者至少1mL的PBS重悬磁珠,然后把离心管放置在磁力架上面静置30s-60s,用枪头小心地吸出上清,注意不要吸取到磁珠,加入1.2倍体积的含有200IU/mL rhIL-2的1640培养基待用。对Ficoll分离的PBMC,使用含有200IU/mL rhIL-2的1640培养基调整T细胞密度至1-1.5×10
T细胞激活:将1640培养基提前在37℃预热,取出液氮罐中的T细胞并放入37℃水浴锅中解冻,将解冻的T细胞转移至离心管中,缓慢加入1640培养基至10mL。600g离心10min,使用1640培养基重悬T细胞并转移至培养容器中,按照磁珠和T细胞数量为1:1的比例加入清洗好后的磁珠,培养24h后得到激活的T细胞。
8、工具箱在细胞表面的锚定策略
(1)于超净台中消化并取出150万的MCF-7细胞。
(2)将取出的细胞使用PBS清洗一遍,均分到三只离心管中1,2,3,500g离心3min。
(3)对得到的3管细胞沉淀进行以下处理:1加入100μL的PBS+5mM Mg
(4)使用PBS清洗三组细胞三次并且在B、C两管中加入500nM与A1互补的荧光链,室温孵育1h后,使用PBS清洗三遍细胞,并使用流式细胞仪检测荧光。
(5)按照同样的方法(第一步孵育时间改为室温1h)测试胆固醇修饰的链A2,并使用流式细胞仪检测荧光。
共聚焦表征DNA在细胞表面锚定
(1)适配体靶向
制备DNA三棱柱折纸并且使用Dox和折纸内部装载Cy3标记链对其标记。按照上述的两步孵育方法(第一步A1处理,第二步折纸处理)处理HepG2和MCF-7,并使用流式细胞仪检测荧光偏移,同时剩余细胞放置在玻璃底的24孔板中使用尼康共聚焦成像系统观察细胞荧光并拍照。
(2)胆固醇修饰
使用Dox和折纸内部装载Cy3标记链对折纸标记。按照上述的两步孵育方法(第一步A1处理,第二步折纸处理)处理MCF-7,并使用流式细胞仪检测荧光偏移,同时尼康共聚焦显微镜观察细胞荧光并拍照。
9、细胞死亡控制
免疫毒素的锚定及纳米工具箱的响应:
用BHQ1部分修饰工具箱中的I-motif,以淬灭免疫毒素-DNA上用FAM标记的互补DNA上的荧光。被选择作为对照的失活的I-motif对pH变化没有反应,其核心序列(富含C)被去除。然后,使用它合成和纯化了失活工具箱。收集了足够的MCF-7细胞(MUC1高表达)和HepG2细胞(MUC1低表达)(每个样品为1×10
纳米工具箱响应不同pH的毒性:配制好不同pH值的培养基(并加入纳米工具箱),并于超净台中使用0.22μM的滤头过滤除菌。MCF-7和HepG2细胞消化,并接种到96孔板上,每孔接种8000个,当细胞密度达到70%-90%的时候更换不同pH的混合工具箱的培养基。继续培养48h,按照上述方法测量细胞活力。
10、免疫细胞和肿瘤细胞互作控制
T细胞与肿瘤细胞共孵育:经过磁珠激活的T细胞经过600g离心收集并使用新鲜的1640培养基重悬后,使用血球计数板进行计数。A431细胞消化下来离心收集后使用1640培养基重悬并计数。之后将A431和T细胞按照3:1的比例(9000个:3000个)共同孵育在96孔板中,每组3个平行,培养两到三天后,离心去除细胞,测量上清中细胞因子IFN-γ的含量。当测试CD47-PD1及工具箱时,先将其与T细胞孵育后加入A431细胞。
IFN-γ细胞因子的测量:适用标准品梯度稀释,制作标准曲线。收集细胞在96孔板中培养三天后的上清,1000g离心3min去除细胞及细胞碎片。
(1)在平衡到室温的板条上,空白孔加入100μL标准品&标本通用稀释液其余各孔加入100μL标本,胶纸封住板条,在37℃避光孵育90min。
(2)提前准备生物素化抗体工作液,洗板5次。
(3)空白孔加入生物素化抗体稀释液,其余孔加入工作液100μL,胶纸封住板条,在37℃避光孵育60min。
(4)提前准备酶结合物工作液,洗板5次。
(5)空白孔加100μL酶结合物稀释液,其余孔加入酶结合物工作液100μL,胶纸封住板条,在37℃避光孵育30min。
(6)洗板5次,加入100μL/孔显色底物TMB,37℃避光孵育15min。
(7)加入终止液100μL/孔,并测量OD
共聚焦检测细胞粘附:消化并收集A431、HepG2以及扩增好的T细胞,将T细胞使用细胞膜红色荧光探针DiI染色,另外两种细胞使用细胞膜绿色荧光探针DiO染色。并将T细胞和肿瘤细胞按照10:1的比例混合,孵育缓冲液为1×PBS+5mM Mg
之后对工具箱激活T细胞分泌IFN-γ及增强对肿瘤细胞的杀伤进行了测试,先将T细胞接种至孔板中,与工具箱在37℃孵育30min后,再加入A431细胞,细胞活力测定和IFN-γ测试方法如上。
11、结果与分析
11.1实验原理
本发明利用核酸自组装技术用staple链将M13 ssDNA固定成三棱柱折纸结构。为了丰富折纸的应用场景,引入了以蛋白质功能为基础的传感核心(包括pH相应的I-motif开关及功能基团)以及以MUC1适配体/胆固醇修饰(特异性靶向/非特异性锚定)为基础的锚定模块。I-motif开关工作原理为:当pH>响应阈值时,三链结构O/I/L保持稳定状态,开关关闭;当pH<响应阈值时,I折叠形成四链结构,O释放到溶液中,开关打开。在控制细胞死亡模型中,MUC1适配体A1为锚定模块,CV1-PE38免疫毒素(IT)与输出链O偶联物(IT-O)为功能基团,当感知到酸性环境,开关打开,释放IT-O从而对靶细胞MCF-7产生杀伤。同时,由于HepG2阴性细胞的MUC1和CD47均为低表达,因此工具箱不会对其发挥功能,体现了工具箱的定制性和特异性。而在控制细胞激活和黏附模型中,使用胆固醇修饰作为锚定模块A2锚定在T细胞表面,CD47-PD1双功能纳米抗体(BsAb)与O链偶联物BsAb-O作为功能基团。同样感知酸性环境,打开开关释放BsAb-O,通过阻断A431细胞表面高表达的PDL1对T细胞的抑制,同时诱导T细胞对肿瘤细胞的黏附,进一步增加T细胞对A431的杀伤效果。如图1所示。
11.2DNA三棱柱折纸载体的设计、构建和表征
本发明中用到的传感工具箱是以DNA三棱柱折纸结构为基础构建的,为此利用caDNAno设计DNA折纸及折纸上的锚定位点,利用NUPACK设计双链、三链取代反应的DNA序列。本发明设计的DNA折纸一共有178条staple链,详细的序列信息如表4。DNA三棱柱折纸的二维、三维设计图如图2所示,图中71、69、75、77、49、51、55、57、59、61、65和67为折纸内部的锚定位点,用于装载功能基团与传感开关。锚定组件A链在56、54两处将工具箱以共价或非共价方式锚定在细胞表面。
将设计好的序列信息输入到TacoxDNA网站,用来预测三棱柱折纸框架结构,如图3所示。
根据“1、DNA三棱柱折纸载体的设计、构建、表征”的实验方法,合成DNA三棱柱折纸框架并纯化。接下来对其进行结构表征,样品琼脂糖凝胶电泳结果(图4中A)显示,左侧泳道为纯化后折纸结构右侧为M13 ssDNA,左侧样品条带迁移速率明显慢于右侧样品,这一结果表明,整合178条staple链的M13ssDNA形成了更大的分子结构。图4中A样品条带后存在的一条浅条带可能是形成了二聚体结构。本发明还对纯化的折纸框架进行了TEM电镜表征(图4中B),比例尺为50nm,从左到右分别是折纸的侧视图、正视图、俯视图,多角度照片证明三棱柱折纸结构的成功组装。折纸三维图如图4中C所示,以不同颜色展示了折纸内部以及外部的结合位点,用来结合不同的功能性核酸或者蛋白质来组装成完整工具箱。深灰色位点用来锚定传感核心,橘色位点用来连接锚定模块。
11.3传感开关的设计、优化以及功能验证
DNA传感工具箱可以响应环境pH(H
正常组织内部环境pH约为7.4,实体瘤组织环境pH为5.5-7.0,为了使开关响应实体瘤酸性微环境,接下来本发明对传感开关进行序列优化从而使其可以在pH 6.0时响应。首先对I链核心序列进行优化,富含C的核心序列是I-motif响应pH能力的决定因素。I1、I2、I3的核心序列分别为(C
将优化好的开关组装到折纸框架上,并验证其在折纸内部以及细胞表面的运行情况。三链开关中的L链为折纸框架固有序列,根据1、“DNA三棱柱折纸载体的设计、构建、表征”的实验方法将O1(SH)链-功能基团复合物和I1(Tri-prism)链锚定于折纸内部,其中O1(SH)链修饰淬灭基团,I1(Tri-prism)链修饰Cy3荧光基团。将样品在不同pH的PBS中孵育并测量反应后的荧光强度,结果如图6,在pH 7.4时样品的荧光强度值低于pH 6时,流式测量的细胞荧光偏移更弱。进一步将折纸通过胆固醇修饰固定在细胞表面,并使用共聚焦显微镜观察荧光。在pH 6.0时下细胞表面存在明显强于pH 7.4条件下的荧光。该结果表明,在pH7.4的条件下,I-motif开关关闭,I1(Tri-prism)链和O1(SH)链结合,I1(Tri-prism)链的荧光被O1(SH)链淬灭。在pH 6.0时,I-motif开关打开,I1(Tri-prism)链折叠,O1(SH)链掉落,荧光强度增强。三种实验结果共同证明这一开关在细胞表面的可行性。
11.4工具箱的MUC1适配体特异性靶向
验证了非特异性的胆固醇修饰策略可行性后,使用和“工具箱的胆固醇修饰锚定”同样的方法测试了MUC1适配体对MCF-7细胞(细胞表面高表达MUC1)的特异性靶向能力。流式细胞仪结果显示,MUC1Apt+S1组处理后的细胞有明显的细胞荧光标记,而阴性对照组S1未加适配体,因此无法使细胞携带荧光(图8中A)。随后进一步验证了折纸通过MUC1适配体靶向细胞的可行性,结果如图8中B所示,MUC1Apt+Origami组比Origami组荧光强度更强。共聚焦成像结果进一步验证了工具箱的靶向,如图8中C所示,折纸通过荧光DNA标记从而将其可视化,带有锚定模块(MUC1适配体)的折纸可以靶向到MCF-7细胞表面从而形成荧光环,而不表达MUC1的HepG2细胞表面则不存在代表折纸结构的荧光环,上述三种实验共同证明MUC1适配体可以使折纸特异性靶向MCF-7细胞(高表达MUC1)。
11.5工具箱的胆固醇修饰锚定
为确保工具箱在细胞表面的锚定,首先验证了胆固醇修饰策略的可行性。使用胆固醇修饰DNA(CW7)与Cy3标记反义链S2结合后与细胞孵育,流式分析细胞荧光。结果见图7中B,CW7+S2组有着明显强于S2组的荧光偏移。随后使用DOX和Cy3标记I链对纯化后的折纸结构进行了双重标记,并将标记的折纸结构经过胆固醇修饰DNA处理后与细胞孵育。流式分析结果如图7中C,CW7+Origami组有着明显强于Origami组的荧光偏移。随后我们还使用共聚焦荧光显微镜观察细胞表面荧光并拍照,如图7中A所示,结果为A2(胆固醇修饰)处理过的折纸组在细胞表面显示明亮的荧光环,而未处理的折纸组细胞表面只有微弱荧光,可能为非特异性吸附。三种实验结果共同表明了胆固醇修饰策略可以成功将纳米工具箱锚定在细胞表面,但此实验也存在以下问题,由于DOX对细胞有较强的侵入性,因此共聚焦图像中细胞内部也存在荧光。
11.6质粒构建及表达
本发明选用的功能基团是CV1-PE38、CD47-PD1,这两种蛋白均构建于大肠杆菌DH5α,并在BL21(DE3)中进行表达。其中CV1-PE38是由CV1纳米抗体片段及截短的铜绿假单胞菌外毒素38(PE38)组成,其中CV1纳米抗体是由CD47的结合抗体SIRPα突变并定向进化得到的高亲和力的突变体(Weiskopf K,et al.Engineered SIRPαVariants asImmunotherapeutic Adjuvants to Anticancer Antibodies.Science 341,88-91(2013).)。PE38内化后,催化延伸因子-2(EF-2)中二甲酰胺残基的ADP核糖基化,引起Mcl-1水平迅速下降,导致细胞凋亡,从而杀死肿瘤细胞(Wayne AS,et al.Anti-CD22Immunotoxin RFB4(dsFv)-PE38(BL22)for CD22-Positive Hematologic Malignanciesof Childhood:Preclinical Studies and Phase I Clinical Trial.Clinical CancerResearch 16,1894-1903(2010).)。两者基因序列由公司合成并通过PCR技术在中间添加经典的GS柔性linker构建成融合蛋白,质粒图谱如图9所示。CD47-PD1双特异性纳米抗体则由靶向CD47的纳米抗体(Sockolosky JT,et al.Durable antitumor responses to CD47blockade require adaptive immune stimulation.Proceedings of the NationalAcademy of Sciences 113,E2646-E2654(2016).)和靶向PD1的纳米抗体融合表达,质粒图谱如图9所示。
11.7蛋白质-DNA偶联技术
利用非共价的Ni-NTA策略,该偶联是基于镍介导的NTA与携带His标签的目标蛋白之间的相互作用。两个组氨酸残基和一个NTA分子可以满足镍(II)离子的所有六个配位点。此方法通常用于蛋白质的镍柱亲和色谱纯化中。具体表征结果“CV1-PE38与CV1-PE38-DNA偶联物的表征”,巯基修饰DNA先与马来酰亚胺-C3-NTA基团反应在DNA上标记NTA基团,随后将携带His标签的重组蛋白、Ni
11.8IT与IT-O的表征
采用大肠杆菌表达系统表达IT,并且通过SDS-PAGE成功验证免疫毒素的表达。如图10所示,左右两条泳道分别是镍柱纯化后以及10k超滤管浓缩后的CV1-PE38的蛋白条带,大小为55kD左右,该结果表明CV1-PE38的成功表达。
确定了阳性细胞MCF-7(CD47高表达)和阴性细胞(CD47低表达)HepG2,首先表征了CV1-PE38的靶向性,如图11所示,使用抗His标签的荧光标记二抗对CV1-PE38标记,并测试了免疫毒素及其偶联物对MCF-7细胞的靶向能力。结果如图11所示,IT-O组(偶联物)与IT组(CV1-PE38)处理过的细胞均携带大量荧光,但是IT-O组荧光偏移相对较弱,推测可能是IT-O结合部位和荧光标记二抗的结合部位重叠导致。随后评估了浓度为50nM、200nM条件下的CV1-PE38对于两种细胞的杀伤能力,结果显示50nM和200nM的CV1-PE38都能达到80%左右的细胞杀伤,但是同浓度的CV1-PE38几乎不杀伤HepG2细胞,全部结果表明CV1-PE38可以特异性杀伤高表达CD47的MCF-7细胞。
随后采用Ni-NTA策略制成蛋白质核酸偶联物(IT-O/CV1-PE38-O),O链采用FAM荧光标记。结果如图12的A、B所示,偶联物在200nM的浓度下有着极强靶向MCF-7(阳性细胞)的能力,同时HepG2(阴性细胞)无荧光偏移,该结果证实偶联物的特异性靶向。进一步测试了偶联物对两种细胞的细胞毒性,结果如图12的C所示,与上述11.9的结果相符。
11.9细胞死亡控制
在验证了工具箱的锚定模块以及传感模块的正常功能后,本发明表征了工具箱的整体运行情况,并通过荧光共聚焦显微镜观察并拍照。如图13所示,折纸内部的I链携带淬灭基团,偶联物的O链携带荧光基团,I/O链结合状态荧光淬灭,而pH为6.0时,I链和O链分离,O链的荧光恢复。这一部分的结果通过细胞表面的荧光可视化纳米工具箱的整个工作流程,折纸使用Dox嵌入双链之间,对其进行染色,但所有结果都显示出红色荧光的侵入性,推测是Dox未洗干净,导致Dox荧光侵入到细胞内部,因此去掉了红色荧光通道,只考虑IT-O的绿色荧光结果(图14)。本发明合成了一条删去了富含C的核心序列的I-motif,并制备成CV1-PE38的纳米工具箱(Toolbox*)作为对照,制备无响应模块的工具箱作为对照。图中结果显示A1+Toolbox(pH 7.4)组无绿色荧光,而A1+Toolbox(pH 6.0)有绿色荧光,同时Toolbox(pH 6.0)组没有绿色荧光,说明了工具箱完全锚定在细胞表面,并且当pH 6.0时,开关打开释放偶联物靶向细胞表面,使细胞携带绿色荧光。A1+Toolbox*(pH 6.0)组的细胞表面不存在绿色荧光,与A1+Toolbox(pH 6.0)组对比可知,无开关的工具箱不会释放内部的偶联物,使细胞携带绿色荧光,同时也说明所有的传感开关都位于折纸内部,否则游离状态的I/O-CV1-PE38偶联物在pH为6.0条件下也会带有绿色荧光并靶向细胞表面。上述结果证明了传感开关在工具箱内部,传感工具箱锚定在细胞表面以及整个工具箱响应pH后释放功能基团并且发挥作用的全流程。
最后本发明测试了工具箱响应pH产生的细胞特异性杀伤,将纯化后纳米工具箱与MCF-7细胞及HepG2在不同pH值的条件下孵育,结果如图15所示。pH 7.8(DMEM培养基的pH值)时装载工具箱的MCF-7细胞,细胞活力大概下降了20%,但环境pH为6.0时,细胞活力下降60%左右,而装载工具箱的HepG2细胞在两种pH条件下细胞活力无明显变化。该结果说明,传感工具箱可以响应酸性环境释放内容物对细胞产生特异性杀伤,但pH 7.8时装载工具箱的MCF-7细胞活力下降20%,推测工具箱稳定性差48小时孵育时间内部分解体,内容物泄露使细胞活力下降。
11.10CD47-PD1的功能表征
构建CD47-PD1表达质粒并且将其在大肠杆菌BL21(DE3)中表达,经镍柱纯化、浓缩后,SDS-PAGE表征CD47-PD1蛋白,图16中的A结果显示,CD47-PD1浓缩后的条带在30kD左右(红框圈出),证明了此功能基团的成功表达。随后,对其进行功能表征,流式细胞仪分析CD47纳米抗体的靶向性及PD1纳米抗体的靶向性,选用的靶细胞为高表达CD47的A431以及高表达PD1的T细胞。使用Ni-NTA策略制备成核酸蛋白偶联物后的流式结果如图16中的B、C所示,纳米抗体处理后细胞有较强的荧光偏移,证明了CD47-PD1的靶向能力。
随后进一步通过共聚焦荧光显微镜观察CD47-PD1对A431和T细胞相互作用的影响。A431(绿色)和T细胞(红色)混合后,按照肿瘤细胞:免疫细胞比例为1:10室温孵育1小时,两种细胞不会连接,当加入CD47-PD1后,两种细胞有明显的细胞黏附,而阴性细胞HepG2(CD47低表达)和T细胞的细胞黏附则不受CD47-PD1影响。见图17。
本发明测试CD47-PD1-O(αCD47/PD1-O)对T细胞的激活能力,测试的指标为IFN-γ分泌量以及细胞毒性。经过调研,A431是高表达PDL1的肿瘤细胞并对T细胞有较强的抑制,可以抑制其分泌IFN-γ,而HepG2不表达PDL1作为阴性对照。纳米抗体对T细胞分泌IFN-γ能力的影响如图18中A所示,磁珠激活后T细胞有着极高的IFN-γ的分泌能力,但和A431共孵育后,分泌量下降,因为其通过PDL1/PD1途径抑制T细胞。但经过CD47-PD1处理后的混合体系,IFN-γ分泌量有所提高,该结果表明双功能抗体及其核酸蛋白偶联物能促进T细胞分泌IFN-γ。同时,本发明也测试了T细胞对A431和HepG2的杀伤能力,如图18中B显示,磁珠激活的T细胞对A431无明显杀伤,这是因为其活性被抑制,当加入CD47-PD1-O后,增加了T细胞的肿瘤杀伤能力,A431的细胞活力下降至50%左右。而对于阴性细胞HepG2,T细胞在有无CD47-PD1的情况下对此细胞都有较强杀伤,经过48h培养后细胞活力在40%以下。该结果表明CD47-PD1可以增加T细胞对肿瘤细胞杀伤能力以及促进T细胞分泌IFN-γ。
11.11免疫细胞和肿瘤细胞互作控制
在验证了工具箱内部传感核心以及胆固醇锚定策略的可行性后,本发明构建了完整的传感工具箱并将其锚定在T细胞表面,通过不同pH的细胞培养基来控制纳米工具箱状态,进而控制免疫细胞和肿瘤细胞相互作用,该实验结果通过荧光共聚焦显微镜观察并拍照得到。如图19所示,混合T细胞(红色)和A431细胞(绿色)后,两种细胞无黏附,经过工具箱处理后的T细胞在pH 7.4的条件下和A431混合后也无细胞黏附。当调整pH值到6.0时,两种细胞在CD47-PD1的作用下产生细胞黏附,甚至一个A431细胞连接了多个T细胞,该结果验证了工具箱响应pH变化控制细胞黏附的能力。
随后本发明表征了不同pH下无传感核心的工具箱(Toolbox*)及工具箱对T细胞的激活能力和杀伤能力。图20中A为工具箱杀伤能力控制,pH 6.0时T细胞对A431的杀伤能力增强、A431细胞活力降低。无传感核心的工具箱(Toolbox*)在不同pH条件下A431细胞活力无明显变化,该结果表明,纳米工具箱可通过响应pH值释放功能基团激活T细胞对肿瘤细胞的杀伤。图20中B则是不同的pH条件下纳米工具箱对T细胞激活的控制,由于IFN-γ对酸性敏感,因此后续培养环境需要将pH维持在7左右,使用工具箱和T细胞在pH为6的培养基中孵育30min后,用偏碱性的培养基调整pH到7左右,之后与A431孵育,注意3天时间内培养基pH不要过低,过低会影响IFN-γ的含量,最后用ELISA试剂盒测试IFN-γ含量。当pH为6时,工具箱响应并释放CD47-PD1用于阻断PD1/PDL1通路,避免了A431对其抑制作用,因此该组有较高的IFN-γ水平。同时失活工具箱组由于不释放功能基团,IFN-γ水平没有明显变化,上述结果共同表明传感工具箱对免疫细胞和肿瘤细胞相互作用(激活及细胞黏附)的控制。
以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
- 一种活细胞内原位加速的DNA马达及其制备方法和应用
- 一种三氧化钨纳米空心球半导体材料及其制备方法、一种气敏传感器及其制备方法和应用
- 一种DNA生物传感器及其应用于测定DNA的方法
- 一种轨道交通用粘胶基炭纤维表面涂层吸声板的制备方法
- 一种低表面处理低VOC通用型环氧底漆的制备方法
- 一种靶向调控细胞膜表面EGFR二聚化的DNA结构及其制备方法和应用
- 一种DNA探针、通用电化学传感平台及其检测方法与应用