一类化合物及其用于新型冠状病毒肺炎的医药用途
文献发布时间:2023-06-19 11:19:16
技术领域
本发明涉及药物化学和药物治疗学领域,具体涉及一类化合物及其用于新型冠状病毒肺炎的医药 用途,属于药学技术领域。
背景技术
自从SARS-CoV-2引起的COVID-19爆发以来,它已夺走了全球数百万人的生命。此外,尽管先 前有人畜共患病暴发,但仍未批准针对紧密相关的冠状病毒,SARS-CoV-1或MERS-CoV的抗病毒 药物或疫苗。对于COVID-19的抗病毒药物的设计和开发,了解复制周期和关键基因组要素仍然至关 重要。冠状病毒是有包膜的,正义的,单链RNA病毒。两种病毒半胱氨酸蛋白酶3CLpro和PLpro 扮演关键的角色。3CLpro在所有冠状病毒中高度保守,并且在介导病毒复制和转录中起着重要作用, 因此被认为是理想的蛋白目标是开发广谱抗病毒药物。Cys145和His41是3CLpro催化位点的关键残 基。此外,3CLpro活性位点上有几个口袋,例如谷氨酰胺特异性S1位点,疏水性S2位点和小的S4 位点,这些共同为药物设计提供了充足的机会。SARS-CoV-2的PLpro可以抑制病毒复制并重新激活 固有的免疫应答。与SARS-CoV PLpro相似,SARS-CoV-2的PLpro的活性位点包含经典的 Cys-His-Asp三元催化分子,Trp106的侧链位于氧阴离子孔内,并建议吲哚环氮参与在整个催化过 程中产生的带负电荷的四面体中间体的稳定化。
发明内容
LY1是一种新型有效的小分子抑制剂,是抗病毒候选新药。化学名为3-((2-(哌嗪-1-基)苯基) 氨基)-5H-萘并[1,8-cd]异噻唑-5-酮1,1-二氧化物,研究表明LY1可以有效抑制COVID-19新冠肺炎 的病毒活性,同时在多次有效剂量下,它不会引起明显的毒性。结构式如下:
因此LY1可以通过抑制3CLpro和PLpro的活性,可能成为COVID-19新冠肺炎的治疗药物。
本发明合成一系列结构类似,对新冠肺炎有效的化合物。
本发明提供一系列化合物的制备方法,该制备方法高效实用经济、生产周期短,收率较高,易于 工业化生产。
本发明验证了系列化合物抗新冠肺炎活性,对化合物LY1进行了系统的验证,为抗新冠药物提 供潜在药物分子。
技术方案:为解决上述技术问题,本发明采用的技术方案为:
一种化合物,所述化合物的结构式如下
本发明采用的技术方案为:
第一方面,提供一类化合物,为式I或式II所示的化合物,或其药学上可接受的盐或酯:
其中,A环可以是五、六、七元饱和或者不饱和环,环内存在一到两个杂原子,杂原子指N、O、 S;
R1为一个或多个取代基,各自独立地选自氢、C1~C3烷基、羟基、C1~C3烷氧基、硝基、氨 基、羧基或C1~C3烷氧酰基;
R2为一个或者两个取代基,选自氢、卤素、C1~C3烷基、C1~C3烷氧基、所述卤素指氟、氯、 溴或碘。
R3为取代基,选自氢、卤素、C1~C3烷基、羟基、C1~C3烷氧基、硝基、氨基、羧基或C1~ C3烷氧酰基,所述卤素指氟、氯、溴或碘。
R4为一个或多个取代基,选自氢、卤素、C1~C3烷基、羟基、C1~C3烷氧基、硝基、氨基、 羧基或C1~C3烷氧酰基,芳香族、杂芳族、环或杂环,所述卤素指氟、氯、溴或碘、
在一些实施例中,A环吡咯基、咪唑基、吡唑基、呋喃基、四氢呋喃基、噻吩基、四氢呋喃基、 噻唑基、苯环、、吡嗪基、嘧啶基、哒嗪基、
所述R2为一个或者两个取代基,选自氢、卤素、C1~C3烷基、C1~C3烷氧基、或C1~C3 烷氧酰基,所述卤素指氟、氯、溴或碘,作为优选为甲基。
所述R3为取代基,选自氢、卤素、C1~C3烷基、羟基、C1~C3烷氧基、硝基、氨基、羧基 或C1~C3烷氧酰基,所述卤素指氟、氯、溴或碘,作为优选为氢、甲基、羟基、卤素。
所述R4为一个或多个取代基,选自氢、卤素、C1~C3烷基、羟基、C1~C3烷氧基、硝基、 氨基、羧基或C1~C3烷氧酰基,芳香族、杂芳族、环或杂环,
在上述优选的基础上,化合物为3-((2-(哌嗪-1-基)苯基)氨基)-5H-萘并[1,8-cd]异噻唑-5- 酮1,1-二氧化物,简称化合物LY1,结构式如下
药理实验证明,该化合物对新冠病毒有明显的抑制作用。
本发明的目的之一在于提供式I/式II通式所示的化合物的制备方法,合成路线如下:
本发明的另一目的在于提供化合物LY1的制备方法,合成路线如下:化合物LY1的制备方法, 其特征在于,合成路线如下:
包括:
(1)(化合物1)1-萘磺酰氯经取代反应制得(化合物2)1-萘磺酰胺;
(2)(化合物2)1-萘磺酰胺经氧化反应制得(化合物3)5,8-二氧代-二氢萘;
(3)(化合物3)5,8-二氧代-二氢萘再与(化合物4)叔丁基4-(2-氨基苯基)哌嗪-1-甲酸酯发 生取代反应得到(化合物5)(4-(2-((1,4-二氧代-5-氨磺酰基-1,4-二氢萘-2-基)氨基)苯基)哌 嗪-1-羧酸叔丁酯);
(4)(化合物5)(4-(2-((1,4-二氧代-5-氨磺酰基-1,4-二氢萘-2-基)氨基)苯基)哌嗪-1- 羧酸叔丁酯)氨基脱保护得到目标化合物(化合物LY1)4-((2-(哌嗪-1-基)苯基)氨)-5H-萘并[1,8-cd] 异噻唑-5-酮1,1-二氧化物。
进一步的,步骤(1)具体是指:将化合物1加入到盛有溶剂的反应器中,搅拌,在冰浴中滴加 0℃的氨水,滴加完毕后,薄层跟踪检测反应,反应完成后,减压蒸馏,析出固体,过滤,真空干燥, 得化合物2。
优选的,所述的溶剂为非质子性溶剂中的一种或几种。
步骤(2)具体是指:将(化合物2)1-萘磺酰胺加入盛有有机溶剂的反应器中,控温65℃-70℃ 并搅拌;将十倍当量硫酸高铈用2mol/L的稀硫酸溶解,控温65℃-70℃并搅拌,缓慢滴加入至体系中, 从滴加开始计时,25-35分钟后停止反应,放置冷却后抽滤,随后在滤液中加入二氯甲烷萃取,取二 氯甲烷层,真空干燥,得5,8-二氧代-二氢萘。作为更优选,温度为65℃,反应时间为27分钟。
进一步的,步骤(2)中,所述有机溶剂为冰乙酸、三氟醋酸中的一种或几种。
步骤(3)具体是指:将化合物3、化合物4与催化剂用冰乙酸溶解,常温下搅拌反应25-30小时; 减压蒸馏除去溶剂,得粗品,粗品经纯化得化合物5;
所述催化剂为一水合乙酸铜、三氯化铈、三乙胺中的一种或几种;更优选为一水合乙酸铜。
步骤(4)具体是指:化合物5脱去氨基保护得到化合物LY1;具体包括:用有机溶剂二氯甲烷 溶解化合物5,通入盐酸气体,室温搅拌,薄层跟踪检测反应;反应结束后,过滤,即得化合物LY1。
步骤(4)中,所述有机溶剂为二氯甲烷、氯仿,二氧六环中的一种或几种。
优选的,步骤(3)中,粗品经纯化得化合物5,包括:
在粗品中加入无水乙醇,110℃加热沸腾,液相监测反应产物全部转化后,停止加热,自然冷却 搅拌6h,过滤得滤饼,柱层析后,加入10倍当量的有机溶剂完全溶解后,滴加入25倍当量的醇类 有机溶剂,旋蒸后除去有机溶剂,待晶体全部析出,过滤得化合物5。
进一步的,纯化过程中,所述有机溶剂为二氯甲烷、乙酸乙酯中的一种或几种,优选为二氯甲烷; 醇类有机溶剂为甲醇、乙醇、异丙醇中的一种或几种,优选为异丙醇。
本发明提供的化合物LY1系列化合物的制备纯化方法,制备方法简单,成本低,收率高,以萘 磺酰氯为原料,以较低的成本方便地制备了有良好抗肿瘤活性的化合物LY1。
第三方面,提供所述的化合物在制备预防和/或治疗与新型冠状病毒肺炎有关疾病的药物中的应 用。
该化合物通过高特异性来抑制3CLpro和PLpro的活性,用作治疗新型冠状病毒肺炎的药物。
能够作为治疗包括结直肠癌,肺癌、新型冠状病毒肺炎在内的对STAT3抑制剂类药物或者吉非 替尼产生耐药性的疾病的药物。
其中化合物LY1效果尤佳。
附图说明
图1为:通过定量实时RT-PCR评估LY1对新冠病毒繁殖的功效,化合物LY1表现出良好的抗 病毒活性;
图2为:MTT法,化合物LY1的细胞毒性通过MTT法评估,化合物LY1的CC50值高于15μM。 如图2所示。实验结果表明,LY1在体外具有优秀的抗病毒活性和较小的毒性;
图3为:荧光共振能量转移(FRET)蛋白酶测定结果表明LY1能有效抑制SARS-CoV-23CLpro 和PLpro的活性;
图4为:表面等离振子共振(SPR)分析结果表明LY1主要靶向3CLpro蛋白。
具体实施方式
为了进一步阐明本发明,下面给出一系列实施例,这些实施例完全是例证性的,它们仅用来对本 发明具体描述,不应当理解为对本发明的限制。
一、化合物及其制备
前体准备
1)、制备1-萘磺酰胺
将1-萘磺酰氯(5g,21.9mmol)加入到盛有200ml丙酮的1L圆底烧瓶中,搅拌,在冰浴中滴加240 ml 0℃的氨水,滴加完毕后,薄层跟踪检测反应,反应完成后,减压蒸馏出有机溶剂和氨水分解出的 氨气,析出固体,过滤,真空干燥,得1-萘磺酰胺(4.35g,收率96.7%)。该化合物无需进一步纯化, 直接用于下一步反应。实验数据如下:
Mp 147~149℃.
2)、制备5,8-二氧代-二氢萘
将无水硫酸高铈(160g,677.45mmol)用750ml 2mol/L稀硫酸溶解,控温65℃。将1-萘磺酰胺 (10g,48.49mmol)加入盛有200ml冰乙酸的500ml圆底烧瓶中,控温65℃,该温度下搅拌,缓慢滴 加入至硫酸高铈水相体系中,从滴加开始计时,薄层色谱监测,27分钟后停止反应,待反应液冷却后, 过滤,滤液用1200ml二氯甲烷萃取,干燥除水后低压旋蒸,得到淡黄色固体,真空干燥,得5,8-二 氧代-二氢萘(6.72g,收率58.43%)。直接用于下一步反应无需纯化。实验数据如下:
Mp 186~188℃.HR-MS(ESI)calcd for C
实施例1
1.1制备(4-(2-((1,4-二氧代-5-氨磺酰基-1,4-二氢萘-2-基)氨基)苯基)哌嗪-1-羧酸叔丁酯)
将5,8-二氧代-二氢萘(56.5g,238.17mmol)、叔丁基4-(2-氨基苯基)哌嗪-1-甲酸酯(60g, 217.90mmol)与一水合乙酸铜(5.6g,28.05mmol)加入到盛有1200ml冰乙酸的2L圆底烧瓶中,25℃反应, 该温度下搅拌反应26小时;减压蒸馏除去溶剂,得粗品。
在粗品中加入1500ml的无水乙醇,110℃加热沸腾,液相监测反应产物全部转化后,停止加热, 自然冷却搅拌6h,过滤得滤饼,柱层析后除去一般杂质,后加入10倍当量的二氯甲烷完全溶解后,缓 慢滴加25倍当量的异丙醇,33℃低压旋蒸后除去相同量的二氯甲烷,待晶体全部析出,过滤,得紫红 色晶体,低压干燥得4-(2-((1,4-二氧代-5-氨磺酰基-1,4-二氢萘-2-基)氨基)苯基)哌嗪-1-羧酸 叔丁酯,纯度为97%.(67.53g,收率83%)。
实验数据如下:
Mp 189~190℃.
1.2制备4-((2-(哌嗪-1-基)苯基)胺)-5H-萘并[1,8-cd]异噻唑-5-酮1,1-二氧化物
将(4-(2-((1,4-二氧代-5-氨磺酰基-1,4-二氢萘-2-基)氨基)苯基)哌嗪-1-羧酸叔丁酯)(2g, 3.78mmol)加入到盛有2ml二氯甲烷的50ml圆底烧瓶中,随后通入盐酸气,薄层跟踪检测反应;反应 结束后,过滤得滤饼,减压干燥分离出4-((2-(哌嗪-1-基)苯基)胺)-5H-萘并[1,8-cd]异噻唑-5-酮1,1- 二氧化物(1.95g,收率97%)。实验数据如下:
Mp 200~201℃.
2.1制备(1,4-二氧代5-氨磺酰基-1,4-二氢萘-2-基)氨基)-烟酸
将5,8-二氧代-二氢萘1(237mg,1mmol)、5-氨基烟酸(172mg,1.2mmol).与一水合乙酸铜(20mg, 0.1mmol)加入到盛有5ml冰乙酸的25ml圆底烧瓶中,加热回流,该温度下搅拌反应3小时;减压蒸 馏除去溶剂,液相色谱分离出3-(萘-2-基氨基)-5H-萘[1,8-cd]异噻唑-5-酮1,1-二氧化物(221mg,收 率71%)。
实验数据如下
实施例3
3.1制备(4-(2-((1,4-二氧代-5-氨磺酰基-1,4-二氢萘-2-基)氨基)苯基)哌嗪-1-羧酸叔丁酯)
将5,8-二氧代-二氢萘(0.5g,2.11mmol)、叔丁基4-(2-氨基苯基)哌嗪-1-甲酸酯(0.787g,2.53mmol) 与一水合乙酸铜(42mg,0.21mmol)加入到盛有12ml冰乙酸的25ml圆底烧瓶中,118℃加热回流,该 温度下搅拌反应3小时;减压蒸馏除去溶剂,液相色谱分离出化合物(4-(2-((1,4-二氧代-5-氨磺 酰基-1,4-二氢萘-2-基)氨基)苯基)哌嗪-1-羧酸叔丁酯)(0.568g,收率51%)。实验数据如下:
Mp 189~190℃.
3.2制备4-((2-(哌嗪-1-基)苯基)胺)-5H-萘并[1,8-cd]异噻唑-5-酮1,1-二氧化物
将(4-(2-((1,4-二氧代-5-氨磺酰基-1,4-二氢萘-2-基)氨基)苯基)哌嗪-1-羧酸叔丁酯)(200 mg,0.378mmol)加入到盛有2ml二氯甲烷的10ml圆底烧瓶中,随后加入2ml三氟醋酸,室温搅拌, 薄层跟踪检测反应;反应结束后,减压蒸馏除去溶剂,液相色谱分离出化合物制备4-((2-(哌嗪-1-基) 苯基)胺)-5H-萘并[1,8-cd]异噻唑-5-酮1,1-二氧化物(149mg,收率95%)。实验数据如下:
Mp 200~201℃.
实施例4
制备7-((4-羟苯基)氨基)-5,8-二氧-5,8-二氢萘-1-磺酰胺
将5,8-二氧代-二氢萘(100mg,0.421mmol)、4-氨基苯酚(55.2mg,0.506mmol)与一水合乙酸 铜(16.83mg,0.084mmol)加入25mL圆底烧瓶中,加入5ml冰乙酸,使之溶解,在室温下反应3小时, 反应完全后,减压蒸馏除去溶剂,液相色谱分离得到7-((4-羟苯基)氨基)-5,8-二氧-5,8-二氢萘-1- 磺酰胺。
实验数据如下.
实施例5
5.1制备3-((4-(二乙氨基)苯基)氨基)-5H-萘并[1,8-cd]异噻唑-5-酮1,1-二氧化物
将5,8-二氧代-二氢萘3(300mg,1.26mmol)、N,N-二乙基对苯二胺4(249.26mg,1.52mmol)与 一水合乙酸铜(20mg,0.1mmol)加入到盛有15ml冰乙酸的100ml圆底烧瓶中,室温下搅拌24小时; 减压蒸馏除去溶剂,加入无水乙醇加热,冷凝回流,将大部分溶剂蒸去后过滤,柱层析分离滤饼出3- ((4-(二乙氨基)苯基)氨基)-5H-萘并[1,8-cd]异噻唑-5-酮1,1-二氧化物(37mg,收率7.67%)。
实施例6
6.1制备3-((2-氨基苯基)氨基)-5H-萘[1,8-cd]异噻唑-5-酮1,1-二氧化物
将5,8-二氧代-二氢萘1(237mg,1mmol)、1,2-二氨基苯胺(130mg,1.2mmol)。与一水合乙酸 铜(20mg,0.1mmol)加入到盛有10ml冰乙酸的50ml茄形瓶中,室温搅拌反应16小时;减压蒸馏 除去溶剂,液相色谱测得3-((2-氨基苯基)氨基)-5H-萘[1,8-cd]异噻唑-5-酮1,1-二氧化物(234mg, 收率72%)
实验数据如下;黄色粉末状固体mp(262℃-267℃).
HRMS(ESI)of C
实施例7
7.1制备3-(4-氟-2-(三氟甲基)苄基)-5H-萘[1,8-cd]异噻唑-5-酮1,1-二氧化物
将5,8-二氧代-二氢萘1(100mg)、4-氟-2-(三氟甲基)苯胺(90.6mg).与一水合乙酸铜(28.4mg)加 入到盛有5ml冰乙酸的25ml圆底烧瓶中,加热回流,该温度下搅拌反应34小时;反应结束后,得到产 物粗产品47.6mg(收率47.6%)。随后加入DCM溶解后加入5g100目硅胶制砂,制砂结束后进行柱层 析分离纯化。分离结束后得到初步纯化产物,进LC-MS中分析得到里面有397分子量产物,可以进一 步纯化。进一步柱层析纯化得到纯化后产物3-(4-氟-2-(三氟甲基)苄基)-5H-萘[1,8-cd]异噻唑-5- 酮1,1-二氧化物26.2mg(收率26.2%)。
实施例8
8.1制备3-((6-氟吡啶-3-基)氨基)-5H-萘并[1,8-cd]异噻唑-5-酮1,1-二氧化物
将5,8-二氧代-二氢萘(100mg,0.421mmol)、2-氟-5氨基吡啶(56.71mg,0.506mmol)与一水合 乙酸铜(16.83mg,0.084mmol)加入25mL圆底烧瓶中,加入5ml冰乙酸,使之溶解,在室温下反应3小 时,反应完全后,减压蒸馏除去溶剂,液相色谱分离得到3-((6-氟吡啶-3-基)氨基)-5H-萘并[1,8-cd] 异噻唑-5-酮1,1-二氧化物。
实验数据如下
实施例9
9.1制备N-甲基萘-1-磺酰胺
将1-萘磺酰氯(5g,21.9mmol)加入到盛有200ml丙酮的1L圆底烧瓶中,搅拌,在冰浴中滴加240 ml 0℃的甲胺,滴加完毕后,薄层跟踪检测反应,反应完成后,减压蒸馏出有机溶剂和氨水分解出的 氨气,析出固体,过滤,真空干燥,得N-甲基萘-1-磺酰胺(4.82g,收率95.66%)。该化合物无需进一 步纯化,直接用于下一步反应。
9.2制备N-甲基-5,8-二氢萘-1-磺酰胺
将无水硫酸高铈(160g,677.45mmol)用750ml 2mol/L稀硫酸溶解,控温65℃。将N-甲基萘-1- 磺酰胺(10g,48.49mmol)加入盛有200ml冰乙酸的500ml圆底烧瓶中,控温65℃,该温度下搅拌, 缓慢滴加入至硫酸高铈水相体系中,从滴加开始计时,薄层色谱监测,27分钟后停止反应,待反应液 冷却后,过滤,滤液用1200ml二氯甲烷萃取,干燥除水后低压旋蒸,得到淡黄色固体,真空干燥,得 N-甲基-5,8-二氢萘-1-磺酰胺(6g,收率54.22%)。直接用于下一步反应无需纯化
9.3 4-(2-(((8-(N-甲基氨磺酰基)-1,4-二氧代-1,4-二氢萘-2-基)氨基)苯基)哌嗪-1-羧酸叔丁 酯
将N-甲基-5,8-二氢萘-1-磺酰胺(56.5g,238.17mmol)、叔丁基4-(2-氨基苯基)哌嗪-1-甲酸酯(60g, 217.90mmol)与一水合乙酸铜(5.6g,28.05mmol)加入到盛有1200ml冰乙酸的2L圆底烧瓶中,25℃反应, 该温度下搅拌反应26小时;减压蒸馏除去溶剂,得粗品。
在粗品中加入1500ml的无水乙醇,110℃加热沸腾,液相监测反应产物全部转化后,停止加热, 自然冷却搅拌6h,过滤得滤饼,柱层析后除去一般杂质,后加入10倍当量的二氯甲烷完全溶解后,缓 慢滴加25倍当量的异丙醇,33℃低压旋蒸后除去相同量的二氯甲烷,待晶体全部析出,过滤,得紫红 色晶体,低压干燥得4-(2-(((8-(N-甲基氨磺酰基)-1,4-二氧代-1,4-二氢萘-2-基)氨基)苯基) 哌嗪-1-羧酸叔丁酯.(54.33g,收率78.32%)。
9.4制备N-甲基-5,8-二氧杂-7-((2-(哌嗪-1-基)苯基)氨基)-5,8-二氢萘-1-磺酰胺
将(4-(2-((1,4-二氧代-5-氨磺酰基-1,4-二氢萘-2-基)氨基)苯基)哌嗪-1-羧酸叔丁酯)(2g, 3.78mmol)加入到盛有20ml二氯甲烷的50ml圆底烧瓶中,随后通入盐酸气,薄层跟踪检测反应;反应 结束后,过滤得滤饼,减压干燥分离出N-甲基-5,8-二氧杂-7-((2-(哌嗪-1-基)苯基)氨基)-5,8- 二氢萘-1-磺酰胺(1.82g,收率98%)。实验数据如下:
二、药理部分(新型冠状病毒肺炎)
1.质粒
将3CLpro的开放阅读框克隆到Nde I和Xho I多个克隆位点之间的pET28a载体中。该构建体确 保了表达的蛋白可以被3CLpro自身自动切割,并可以用PreScission蛋白酶处理以去除6ⅹHis标签。 对于PLP构建,将密码子优化的SARS-CoV-2nsp3在aa746-1062部分中插入到pET28a的Nde I和 EcoR I中,该肽与N端的SAVLQ蛋白序列融合。将第一个氨基酸(Glu)突变为Ser,以促进3CLpro 对PLP的消化,以去除N端His标签。所有突变体均使用KOD-plus-neo(TOYOBO,日本大阪)产 生。
2.重组蛋白的生产与纯化
将3CLpro和PLP的构建体分别转化到大肠杆菌BL21(DE3)(Novagen)中进行表达。有关详 细信息,将单个克隆在含有卡那霉素(50μg/mL)的LB培养基中于37℃预培养过夜,以产生种子 培养物。然后,以0.5%的异丙基-D-硫代半乳糖苷(IPTG)的比例,以1%的比例接种种子培养物以 产生足够的培养物以表达蛋白质。诱导表达12小时后,通过在4℃以5000rpm离心10分钟收获细 胞。然后,在通过超声处理裂解之前,将沉淀重悬于缓冲液A(20mM HEPES,500mM NaCl,pH 7.5) 中。超声处理后,通过在4℃下以18000rpm超离心1.5h来澄清裂解物,并使用HisTrap FF柱(GE Healthcare)将上清液用于蛋白质纯化。将获得的蛋白质在4℃下用PreScission蛋白酶以20:1的摩 尔比对3CLpro裂解一整夜,然后在用缓冲液B(20mM HEPES,100mM NaCl,1mM DTT,pH 7.5) 平衡的Superdex 200柱(GEHealthcare)上通过凝胶过滤色谱法进一步纯化。之后,使用Amicon(10 kDa临界值,Millipore)将洗脱的级分浓缩至15mg/ml。然后将浓缩的蛋白质储存在-80℃,用于结 晶和活性测定。对于PLP纯化,以20:1的摩尔比使用3CLpro来去除N端His标签。
3.SARS-CoV-2 3CLpro和PLP的抑制分析
分别使用Dabcyl-KLSAVLQSGFRKM-Edans-NH2和CBZ-RLRGG-AMC作为荧光底物,进行了 荧光共振能量转移(FRET)蛋白酶测定。为了进行3CLpro抑制分析,将100nM 3CLpro重组蛋白(化 合物的系列稀释液)在反应缓冲液1(50mM Tris-HCl,1mM EDTA,pH 7.3)中混合,共孵育50h, 混合后的总体积为50μL,加入20μM底物以引发反应。然后立即用BioTekSynergy4读板器每3秒钟 测量320nm激发和405nm发射的荧光信号。对于PLP抑制测定,进行了类似的步骤,同时在340nm (激发)和450nm(发射)监测荧光信号。使用GraphPad Prism5.0软件(GraphPad Software,Inc., San Diego,CA,USA)来计算IC50曲线。来自三个独立实验的IC 50值表示为平均值±SD。
4.LY1的抗病毒测定
非洲绿猴肾Vero E6细胞系获自美国典型培养物保藏中心(ATCC,编号1586),并保存在Dulbecco 改良的Eagle培养基(DMEM;Gibco Invitrogen)中,并补充了10%胎牛血清(FBS;Gibco Invitrogen) 和1%青霉素-链霉素(Gibco Invitrogen)。将SARS-CoV-2的临床分离株(nCoV-2019BetaCoV/WIV04 /2019)在Vero E6细胞中繁殖,并按照先前的描述确定病毒滴度(11)。所有感染实验均在生物安全 等级3(BLS-3)下进行。
5.RT-PCR
通过定量实时RT-PCR评估LY1对病毒产量的功效。将Vero E6细胞接种在96孔板中,并以0.05 的MOI感染SARS-CoV-2。感染2小时后,去除含有病毒的培养基,然后用0.1%DMSO作为对照或 2.5-15μMLL1处理细胞。处理48小时后,使用实时荧光定量PCR检测细胞上清液中病毒RNA的拷 贝数,该反应由RdRP,N和E的表达指示。
表面等离子体共振分析
6.SPR分析
SPR分析是在BiaCore T200系统(GE Healthcare,瑞典)上进行的。将目标蛋白稀释在10mM 乙酸钠中,并通过胺偶联方法固定在CM5传感器芯片上。然后将样品在运行缓冲液(PBS)中稀释。 然后,将经过分析物的药物以30μL/min的流速通过参比通道和活性通道注入。关联和解离时间均设 置为120s。在Biacore T200评估软件上,通过稳态亲和模型通过全局拟合进行亲和拟合,以获得平 衡解离常数K
实施例1.LY1体外抗病毒活性
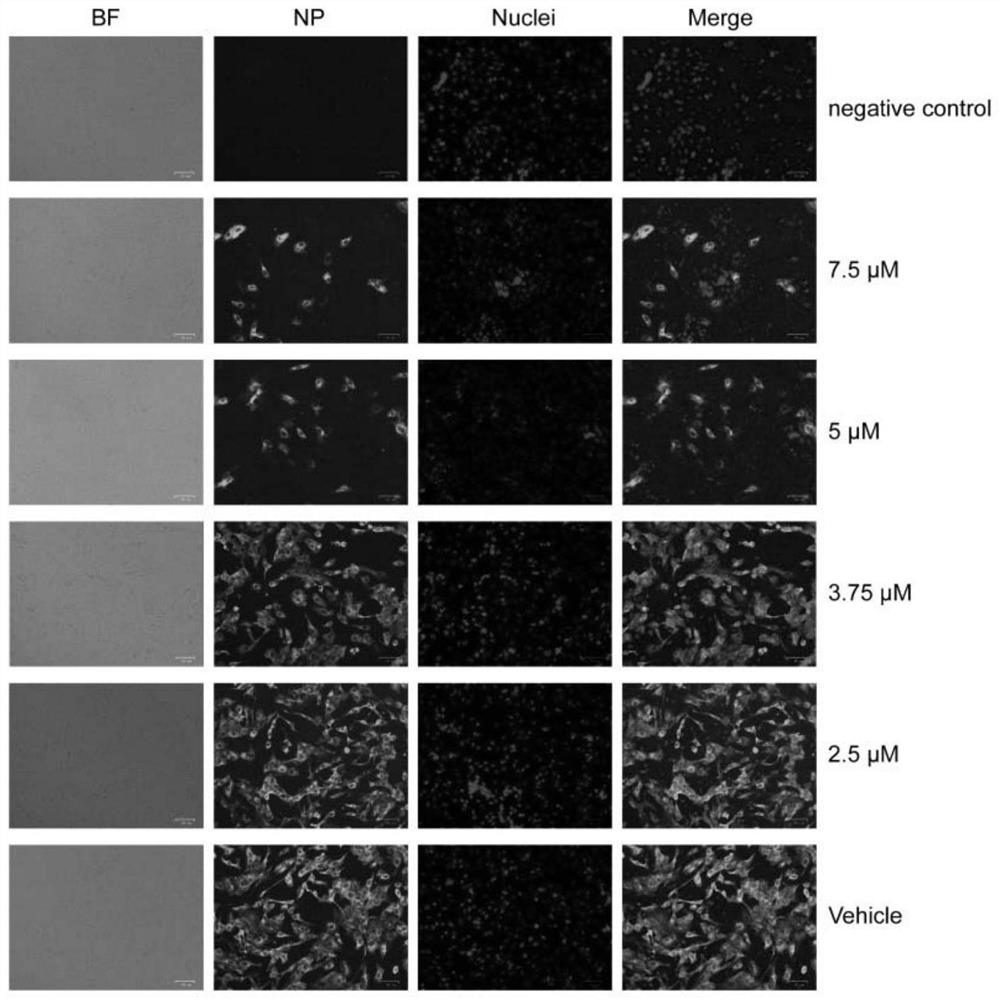
并行使用免疫荧光染色测定法和通过定量RT-PCR评估的抗病毒作用,以证明LY1的体外抗病毒 能力。我们发现在组装好的化合物库中,化合物LY1表现出强大的抗病毒活性,在30μM时抑制率 达到100%。使用免疫荧光染色测定法,用抗核蛋白(NP)抗体和DAPI染色,在显微镜下观察到结 果LY1使得NP明显减少,如图1所示。通过定量实时RT-PCR,化合物LY1表现出良好的抗病毒活 性,EC50值为3.93μM,见图2。化合物LY1的细胞毒性通过MTT法评估,化合物LY1的CC50值 高于15μM。如图2所示。实验结果表明,LY1在体外具有优秀的抗病毒活性和较小的毒性。
实施例2.LY1抑制3CLpro或PLpro的活性
从大肠杆菌(E.coli)发酵液纯化得到重组SARS-CoV-2 3CLpro和PLpro。为了测量化合物LY1 对3CLpro或PLpro的抑制能力,分别使用Dabcyl-KLSAVLQSGFRKM-Edans-NH2和CBZ-RLRGG-AMC作为荧光底物,进行了荧光共振能量转移(FRET)蛋白酶测定。结果如图3所示, 当浓度为10μM时,对3CLpro和PLpro的抑制效果分别达到94.4%和88.6%。结果表明化合物LY1 能有效抑制SARS-CoV-2 3CLpro和PLpro的活性。我们使用了基于荧光共振能量转移(FRET)的裂 解测定法来确定IC
实施例3.LY1主要靶向3CLpro蛋白
为了进一步确定3CLpro和PLpro作为潜在的靶标,我们然后通过表面等离振子共振(SPR)分 析来表征3CLpro和PLpro与化合物LY1之间的亲和力。K
- 一类化合物及其用于新型冠状病毒肺炎的医药用途
- 一类三唑类化合物、制备方法及其医药用途