一种基于微波作用下合成的碳气凝胶催化剂及其合成方法和应用
文献发布时间:2023-06-19 12:21:13
技术领域
本发明属于材料制备技术领域,具体涉及一种基于微波作用下合成的碳气凝胶催化剂及其合成方法和应用。
背景技术
双氧水(过氧化氢)是一种重要的化学物质,在工业、医药、环保、军工、食品和环境等领域有着广泛的应用。他既有氧化性又有还原性并且使用后无二次污染,被定义为绿色化工产品。目前工业上大规模生产双氧水的最成熟的方法是蒽醌法,但蒽醌法不仅步骤繁琐,还会用到一些有机溶剂,对环境造成二次污染,因此寻找一种绿色高效的方法生产双氧水迫在眉睫。
在电化学氧还原的反应(ORR)中,有两种反应途径:
O
O
即反应转移4e-途径产生H
目前电催化氧化还原反应的高效催化剂,大部分都是选择4e-途径;只有贵金属及其合金、单原子催化剂、碳基材料、金属配合物等可以催化选择2e-途径,却价格昂贵,并且要精确控制催化剂的结构等,制备条件苛刻。
碳气凝胶作为一种新型的碳材料,具有低密度,质量轻、比表面积大、孔隙率大、表面疏水、导电性较好等性质,在吸附、能量的转化与储存、电容器和催化剂等领域具有广泛的应用。
目前制备碳气凝胶常用方法是常温常压干燥法,超临界干燥法等,超临界干燥法在理论上能够消除干燥过程中的表面张力,但其在高温高压环境下操作具有一定的危险性,且溶剂置换过程的周期长,需要将湿凝胶中的水替换为超临界干燥的介质(如二氧化碳、石油醚或乙醇等),再将干燥介质排出,这一过程需要极其缓慢以避免破环介质的超临界状态。近年来研究比较火热的常压干燥法是将湿凝胶放入低表面张力系数的溶剂中反复浸泡,将湿凝胶中的水置换为低表面张力溶剂,在这个过程中不仅耗时长,而且需要使用数十倍乃至数百倍凝胶体积的溶剂,这些有机溶剂如乙醇、丙酮等多种有机溶剂,易挥发,有毒性且难回收,对环境造成较大的污染。
发明内容
针对现有技术中存在的上述问题,本发明的目的在于提供一种基于微波作用下合成的碳气凝胶催化剂及其合成方法和应用,本发明利用微波合成法代替常温搅拌法,优点在于操作简单、合成高效,且制得的催化剂具有较好的疏水性、稳定性和电催化性能,在应用于制备双氧水中具有良好的应用前景。
所述的一种基于微波作用下合成碳气凝胶催化剂的方法,其特征在于包括以下步骤:以间苯二酚和甲醛为反应原料,以水为溶剂,将反应原料加入水中并调节pH至5~7之间后,将反应液体系置于微波辐射环境下进行反应,反应结束后烘干,所得固体转移至管式炉中于氮气气氛下焙烧,即制得所述碳气凝胶催化剂产品。
所述的一种基于微波作用下合成碳气凝胶催化剂的方法,其特征在于具体包括以下步骤:
1)向烧杯中加入间苯二酚和去离子水溶剂,搅拌,加入甲醛溶液并搅拌均匀,然后用浓度为0.5~2mol/L的氢氧化钠溶液调节pH至5~7之间;
2)将步骤1)所得反应液体系转移至反应管中,再将反应管放置在微波合成仪中,开启微波合成仪进行微波辐射加热,使反应液体系升温至70~90℃下反应,微波辐射时间控制在10~30min,使反应管中反应形成湿凝胶;
3)步骤2)反应结束后,将合成的湿凝胶从反应管转移到培养皿中,然后放入烘箱中烘干,干燥好的固体转移到瓷舟中,然后将瓷舟放置于管式炉中于氮气气氛下焙烧,焙烧结束后冷却至室温,用研钵充分研磨,即得最终的碳气凝胶催化剂。
所述的一种基于微波作用下合成碳气凝胶催化剂的方法,其特征在于步骤1)中,甲醛溶液的质量浓度在30~40%,优选为35~37%;间苯二酚与甲醛的摩尔比为0.33~0.7:1,优选为0.5:1;间苯二酚的质量与去离子水溶剂的体积之比是1:3~8,优选为1:5,质量的单位是g,体积的单位是mL。
所述的一种基于微波作用下合成碳气凝胶催化剂的方法,其特征在于步骤1)中,氢氧化钠溶液的浓度为1mol/L,用氢氧化钠溶液调节pH至5.8~6.2。
所述的一种基于微波作用下合成碳气凝胶催化剂的方法,其特征在于步骤2)中,微波合成仪的运行功率控制在100~300W之间,优选200W;反应温度控制在85℃,微波辐射时间控制在15min。
所述的一种基于微波作用下合成碳气凝胶催化剂的方法,其特征在于步骤3)中,烘箱中干燥的温度在50~80℃,优选为60℃;干燥时间在8~18h,优选为12h。
所述的一种基于微波作用下合成碳气凝胶催化剂的方法,其特征在于步骤3)中,管式炉中焙烧的过程为:从室温以3~10℃/min的升温速率升温至500~900℃,优选为600℃,然后恒温焙烧1~5h,优选为3h,最后自然降温至室温,即制得催化剂产品;其中,管式炉中焙烧的升温速率优选为5℃/min。
按照上述方法合成的碳气凝胶催化剂。
所述的碳气凝胶催化剂在电催化氧还原制备双氧水中的应用。
通过上述技术制备的催化剂,与现有的催化剂相比,具备以下有益效果:
本发明采用微波法制备碳气凝胶催化剂的过程,微波辐射被用作有机干凝胶合成中的加热源,使得所有阶段(凝胶化、老化和干燥)在一个简单快速的装置中进行操作简单,其耗时短,所制备得催化剂具有较好的选择性、优异的循环稳定性,良好的电化学活性。
附图说明
图1为本发明实施例1微波合成10min制备的10-CXGs-600的SEM图;
图2为本发明实施例2微波合成15min制备的15-CXGs-600的SEM图;
图3为本发明实施例3微波合成20min制备的20-CXGs-600的SEM图;
图4为本发明实施例4微波合成25min制备的25-CXGs-500的SEM图;
图5为本发明实施例5微波合成25min制备的25-CXGs-600的SEM图;
图6为本发明实施例6微波合成30min制备的30-CXGs-500的SEM图;
图7为本发明实施例7微波合成30min制备的30-CXGs-800的SEM图;
图8为本发明实施例8微波合成30min制备的30-CXGs-600的SEM图;
图9为本发明对比例1常温搅拌制备的CXGs-600的SEM图;
图10为本发明对比例2常温搅拌制备的CXGs-800的SEM图;
图11为本发明对比例3常温搅拌制备的CXGs-500的SEM图;
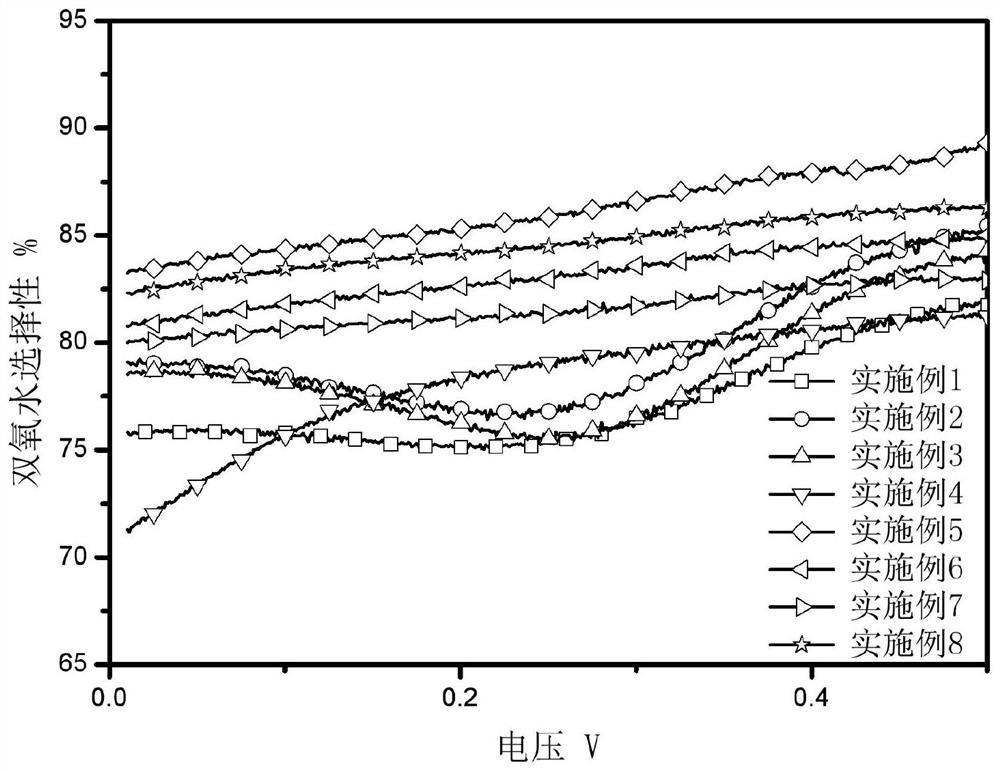
图12为分别以实施例1-8的催化剂进行电催化反应时,反应的双氧水选择性与电压之间的关系曲线对比图;
图13为分别以实施例1-8的催化剂进行电催化反应时,反应的电子转移数与电压之间的关系曲线对比图;
图14为分别以对比例1-3的催化剂进行电催化反应时,反应的双氧水选择性与电压之间的关系曲线对比图;
图15为分别以对比例1-3的催化剂进行电催化反应时,反应的电子转移数与电压之间的关系曲线对比图;
图16为催化剂25-CXGs-600的疏水性图;
图17为催化剂25-CXGs-600的稳定性图。
具体实施方式
下面结合具体实施例对本发明作进一步说明,但本发明的保护范围并不限于此。
实施例1
微波合成10min制备10-CXGs-600,包括以下步骤:
称取质量2.20g的间苯二酚置于50mL的烧杯中,加入10.8mL的水室温下搅拌10min,搅拌转速为700rpm,用移液管取4mL的甲醛溶液(质量浓度37%)加入上述溶液中,搅拌10min,转速700rpm,滴加1mol/L的氢氧化钠溶液调节pH值在6左右。然后将所得反应液体系转移至反应管中,再将反应管放置在微波合成仪中,开启微波合成仪进行微波辐射加热(微波合成仪的运行功率控制在200W,以下实施例的微波功率与实施例1中相同),使反应液体系升温至85℃、微波辐射时间10min。然后将合成的湿凝胶从反应管转移到培养皿中,放入烘箱60℃干燥12h。然后转移到瓷舟,放在管式炉中以氮气作为保护气体,从室温以5℃/min的升温速率升温至600℃,然后在600℃下恒温煅烧3h,随后冷却至室温。将煅烧后的块状研磨成粉末状,得到10-CXGs-600。
实施例2
微波合成15min制备15-CXGs-600,包括以下步骤;
称取质量2.20g的间苯二酚置于50mL的烧杯中,加入10.8mL的水室温下搅拌10min,搅拌转速为700rpm,用移液管取4mL的甲醛溶液(质量浓度37%)加入上述溶液中,搅拌2min,转速700rpm,滴加1mol/L的氢氧化钠溶液调节pH值在6左右。然后将所得反应液体系转移至反应管中,再将反应管放置在微波合成仪中,开启微波合成仪进行微波辐射加热,使反应液体系升温至85℃、微波辐射时间15min。然后将合成的湿凝胶从反应管转移到培养皿中,放入烘箱60℃干燥12h。然后转移到瓷舟,放在管式炉中以氮气作为保护气体,从室温以5℃/min的升温速率升温至600℃,然后在600℃下恒温煅烧3h,随后冷却至室温。将煅烧后的块状研磨成粉末状,得到15-CXGs-600。
实施例3
微波合成20min制备20-CXGs-600,包括以下步骤:
称取质量2.20g的间苯二酚置于50mL的烧杯中,加入10.8mL的水室温下搅拌10min,搅拌转速为700rpm,用移液管取4mL的甲醛溶液(质量浓度37%)加入上述溶液中,搅拌10min,转速700rpm,滴加1mol/L的氢氧化钠溶液调节pH值在6左右。然后将所得反应液体系转移至反应管中,再将反应管放置在微波合成仪中,开启微波合成仪进行微波辐射加热,使反应液体系升温至85℃、微波辐射时间20min。然后将合成的湿凝胶从反应管转移到培养皿中,放入烘箱60℃干燥12h。然后转移到瓷舟,放在管式炉中以氮气作为保护气体,从室温以5℃/min的升温速率升温至600℃,然后在600℃下恒温煅烧3h,随后冷却至室温。将煅烧后的块状研磨成粉末状,得到20-CXGs-600。
实施例4
微波合成25min制备25-CXGs-500,包括以下步骤:
称取质量2.20g的间苯二酚置于50mL的烧杯中,加入10.8mL的水室温下搅拌10min,搅拌转速为700rpm,用移液管取4mL的甲醛溶液(质量浓度37%)加入上述溶液中,搅拌10min,转速700rpm,滴加1mol/L的氢氧化钠溶液调节pH值在6左右。然后将所得反应液体系转移至反应管中,再将反应管放置在微波合成仪中,开启微波合成仪进行微波辐射加热,使反应液体系升温至85℃、微波辐射时间25min。然后将合成的湿凝胶从反应管转移到培养皿中,放入烘箱60℃干燥12h。然后转移到瓷舟,放在管式炉中以氮气作为保护气体,从室温以5℃/min的升温速率升温至500℃,然后在500℃下恒温煅烧3h,随后冷却至室温。将煅烧后的块状研磨成粉末状,得到25-CXGs-500。
实施例5
微波合成25min制备25-CXGs-600,包括以下步骤:
称取质量2.20g的间苯二酚置于50mL的烧杯中,加入10.8mL的水室温下搅拌10min,搅拌转速为700rpm,用移液管取4mL的甲醛溶液(质量浓度37%)加入上述溶液中,搅拌10min,转速700rpm,滴加1mol/L的氢氧化钠溶液调节pH值在6左右。然后将所得反应液体系转移至反应管中,再将反应管放置在微波合成仪中,开启微波合成仪进行微波辐射加热,使反应液体系升温至85℃、微波辐射时间25min。然后将合成的湿凝胶从反应管转移到培养皿中,放入烘箱60℃干燥12h。然后转移到瓷舟,放在管式炉中以氮气作为保护气体,从室温以5℃/min的升温速率升温至600℃,然后在600℃下恒温煅烧3h,随后冷却至室温。将煅烧后的块状研磨成粉末状,得到25-CXGs-600。
实施例6
微波合成30min制备30-CXGs-500,包括以下步骤:
称取质量2.20g的间苯二酚置于50mL的烧杯中,加入10.8mL的水室温下搅拌10min,搅拌转速为700rpm,用移液管取4mL的甲醛溶液(质量浓度37%)加入上述溶液中,搅拌10min,转速700rpm,滴加1mol/L的氢氧化钠溶液调节pH值在6左右。然后将所得反应液体系转移至反应管中,再将反应管放置在微波合成仪中,开启微波合成仪进行微波辐射加热,使反应液体系升温至85℃、微波辐射时间30min。然后将合成的湿凝胶从反应管转移到培养皿中,放入烘箱60℃干燥10h。然后转移到瓷舟,放在管式炉中以氮气作为保护气体,从室温以3℃/min的升温速率升温至500℃,然后在500℃下恒温煅烧4h,随后冷却至室温。将煅烧后的块状研磨成粉末状,得到30-CXGs-500。
实施例7
微波合成30min制备30-CXGs-800,包括以下步骤:
称取质量2.20g的间苯二酚置于50mL的烧杯中,加入10.8mL的水室温下搅拌10min,搅拌转速为700rpm,用移液管取4mL的甲醛溶液(质量浓度37%)加入上述溶液中,搅拌10min,转速700rpm,滴加1mol/L的氢氧化钠溶液调节pH值在6左右。然后将所得反应液体系转移至反应管中,再将反应管放置在微波合成仪中,开启微波合成仪进行微波辐射加热,使反应液体系升温至85℃、微波辐射时间30min。然后将合成的湿凝胶从反应管转移在培养皿中,放入烘箱60℃干燥8h。然后转移到瓷舟,放在管式炉中以氮气作为保护气体,从室温以5℃/min的升温速率升温至800℃,然后在800℃下恒温煅烧5h,随后冷却至室温。将煅烧后的块状研磨成粉末状,得到30-CXGs-800。
实施例8
微波合成30min制备30-CXGs-600,包括以下步骤:
称取质量2.02g的间苯二酚置于50mL的烧杯中,加入10.8mL的水室温下搅拌10min,搅拌转速为700rpm,用移液管取4mL的甲醛溶液(质量浓度37%)加入上述溶液中,搅拌10min,转速700rpm,滴加1mol/L的氢氧化钠溶液调节pH值在6左右。然后将所得反应液体系转移至反应管中,再将反应管放置在微波合成仪中,开启微波合成仪进行微波辐射加热,使反应液体系升温至85℃、微波辐射时间30min。然后将合成的湿凝胶从反应管转移在培养皿中,放入烘箱60℃干燥8h。然后转移到瓷舟,放在管式炉中以氮气作为保护气体,从室温以5℃/min的升温速率升温至600℃,然后在600℃下恒温煅烧5h,随后冷却至室温。将煅烧后的块状研磨成粉末状,得到30-CXGs-600。
对比例1
常温搅拌制备CXGs-600,包括以下步骤:
称取质量2.20g的间苯二酚置于50mL的烧杯中,加入10.8mL的水室温下搅拌10min,搅拌转速为700rpm,用移液管取4mL的甲醛溶液(质量浓度37%)加入上述溶液中,搅拌10min,转速700rpm,滴加1mol/L的氢氧化钠溶液调节pH值在6左右。常温搅拌24h,将合成的湿凝胶从烧杯转移在培养皿中,放入烘箱60℃干燥8h。然后转移到瓷舟,放在管式炉中以氮气作为保护气体,从室温以5℃/min的升温速率升温至600℃,然后在600℃下恒温煅烧5h,随后冷却至室温。将煅烧后的块状研磨成粉末状,得到CXGs-600。
对比例2
常温搅拌制备CXGs-800,包括以下步骤:
称取质量2.20g的间苯二酚置于50mL的烧杯中,加入10.8mL的水室温下搅拌10min,搅拌转速为700rpm,用移液管取4mL的甲醛溶液(质量浓度37%)加入上述溶液中,搅拌10min,转速700rpm,滴加1mol/L的氢氧化钠溶液调节pH值在6左右。常温搅拌24h,将合成的湿凝胶从烧杯转移在培养皿中,放入烘箱60℃干燥8h。然后转移到瓷舟,放在管式炉中以氮气作为保护气体,从室温以5℃/min的升温速率升温至800℃,然后在800℃下恒温煅烧5h,随后冷却至室温。将煅烧后的块状研磨成粉末状,得到CXGs-800。
对比例3
常温搅拌制备CXGs-500,包括以下步骤:
称取质量2.02g的间苯二酚置于50mL的烧杯中,加入10.8mL的水室温下搅拌10min,搅拌转速为700rpm,用移液管取4mL的甲醛溶液(质量浓度37%)加入上述溶液中,搅拌10min,转速700rpm,滴加1mol/L的氢氧化钠溶液调节pH值在6左右。常温搅拌24h,将合成的湿凝胶从烧杯转移在培养皿中,放入烘箱60℃干燥8h。然后转移到瓷舟,放在管式炉中以氮气作为保护气体,从室温以5℃/min的升温速率升温至500℃,然后在500℃下恒温煅烧5h,随后冷却至室温。将煅烧后的块状研磨成粉末状,得到CXGs-500。
从实施例1-8和对比例1-3的实验过程来看,相比较于常规的常温合成碳气凝胶,微波合成的碳气凝胶催化剂的合成时间大大缩短。
实施例1-8制备的催化剂的SEM图分别如图1-8所示。图1~8展示了在不同微波合成条件下制得的催化剂的SEM图形貌,可以看出:在微波时间10~30min、碳化温度在500~800℃时,制得的催化剂均为球状,且球的尺寸比较均匀,在3~5nm,升高碳化温度并没有明显破坏催化剂的形貌。
对比例1-3制备的催化剂的SEM图分别如图9-11所示。图9~11展示了在常温搅拌条件下制得的催化剂的SEM图形貌,可以看出:常温搅拌制备的催化剂呈现多孔的纳米块状,块状边缘有很多的缺陷。
对比图1~8的表征结果以及图9~11的表征结果可以看出,两种方法合成的催化剂形貌截然不同。相对于对比例1-3的制备方法,实施例1-8采用微波合成方法获得的催化剂具有更好的形貌结构,由此得出结论:相比较于常规的常温合成碳气凝胶,微波合成的碳气凝胶催化剂的不仅合成时间大大缩短,而且很可能具备更好的电催化性能。两种合成方法的不同,造成制得的催化剂的形貌具有巨大差异性的原因推测是:反应液体系在微波辐射下加热反应的方法,甲醛和间苯二酚作为加热介质,其导电率和极化率比较好,另外甲醛分子小、极性相对较大,这使得它更容易吸收微波而被加热,在合成的过程中,从前驱体直接合成球状,不会经过块状的过程。
应用实施例1:
分别验证实施例1-8和对比例1-3的催化剂的电催化性能:
分别以实施例1-8和对比例1-3的催化剂,制备催化剂浆液:取催化剂4.0mg、100μL杜邦5%的nafion溶液和900μL无水乙醇,超声30min分散均匀,分别得到利用实施例1-8和对比例1-3的催化剂制备的相应的催化剂浆液。将5μL催化剂浆液涂在旋转环盘电极的圆形玻碳区域,干燥后形成工作电极。
采用电化学工作站作为电化学发生装置,以涂覆有催化剂的旋转环盘电极作为工作电极,铂丝作为对电极,饱和甘汞作为参比电极,其中铂环端的电压E
利用Koutecky-Levich(K-L)方程来研究阴极氧还原的电子数和双氧水的选择性,计算公式分别如式(3)和式(4)所示:
式(3)中,n表示阴极氧还原的转移电子数;I
和(4)式,%H
在测试过程中,分别以实施例1-8的催化剂进行电催化反应时,在不同电压条件下的对双氧水选择性情况结果如图12所示,在不同电压条件下的转移电子数结果如图13所示。分别以对比例1-3的催化剂进行电催化反应时,在不同电压条件下的对双氧水选择性情况结果如图14所示,在不同电压条件下的转移电子数结果如图15所示。
图12~15是催化剂的电催化化学性能测试结果汇总,可以看出:实施例5的催化剂对二电子双氧水的选择性最好,最高可达90%,也就是在微波合成时间25min,碳化温度在600℃的条件下制得的催化剂效果最好。即催化剂的制备过程中,微波合成时间需要选择合理的范围,这是因为微波合成时间长、碳化温度高时,形成的水凝胶有大量的交联结构,高温碳化时会使结构坍塌,破坏活性位,不利于氧还原反应;而微波时间短、碳化温度低时凝胶的还没完全,甲醛与间苯二酚还没达到胶凝点,导致合成的结构弱支化,未完全凝胶化,机械强度较弱同时对活性位也有影响,所制备的水凝胶的电催化效果不好。
图16是25-CXGs-600的疏水性图,取0.5g实施例5的催化剂置于桌面上,滴5水滴到催化剂上,小水珠会融合成大水珠,若催化剂为亲水则也会溶于水中,从图16中可以看出,催化剂是包覆在水珠的表面上而不是与水相溶。
应用实施例2(测试催化剂的使用寿命):
以实施例5的25-CXGs-600材料作为催化剂,制备催化剂浆液:取催化剂4.0mg、100μL杜邦5%的nafion溶液和900μL无水乙醇,超声30min分散均匀,得到催化剂浆液。
双氧水的寿命测试(电流i-时间t):将5μL催化剂浆液涂在旋转环盘电极的圆形玻碳区域,干燥后形成工作电极。采用电化学工作站作为电化学发生装置,以涂覆有催化剂的旋转环盘电极作为工作电极,铂丝作为对电极,饱和甘汞作为参比电极,其中铂环端的电压E
图17反映的是25-CXGs-600的电催化反应稳定性图,在反应60个小时后,电流密度仍然是保持不变。因此,可以得出结论,通过调控微波合成的时间和碳化温度可以促进间苯二酚与甲醛的凝胶化,使得制备的碳气凝胶具有较好的稳定性,且形貌更容易在反应中暴露活性位,促进电催化氧还原过程,达到很好的催化效果。
本说明书所述的内容仅仅是对发明构思实现形式的列举,本发明的保护范围不应当被视为仅限于实施例所陈述的具体形式。
- 一种基于微波作用下合成的碳气凝胶催化剂及其合成方法和应用
- 一种合成气合成高碳醇的Cu-Fe基催化剂及其制备方法和其在合成气合成高碳醇工艺中的应用