一种m6A高通量测序数据的分析方法和系统
文献发布时间:2023-06-19 18:53:06
技术领域
本发明属于生物信息技术领域,具体地,涉及一种m6A高通量测序数据的分析方法和系统。
背景技术
RNA修饰作为继DNA修饰和组蛋白修饰后又一种比较重要的生物学现象,在RNA序列不发生改变的情况可以影响基因表达和蛋白质合成。已知RNA上含有170多种修饰,其中m6A修饰是丰度最高的RNA碱基修饰之一,并成为近年来出现的一种控制真核细胞基因表达的新的调控机制。这种发生在腺嘌呤A碱基上N第六位的甲基修饰,在信使RNA(mRNA)、长链非编码RNA(lncRNA)、小RNA(miRNA)、环状RNA(circRNA)、核糖体RNA(rRNA)中均广泛存在。作为一种可逆的表观遗传修饰,m6A影响修饰RNA分子的命运,并在几乎所有重要的生物过程中发挥重要作用。
lncRNA是一组内源性RNA分子,它们不翻译成蛋白质,但在调节基因表达方面具有特殊功能,可分为长度超过200nt的lncRNA和长度小于200nt的小ncRNA。除了蛋白质编码的mRNA外,在ncRNA中还发现了m6A修饰,包括lncRNA、circRNA、miRNA等,并被证明m6A对这些RNA的表达和功能很重要。
近些年m6A修饰已经从基础科学研究领域慢慢拓展到了各种下游应用场景中。包括各种肿瘤细胞靶向药开发、免疫治疗抑制剂开发、粮食增产等几乎涵盖医学和农学的各个领域研究都需要研究者前期对样本中潜在的RNA修饰信息进行检测。采用高通量测序的方法可以对样本中所有RNA的修饰信息进行检测。其中基于m6A抗体IP原理的m6A-seq测序方法是目前最为普遍的实验方法,每天也产生了大量的m6A-seq数据,需要一种能够同时挖掘mRNA和lncRNA中m6A修饰的生物信息学分析方法就尤为重要。
发明内容
为了解决上述技术问题,本发明旨在提供一种可同时分析mRNA和lncRNA的m6A-seq高通量测序的生物信息分析方法。为此,本发明采用的技术方案如下:
本发明第一方面提供一种m6A-seq高通量测序数据的分析方法,包括以下步骤:
S1,m6A-seq高通量测序数据进行处理,过滤掉不合格的序列,其中,所述m6A-seq测序数据来自至少一对配对的IP样本和Input样本。其中IP指的是m6A测序中免疫沉淀immunoprecipitation的缩写,Input是m6A实验中的一种阳性对照。Input样本不进行IP实验,可以帮助判断m6A的IP效果,通过Input和IP对比,判断m6A修饰程度大小及实验效果;
S2,利用包括核糖体RNA序列和/或支原体RNA序列的数据库过滤掉污染序列;
S3,将过滤后的序列与样本来源物种参考基因组进行比对,得到能比对到所述参考基因组上的Reads;
S4,全基因组层面Peak Calling与注释:对能比对到所述参考基因组上的Reads进行全基因组层面的peak鉴定,得到peak在基因组上的位置及peak长度和Reads数量信息,对Peak在基因组上的位置进行注释,并对Reads在转录起始位置和终止位置分布进行分析;
S5,差异Peak分析:取IP样本和Input样本交集的Peak进行差异Peak分析,得到具有差异的Peak,将具有差异的Peak对应的相关基因采用负二项式分布法进行GO和KEGG功能富集分析;
S6,Peak序列motif分析:对步骤S4中得到的单个样本Peak以及步骤S5中得到的具有差异的Peak进行motif分析;
S7,mRNA与lncRNA表达丰度分析:对样本中mRNA以及lncRNA的进行定量分析,得到每个mRNA和lncRNA在样本中的原始值Counts以及归一化表达量FPKM值;
S8.差异mRNA和lncRNA表达谱分析:对IP样本和Input样本中的每个mRNA和lncRNA进行差异分析,并对所有差异相关基因采用负二项式分布法进行GO和KEGG功能富集分析。
在本发明的一些实施方案中,步骤S1中,所述不合格的序列包括:①带有接头的reads;②含有N碱基大于5%的reads;③质量值Q≤10的碱基数占整个read的20%以上的reads;④碱基均为A或N的reads。
在本发明的一些实施方案中,步骤S4中,所述Peak鉴定的方法为:
S41,对外显子区域进行扫描,获得候选Peak区域;
S42,假定基因位于外显子窗口w=[w
X
X
其中,X
S43,使用C-test对候选Peak区域的X1,w~Poisson(n1λ1,w)和X0,w~Poisson(n0λ0,w)两个Poisson的平均值比值c=λ1,w/λ0,w进行比较,并计算p-value;
S44,使用Fisher检验计算候选Peak区域的p-value,小于p-value的区域为Peak,其中,p-value<0.05。
在本发明的一些实施方案中,步骤S7中,FPKM值计算公式如下:
其中,total exon Fragments表示总外显子片段reads数,Mappedreads表示能比对到基因组上的Reads百万数,exon length表示外显子的kb长度。
根据权利要求1所述的分析方法,其特征在于,步骤S8中,所述采用负二项式分布法进行GO和KEGG功能富集分析,公式为:
其中,pvalue of enriched GO/KEGG代GO/KEGG功能富集分析数值,TB代表所有背景基因,TS代表所有差异表达基因,B代表所有带有GO或KEGG注释的基因,S代表所有所有带有GO或KEGG注释的差异表达基因。
本发明的第二方面提供一种m6A-seq测序数据的分析系统,包括以下模块:
数据输入模块,用于接受m6A-seq高通量测序数据输入并进行预处理;
比对模块;与数据输入模块,用于将预处理后的序列与样本来源物种参考基因组进行比对,得到能比对到所述参考基因组上的Reads;
Peak Calling与注释模块,与所述比对模块连接,用于对能比对到所述参考基因组上的Reads进行全基因组层面的peak鉴定,得到peak在基因组上的位置及peak长度和Reads数量信息,对Peak在基因组上的位置进行注释,并对Reads在转录起始位置和终止位置分布进行分析;
差异Peak分析模块,与所述Peak Calling与注释模块连接,用于取IP样本和Input样本交集的Peak进行差异Peak分析,得到具有差异的Peak,并用于将具有差异的Peak对应的相关基因采用负二项式分布法进行GO和KEGG功能富集分析;
Peak序列motif分析模块,与所述差异Peak分析模块连接,用于将得到的单个样本Peak以及具有差异的Peak进行motif分析;
mRNA与lncRNA表达丰度分析模块,与所述比对模块连接,用于对样本中mRNA以及lncRNA的进行定量分析,得到每个mRNA和lncRNA在样本中的原始值Counts以及归一化表达量FPKM值;
差异mRNA和lncRNA表达谱分析模块,与所述mRNA与lncRNA表达丰度分析模块连接,用于对IP样本和Input样本中的每个mRNA和lncRNA进行差异分析,并对所有差异相关基因采用负二项式分布法进行GO和KEGG功能富集分析。
在本发明的一些实施方案中,所述数据输入模块中预处理是指过滤掉不合格的序列和/或污染序列,所述不合格的序列包括:①带有接头的reads;②含有N碱基大于5%的reads;③质量值Q≤10的碱基数占整个read的20%以上的reads;④碱基均为A或N的reads,利用包括核糖体RNA序列和/或支原体RNA序列的数据库过滤掉污染序列。
在本发明的一些实施方案中,所述Peak Calling与注释模块中,所述Peak鉴定的方法为:
S41,对外显子区域进行扫描,获得候选Peak区域;
S42,假定基因位于外显子窗口w=[w1,w2],计算IP和Input比对到窗口w=[w1,w2]内的reads数量:
X1,w~Poisson(n1λ1,w),
X0,w~Poisson(n0λ0,w),
其中,X1,w和X0,w是IP样本和Input样本比对到窗口w=[w1,w2]内的reads数量,n1和n0分别是IP样本和Input样本的reads总数,λ1,w和λ0,w分别表示IP样本和Input样本测序reads落入窗口w的概率;
S43,使用C-test对候选Peak区域的X1,w~Poisson(n1λ1,w)和X0,w~Poisson(n0λ0,w)比值c=λ1,w/λ0,w进行比较,并计算p-value;
S44,使用Fisher检验计算候选Peak区域的p-value,小于p-value的区域为Peak,其中,p-value<0.05。
在本发明的一些实施方案中,所述mRNA与lncRNA表达丰度分析模块中,FPKM值计算公式如下:
其中,total exon Fragments表示总外显子片段reads数,Mapped reads表示能比对到基因组上的Reads百万数,exon length表示外显子的kb长度。
在本发明的一些实施方案中,所述差异mRNA和lncRNA表达谱分析模块中,所述采用负二项式分布法进行GO和KEGG功能富集分析,公式为:
其中,pvalue of enriched GO/KEGG代GO/KEGG功能富集分析数值,TB代表所有背景基因,TS代表所有差异表达基因,B代表所有带有GO或KEGG注释的基因,S代表所有所有带有GO或KEGG注释的差异表达基因。
本发明的有益效果
相对于现有技术,本发明具有以下有益效果:
(1)利用本发明的方法或系统,可以对一种实验方法获得的高通量测序数据同时检测mRNA和lncRNA上的m6A修饰信息和表达量信息,可充分对数据从4个维度进行信息挖掘
(2)本发明的分析方法和系统在Peak Calling时,只扫描外显子区域,不扫描内含子区域,仅仅报告已注释的mRNA和lncRNA的Peak信息,使得信息更加具有针对性。
(3)本发明的分析方法和系统输出了Peak的counts值,会使Peak的丰度值更加精确
附图说明
图1示出了本发明lncRNA的m6A-seq高通量测序数据的生物信息分析流程图。
图2示出了本发明实施例中Reads在基因组上外显子和内含子占比饼状图。
图3示出了本发明实施例中Peak在相对基因位置的分布图。
图4示出了本发明实施例中Peak在不同基因功能原件上的占比饼状图。
图5示出了本发明实施例中Peak对应motif图。
图6示出了本发明实施例中基因表达盒形图。
图7示出了本发明实施例中基因表达密度图。
图8示出了本发明实施例中差异基因热图。
图9示出了本发明实施例中差异基因火山图。
图10本发明实施例中GO term功能富集柱状图。
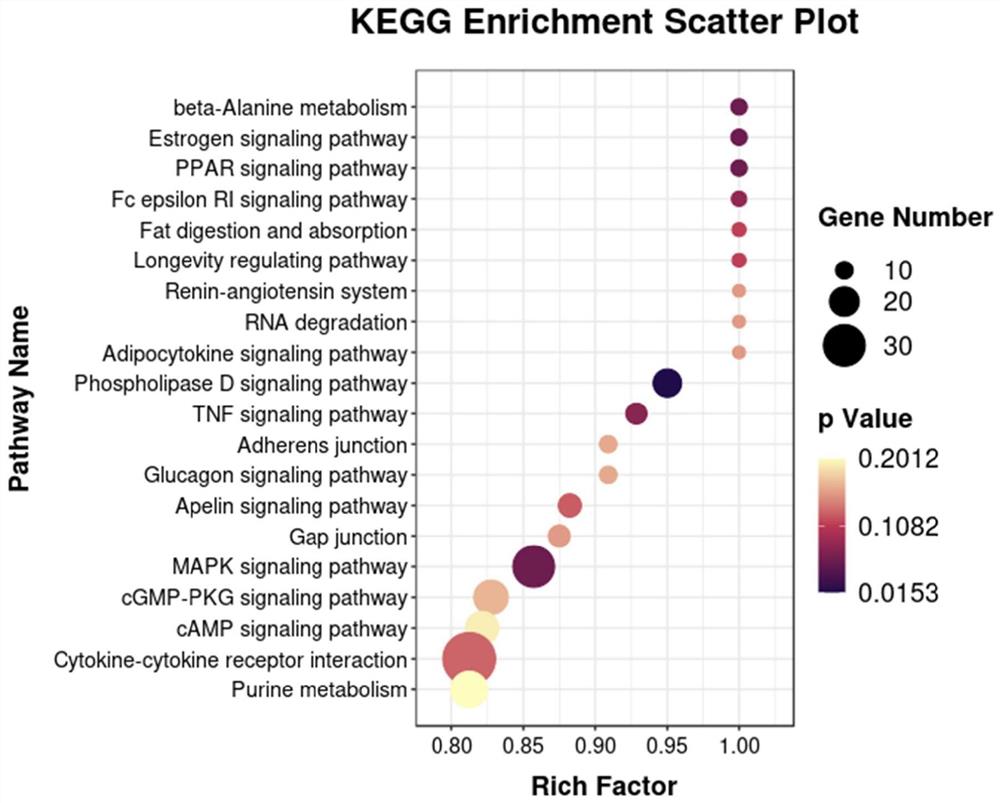
图11本发明实施例中KEGG通路通路富集气泡图。
具体实施方式
除非另有说明、从上下文暗示或属于现有技术的惯例,否则本申请中所有的份数和百分比都基于重量,且所用的测试和表征方法都是与本申请的提交日期同步的。在适用的情况下,本申请中涉及的任何专利、专利申请或公开的内容全部结合于此作为参考,且其等价的同族专利也引入作为参考,特别这些文献所披露的关于本领域中的合成技术、产物和加工设计、聚合物、共聚单体、引发剂或催化剂等的定义。如果现有技术中披露的具体术语的定义与本申请中提供的任何定义不一致,则以本申请中提供的术语定义为准。
本申请中的数字范围是近似值,因此除非另有说明,否则其可包括范围以外的数值。数值范围包括以1个单位增加的从下限值到上限值的所有数值,条件是在任意较低值与任意较高值之间存在至少2个单位的间隔。例如,如果记载组分、物理或其它性质(如分子量,熔体指数等)是100至1000,意味着明确列举了所有的单个数值,例如100,101,102等,以及所有的子范围,例如100到166,155到170,198到200等。对于包含小于1的数值或者包含大于1的分数(例如1.1,1.5等)的范围,则适当地将1个单位看作0.0001,0.001,0.01或者0.1。对于包含小于10(例如1到5)的个位数的范围,通常将1个单位看作0.1。这些仅仅是想要表达的内容的具体示例,并且所列举的最低值与最高值之间的数值的所有可能的组合都被认为清楚记载在本申请中。
术语“包含”,“包括”,“具有”以及它们的派生词不排除任何其它的组分、步骤或过程的存在,且与这些其它的组分、步骤或过程是否在本申请中披露无关。为消除任何疑问,除非明确说明,否则本申请中所有使用术语“包含”,“包括”,或“具有”的组合物可以包含任何附加的添加剂、辅料或化合物。相反,出来对操作性能所必要的那些,术语“基本上由……组成”将任何其他组分、步骤或过程排除在任何该术语下文叙述的范围之外。术语“由……组成”不包括未具体描述或列出的任何组分、步骤或过程。除非明确说明,否则术语“或”指列出的单独成员或其任何组合。
为了使本发明所解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。
实施例
以下例子在此用于示范本发明的优选实施方案。本领域内的技术人员会明白,下述例子中披露的技术代表发明人发现的可以用于实施本发明的技术,因此可以视为实施本发明的优选方案。但是本领域内的技术人员根据本说明书应该明白,这里所公开的特定实施例可以做很多修改,仍然能得到相同的或者类似的结果,而非背离本发明的精神或范围。
除非另有定义,所有在此使用的技术和科学的术语,和本发明所属领域内的技术人员所通常理解的意思相同,在此公开引用及他们引用的材料都将以引用的方式被并入。
那些本领域内的技术人员将意识到或者通过常规试验就能了解许多这里所描述的发明的特定实施方案的许多等同技术。这些等同将被包含在权利要求书中。
下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的仪器设备,如无特殊说明,均为实验室常规仪器设备;下述实施例中所用的试验材料,如无特殊说明,均为自常规生化试剂商店购买得到的。
实施例1一种lncRNA m6A-seq高通量测序数据的生信分析
结合图1,详细的分析流程如下:
1.原始数据产出统计与预处理
由illumina Novaseq 6000或Hiseq系列测序仪使用Paired-End双端测序法产生的原始数据为带有接头序列的fastq格式,该数据格式中包含碱基质量及具体碱基信息,这些原始数据需要进行前期预处理,使用fastq软件过滤掉不合格的序列,具体步骤包括:①带有接头的reads;②去除含有N碱基(N表示为测序仪无法识别的碱基信息)大于5%的reads;③去除低质量Reads(质量值Q≤10的碱基数占整个read的20%以上);④去除碱基均为A或N的reads。
然后统计测序数据的Q20%、Q30%、GC%、有效数据量、有效Reads数等数据,如表1所示。
表1测序序列质控统计表
注:
Sample_ID:样本名
Raw_Reads:原始下机数据的reads数
Raw_Bases:原始下机数据的碱基数
Valid_Reads:有效数据的reads数
Valid_Bases:有效数据的碱基数
Valid_Ratio%:有效reads所占比例
Q20%:质量值≥20的碱基所占比例(测序错误率小于0.01)
Q30%:质量值≥30的碱基所占比例(测序错误率小于0.001)
GC%:GC含量所占的比例
2.污染序列过滤
(1)rRNA和支原体RNA序列下载
从NCBI的NR数据库以及Ensembl数据库,下载所有5s/5.8s/18s/28s rRNA序列,物种涵盖人、动物和植物,包括智人(Homo sapiens)、小鼠(Mus musculus)、大鼠(Rattusnorvegicus)、斑马鱼(Danio rerio)、黑腹果蝇(Drosophila melanogaster)、拟南芥(Arabidopsis thaliala)、水稻(Oryza sativa)、烟草(Nicotiana tabacum)、小麦(Triticum aestivum)等。从NCBI的NR数据库下载关键词为mycoplasma的常见所有支原体RNA序列。所有RNA序列为fasta格式。
(2)创建In-House核糖体及支原体序列自建数据库
从NCBI数据库和Ensembl数据库下载所有核糖体RNA序列后合并成一个大的序列文件,用于后续污染数据过滤。
(3)污染序列过滤
使用bowtie软件,采用默认参数与核糖体及支原体序列自建数据库比对,碱基错配为3,将测序数据中相关污染序列进行过滤。
3.测序数据与基因组比对
(1)下载参考基因组的序列文件(fasta或fa格式)以及注释文件(gtf格式)。
(2)使用HISAT2软件中的extract_exons.py和extract_exons.py,将参考基因组的gtf文件转化成exon文件和ss文件,分别代表外显子信息和可变剪切信息。
(3)使用HISAT2软件中的hisat2-build指令,通过基因组fa文件、exon文件和ss文件,构建基因组index索引文件(ht2格式),用于后期基因组比对。
(4)使用HISAT2软件的hisat2指令,将IP和Input的CleanData文件(fastq格式)中的每条reads与参考基因组index索引文件进行比对分析,结果如表2所示。
表2参考基因组比对Reads统计表
注:
Sample:测序文库名称
Valid reads:质控后的数据
Mapped reads:能比对到基因组上的Reads数
Unique Mapped reads:只能唯一比对到基因组一个位置的reads数
Multi mapped reads:能比对到基因组多个位置的reads数
Reads map to sense strand:Read比对到基因组上正义链的统计数
Reads map to antisense strand:Read比对到基因组上负义链的统计数
Non-splice reads:Read能够end-to-end比对到基因组区域,为整段比对统计
Splice reads:Read无法end-to-end比对到基因组区域,为分段比对统计
将能够比对上基因组的reads输出为Mapped.bam文件。
(5)绘制IP样本和Input基因组上比对率饼状图。
结果如图2所示,结果显示,reads比对上基因组外显子区域(exon)、内含子区域(intron)和基因间区(intergenic)进行统计,超过90%reads都位于外显子区域
4.全基因组层面Peak Calling与注释
(1)使用exomePeak2软件进行Peak Calling分析,输入主要文件包括IP样本和Input样本的bam文件和基因组注释gtf文件。诸多参数中主要参数包括“glm_type=c("Poisson","NB","DESeq2")”、“peak_calling_mode=exon”、“fragment_length=100”。该步骤只对外显子区域进行扫描,不扫描内含子区域,所以Peak Calling模式仅为exon,差异分析默认使用DESeq2。不同扫描长度会影响Peak数量,长度越短Peak数量越多,默认扫描长度为100。由于Input样本的异质性,采用位置Tag密度的方式来对背景进行估计。
X
然后使用C-test对候选Peak区域的IP样本X1,w~Poisson(n1λ1,w)和Input样本X0,w~Poisson(n0λ0,w)两个Poisson的平均值比值c=λ1,w/λ0,w进行比较,并计算p-value。然后使用Fisher检验计算候选peak区域的p-value。默认p-value<0.05,则小于该p值的区域都认为是一个Peak。输出格式为bed格式,主要信息包括染色体编号及区域、Peak长度、基因ID、IP和Input样本Counts数、差异倍数log
(2)使用ChIPseeker软件读取Peak的bed文件后,分别将Peak在染色体上的分布信息和在转录起始位点TSS分布信息进行整体统计,并对Peak具体所对应的基因功能原件进行注释,包括Promoter区域、5’UTR区域、3’UTR区域、CDS区,结果以统计表格形式输出,如表3所示:
表3Peak calling结果输出表
注:
chr:mRNA或lncRNA位于所在染色体的编号,如chr1意味着该转录本对应的基因位于1号染色体上start:mRNA或lncRNA的Peak峰在染色体上的起始位点
end:mRNA或lncRNA的Peak峰在染色体上的终止位点
width:Peak的范围长度,即峰的区域大小
geneId:基因对应的ID号,一般来源于NCBI或ensembl等权威数据库
gene_biotype:mRNA或lncRNA对应编码蛋白的属性,如protein coding、lincRNA等
geneName:mRNA或lncRNA的缩写或简写,也叫gene symbol
GO:全称gene ontology,即对基因进行注释,也叫GO功能注释
KEGG:小k,指的是KEGG代谢通路的名称,用来表示一个特定的生物路径
ReadsCount.Input:Input样本该Peak区域的reads数
ReadsCount.IP:IP样本该Peak区域的reads数
log2fc:IP样本中Peak对input中Peak增加的倍数,在用log2处理
pval:p-value of the peak再进行log10处理
fdr:用fdr值对p-value of the peak进行校正后的数值
annotation:Peak基因组位置功能元件注释,即Peak处于exon还是3'UTR等信息
(3)绘制每个样本m6A在起始位点TSS富集峰图
如图3所示,从图中可以看出,将所有基因上分布的reads归一化处理后,超过50%片段都富集在了5’UTR末端和CDS起始位置的转录起始位点TSS,更有超过80%片段富集在了CDS末端及3’UTR起始位置的转录终止位点TES。符合m6A实验的特征。
5.差异Peak分析
(1)差异Peak分析使用的exomePeak2软件,在分析参数与Peak Calling相同,输出文件为bed格式。具体内容包括差异Peak在染色体的位置、基因或lncRNA的ID号、p-value值、log2FC、Peak基因功能元件注释、Peak增加倍数Fold Enrichment等。默认p-value<0.05且log2FC绝对值>1为差异Peak。结果如表4所示:
表4差异Peak信息统计表
注:
Chr:mRNA或lncRNA位于所在染色体的编号,如chr1意味着该转录本对应的基因位于1号染色体上Start:mRNA或lncRNA的Peak峰在染色体上的起始位点
end mRNA或lncRNA的Peak峰在染色体上的终止位点
width:Peak的范围长度,即峰的区域大小
geneId:mRNA或lncRNA对应的ID号,一般来源于NCBI或ensembl等权威数据库
gene_biotype:mRNA或lncRNA对应的属性,如protein coding,lincRNA等
geneName:mRNA或lncRNA的缩写或简写,也叫gene symbol
ReadsCount.Input.nc:nc组的Input样本所在Peak区域的reads数
ReadsCount.IP.nc:nc组IP样本所在Peak区域的reads数
ReadsCount.Input.t1:t1组的Input样本所在Peak区域的reads数
ReadsCount.IP.t1:t1组IP样本所在Peak区域的reads数
log2fc:nc组合t1组(两组)之间Peak的差异倍数再用log2处理
pval:nc组和t1组(两组)之间Peak进行差异分析后得到的p-value fdr:用fdr值对nc组和t1组(两组)之间Peak差异分析得到的p-value再进行校正后的数值
annotation:Peak基因组位置功能元件注释,即Peak处于exon还是3'UTR等信息
(2)绘制差异Peak在基因功能原件分布占比饼状图
如图4所示,从图中可以看出,超过60%的peak都位于3’UTR区域,也叫UTR3,排在第二的peak超过13%都位于UTR5也就是5’UTR。整体来讲超过70%的peak都位于UTR区域,符合m6A实验的特征。
6.Peak序列motif分析
使用软件HOMER中的findmotifsGenome模块,输出文件为Peak的bed文件和基因组位置注释信息文件(如tss、tts、aug、stop、repeats.rna等文件)。核心参数包括RegionSize=100和Motif length=5。输出网页版版本的Homer de novo Motif Results结果。
结果如图5所示,从图中可以看出,motif分析结构为非常典型的m6A特征motif为RGACH。其中R为G碱基或A碱基,而H则是A碱基、C碱基或U碱基。且pvalue<0.05。
7.mRNA与lncRNA表达丰度分析
(1)使用StringTie软件,输入文件为单个样本Input的bam文件和基因组gtf文件,最后输出merged.gtf文件。根据gtf文件中transcript_type定义信息,如protein_coding、lincRNA、ncRNA等注释里面先对每条RNA进行mRNA和lncRNA注释。
(2)提取merged.gtf文件中每条mRNA和lncRNA的Counts值和归一化后的表达量FPKM值,然后输出表达谱矩阵。其中FPKM值计算公式如下:
其中,total exon Fragments表示总外显子片段reads数,Mappedreads表示能比对到基因组上的Reads数(百万计),exon length表示外显子长度(kb)。
结果如表5所示:
表5mRNA和lncRNA整体表达水平
注:
gene_id:mRNA或lncRNA对应的ID号,一般来源于NCBI或ensembl等权威数据库
gene_name:mRNA或lncRNA的缩写或简写,也叫gene
GO:全称gene ontology,即对基因进行注释,也叫GO功能注释
KEGG:小k,指的是KEGG代谢通路的名称,用来表示一个特定的生物路径
KO_Entry:大K,指的是KEGG在所有物种中具有相似结果或功能
EC:一般特指某种酶的代号。如EC2.7.1.1指的是丝氨酸/苏氨酸激酶
Description:该mRNA或lncRNA的具体描述,包括功能等
FPKM.nc1_input:样本cn1的mRNA或lncRNA归一化表达量FPKM
FPKM.t1_input:样本t1的mRNA或lncRNA归一化表达量FPKM
(3)根据基因和lncRNA的FPKM值,绘制基因表达盒形图、基因表达密度图。
基因表达盒形图如图6所示,从图中可以看出,通过对样本中所有mRNA和lncRNA的表达量FPKM进行log10归一化后,nc1样本整体表达量略高于t1样本
基因表达密度图如图7所示,从图中可以看出,对样本中所有mRNA和lncRNA表达量FPKM进行log10处理后,成为相对表达量。因此可以找到相对表达量大小的规律分布图,其中无论是nc1还是t1表达量高低分布规律相似
8.差异mRNA和lncRNA的表达谱分析
(1)使用软件edgeR,根据组间样本中的每个mRNA和lncRNA的Counts进行差异分析,并计算p-value值,默认p-value<0.05为差异表达的mRNA和lncRNA,结果如表6所示:
表6mRNA和lncRNA差异分析表
注:
gene_id:mRNA或lncRNA对应的ID号,一般来源于NCBI或ensembl等权威数据库
gene_name:mRNA或lncRNA的缩写或简写,也叫gene GO:全称gene ontology,即对基因进行注释,也叫GO功能注释
KEGG:小k,指的是KEGG代谢通路的名称,用来表示一个特定的生物路径
KO_Entry:大K,指的是KEGG在所有物种中具有相似结果或功能
EC:一般特指某种酶的代号。如EC2.7.1.1指的是丝氨酸/苏氨酸激酶
Description:该mRNA或lncRNA的具体描述,包括功能等
FPKM.nc1_input:样本cn1的mRNA或lncRNA归一化表达量FPKM
FPKM.t1_input:样本t1的mRNA或lncRNA归一化表达量FPKM
fc:差异倍数,fold change全称
pval:两组差异分析后得到的p-value qval:对p-value进行FDR校正拿到的q值
(2)根据差异基因的表达量及p-value绘制差异基因热图、火山图
差异基因热图如图8所示,从图中可以看出,两组样本之间将表达量FPKM进行log10处理后,进行相对表达量高低的比较。每一个基因在两组样本之间表达量相对高低对应颜色相关。某一个基因颜色越靠近深蓝色相对表达量月底,颜色越靠近深红色相对表达量越高。
差异基因火山图如图9所示,从图中可以看出,两组样本之间某个基因差异分析后得到的pvalue及差异倍数分别进行-log10和log2处理后,每个点代表一个基因,在X轴和Y轴双象限图中展示。左上角的小点且在虚线外代表着基因表达量降低显著,右上的小点且在虚线外的代表基因表达量显著升高。
(3)对所有差异相关基因,根据其对应GO和KEGG注释信息,采用负二项式分布法进行GO和KEGG功能富集分析,公式为:
其中,pvalue of enriched GO/KEGG代GO/KEGG功能富集分析数值,TB代表所有背景基因,TS代表所有差异表达基因,B代表所有带有GO或KEGG注释的基因,S代表所有所有带有GO或KEGG注释的差异表达基因。
(4)如果pvalue<0.05,则认为该GO或KEGG条目为差异显著富集。最后绘制GO和KEGG富集分析的柱状图、气泡图。
GO富集分析的柱状图如图10所示,从图中可以看出,某个基因功能注释基因数量越高,对应柱子就越高,意味着这个基因功能富集程度越高。
KEGG富集分析的气泡图如图11所示,从图中可以看出,对应的基因通路也叫pathway,富集到的基因越多气泡就越大,对应通路富集的pvalue越小颜色就越近蓝色,意味着该通路富集程度越高。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 一种高通量测序的微生物数据处理方法
- 一种基于高通量测序分析寡核苷酸序列杂质的方法及应用
- 一种高通量测序数据的分析方法、试剂盒及分析系统
- 一种基于特有识别序列的T细胞受体库高通量测序文库构建及测序数据分析方法