一种用于检测川崎病及疗效评估的试剂、试剂盒及应用
文献发布时间:2023-06-19 13:49:36
技术领域
本发明涉及试剂盒制备技术领域,具体涉及一种用于检测川崎病及疗效评估的试剂、试剂盒及应用。
背景技术
川崎病是一种多系统血管发炎症候群,目前病因未明。日本小儿科医师川崎富作于1961年发现了第一例孩童伴随有口腔黏膜、皮肤以及淋巴结病变的案例,自此到1967年,又陆续发现了超过五十例类似的病例;为纪念川崎富作医师的贡献,故称这种疾病为川崎病(Kawasaki disease),也称为黏膜皮肤淋巴腺症候群(mucocutaneous lymph nodesyndrome,MCLS)或川崎症候群(Kawasaki syndrome)。罹患川崎病的病童,85%小于五岁;而男女患者比例约为1.5:1。据流行病学统计,全世界发病率最高的国家为日本,其次为韩国,中国每年新发确诊病例约为200,000个,同时,近年来该病在世界上不同国家和地区的发病率均呈现总体上升趋势。
川崎病是造成小儿后天心脏疾病的首因。多发于春夏季,每年4~6月为高峰期,罹病患童中复发率在1~4%。如果没有及时治疗,大约20~25%的儿童病人会受影响,导致冠状动脉动脉瘤,造成严重的后遗症,而且幸存者常持续出现心内膜或心包膜发炎症状,可能在多年后引发心肌血流异常和猝死。
目前川崎病的病因虽然未明,但大致上存在两种假说:其中之一认为跟感染有关,因为川崎病的临床表征和感染症状相似(如发烧、发炎指数上升等),又好发于年纪较小的孩童、且在固定季节爆发流行(在温带国家以冬天及春天为流行季节,亚洲国家则以春夏天为流行季节)。目前认为可能的感染源有New Haven人类冠状病毒(New Havenhumancoronavirus)、人类博卡病毒(human bocavirus,hboV)、巨细胞病毒(cytomegalovirus)等;另一假说则认为川崎病跟超级抗原(Superantigen)有密切相关,可能是入侵的细菌上的某些成分组成超级抗原,作用在含有VB序列的T细胞接受器,进而引发特殊的免疫反应;这种特殊的免疫反应有比传统免疫反应更强的反应强度,而造成了川崎病。近期的新冠肺炎(COVID-19)大流行,造成在欧美国家川崎病发生率上升6倍之多。因此及时和正确的检测川崎病,预防川崎病所造成的后遗症,避免造成终生不可逆的心脏损害,是越来越重要的临床课题。
目前世界上主流的诊断标准分别为日本川崎病研究委员会和美国心脏学会提出的两种。在中国,综合参考了2005年版的日本标准及2004年版的美国标准,并结合当时临床总结经验形成了国内诊治专家共识,以《川崎病专题讨论会纪要》的形式发表在2007年《中华儿科杂志》。目前川崎病的诊断仍主要靠临床表现,包括以下六大症状:1)发热持续1~2周,抗生素治疗无效;2)双侧结膜充血;3)唇及口腔改变:唇干燥、发红、皲裂,舌乳头隆起(杨莓舌),口腔及咽部粘膜弥漫性充血;4)四肢末端改变:发病初期掌跖发红和手足硬肿,恢复期指(趾)端膜状脱皮;5)躯干部多形性红斑,但无水疱和结痂;6)颈淋巴结非化脓性肿胀,直径可达1.5cm或更大。然而,种种实际临床经验显示,患者表现个体化差异大,不同的诊断标准可能存在差异,即便参考不同的诊断标准,能确定的是,川崎病的诊断是一排他性过程。换言之,针对川崎病的诊断仍尚无特异性诊断方法。在目前尚缺乏特异诊断方法的情况下,临床上对川崎病的诊断相当复杂且滞后。
除此之外,近年来亦发现许多疑似川崎氏症,但又无法完全符合诊断条件者,称为非典型或不完全性川崎氏症(incomplete KD),其大约占全部川崎氏症的15%,这时必须配合辅助性的诊断指标,如白蛋白指数、尿液检查、肝功能指数、白血球数量、血色素、血小板数目、红血球沈降系数(ESR)和C-reactive protein(CRP)发炎指数,并同时排除其他疾病才能加以确认,相当复杂且不易。
有鉴于目前川崎病的检测诊断仍存在着不足之处,相关领域有必要发展出更有效的川崎病诊断方式。
发明内容
本发明的目的在于提供一种用于检测川崎病及疗效评估的试剂、试剂盒及应用。本发明所述试剂能够实现川崎病的检测以及治疗川崎病的治疗机的疗效评估,能够增加川崎病检测灵敏度及准确度。
本发明提供了一种用于检测川崎病和/或疗效评估的试剂,所述试剂包括检测生物标记的生物标记量的试剂,所述生物标记包括CCL23和/或CXCL10。
优选的是,所述试剂的类型包括抗体。
本发明还提供了一种用于检测川崎病和/或疗效评估的试剂盒,所述试剂盒包括上述技术方案所述的试剂和反应试剂。
优选的是,所述试剂盒包括免疫测定试剂盒,所述免疫测定试剂盒包括酶联免疫吸附法检测试剂盒、化学发光法检测试剂盒、免疫层析法检测试剂盒、免疫渗滤法检测试剂盒、蛋白质阵列检测试剂盒、流式细胞计数法检测试剂盒、建立于磁珠上的多重免疫测定法检测试剂盒、蛋白质印迹检测试剂盒、点印迹检测试剂盒、酶联免疫斑点法检测试剂盒或胶体金法检测试剂盒。
优选的是,当所述试剂盒为酶联免疫吸附法检测试剂盒时,所述反应试剂包括样本稀释液、酶标液、洗涤液、显色液和终止液。
优选的是,所述样本稀释液包括含牛血清白蛋白的磷酸盐缓冲溶液;所述酶标液包括辣根过氧化物酶溶液;所述洗涤液包括含聚山梨醇酯的磷酸盐缓冲溶液;所述显色液包括由过氧化氢和3,3',5,5'-四甲基联苯胺所组成的群组;所述终止液包括硫酸。
本发明还提供了上述技术方案所述的试剂在制备诊断个体川崎病的试剂盒中的应用。
优选的是,所述个体包括发热至少3日的个体。
本发明还提供了上述技术方案所述的试剂在制备预测一治疗剂对川崎病个体的疗效的试剂盒中的应用。
优选的是,所述治疗剂包括免疫球蛋白、解热镇痛剂、肿瘤坏死因子抗体和糖皮质激素中的一种或两种以上。
本发明提供了一种用于检测川崎病及疗效评估的试剂。本发明所述试剂能够用于川崎病的检测诊断及疗效评估,(1)相较于主观的临床表征诊断,通过本发明试剂给予生物标记定量定性地检测,可达到科学性客观性诊断。(2)通过本发明检测试剂,可增加川崎病检测灵敏度,有利于提升基层医疗单位的医疗量能;(3)通过本发明检测试剂,可提升川崎症早期诊断的准确度,特别是针对非典型川崎病病患;(4)通过本发明试剂,可对川崎病病患进行治疗剂的疗效预测,助于达成临床精准治疗。
附图说明
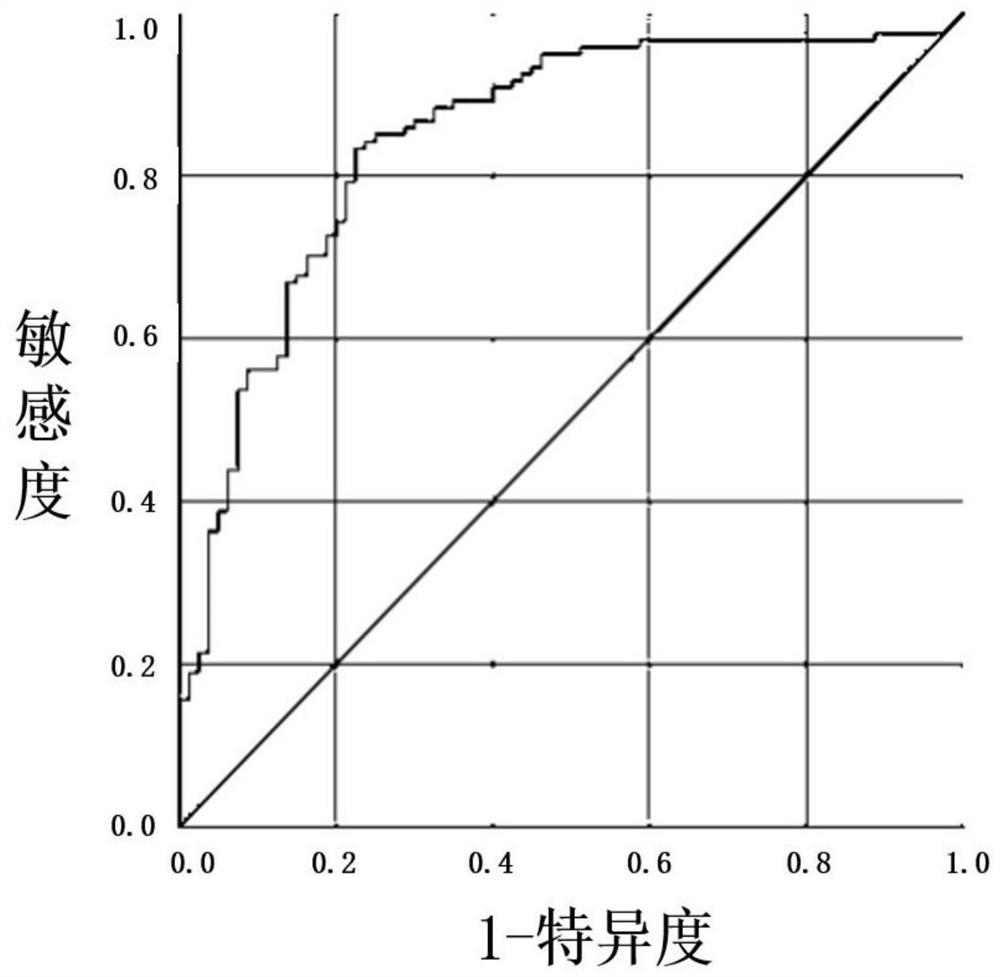
图1为本发明提供的检测个体样品中CXCL10的阳性率与川崎病患病关联性的ROC分析结果;
图2为本发明提供的检测个体样品中CCL23的阳性率与川崎病患病关联性的ROC分析结果。
具体实施方式
本发明提供了一种用于检测川崎病和/或疗效评估的试剂,所述试剂包括检测生物标记的生物标记量的试剂,所述生物标记包括CCL23和/或CXCL10。本发明至少部分基于发明人发现川崎病患者于急性发病时期,CXCL10与CCL23的表达水平都有所提升,特别是在个体的全血样品及/或血清样品中有明显趋向。川崎病患发热3至5天后,CXCL10与CCL23的表达水平提升更为显着,与发热但是非川崎病病患具有差异。本发明所述试剂用于检测个体发热至少3日的样品中生物标记的生物标记量,可以实现个体川崎症(即川崎病)的诊断;本发明所述试剂透过所述生物标记的差异,可用于川崎症个体正在经一治疗剂治疗或是已经所述治疗剂治疗的疗效评估。本发明所述试剂优选通过免疫学检测方法来使用,本发明所述试剂使用方法简单,容易实现川崎病的辅助诊断及疗效判断。免疫学检测方法的优选包括酶联免疫吸附测定法(ELISA)、蛋白质阵列、流式细胞计数法、建立于磁珠上的多重(multiplex)免疫测定法、蛋白质印迹(western blot)、点印迹(dot blot)、酶联免疫斑点法(ELISPOT)、免疫层析法(ICA)、免疫渗滤法(IFA)、胶体金法、及化学发光法。本发明优选采用酶联免疫吸附测定法来检测。
在本发明中,所述试剂的类型优选包括抗体,如单克隆抗体或多克隆抗体。在本发明中,所述试剂优选包括CCL23抗体和/或CXCL10抗体;所述CCL23抗体是可特异性结合至个体样品中CCL23蛋白的单株或多株抗体;所述CXCL10抗体是可特异性结合至个体样品中CXCL10蛋白的单株或多株抗体。本发明优选可依据本领域普通知识制备适用于本发明检测标的蛋白分子的抗体,该抗体可专一性结合至标的蛋白,例如样品中的CXCL10与CCL23。抗体「特异性结合」(specifically bind)至标的为本技术领域中已熟知的用词,且用来判定此种「特异性结合」的方法亦为本技术领域中所熟知。当分子与特定标的抗原比其他标的能够更频繁、更快速地反应或连结,且能持续一段时间及/或具有较佳的亲和力,即称为展现「特异性结合」的分子。若抗体结合至标的抗原时,相较于结合至其他物质能具有较佳的亲和力、结合力、更快速、及/或更长的持续时间,即称该抗体「特异性结合」至该标的。适用于本发明的具体抗体包含可特异性结合至CXCL10的CXCL10抗体以及可特异性结合至CCL23的CCL23抗体。本发明的任何抗体可为单株抗体或多株抗体其中一者。“单株抗体”(monoclonal antibody)指的是同源抗体群集,以及“多株抗体”(polyclonal antibody)指的是异种抗体群集。这两种名词不会限制抗体来源或其制备方式。本发明对CXCL10单株抗体和CCL23单株抗体的来源没有特殊限定,在本发明实施例中,所述CXCL10单株抗体的市售信息优选为:厂家:invitrogen,品名:CXCL10 Polyclonal Antibody,产品编号:PA5-95302或厂家:Abcam,品名:Anti-IP10antibody[6D4],产品编号:ab8098;所述CCL23单株抗体的市售信息优选为:厂家:Sino biological,品名:CCL23/MIP 3Antibody,Rabbit MAb,产品编号:10655-R004或厂家:BioLegend,品名:Purified anti-CCL23(MPIF-1)Antibody,产品编号:684002。
本发明还提供了一种用于检测川崎病和/或疗效评估的试剂盒,所述试剂盒包括上述技术方案所述的试剂和反应试剂。在本发明中,所述试剂盒检测的样品优选包括全血、血清或血浆。
在本发明中,所述试剂盒优选包括免疫测定试剂盒,所述免疫测定试剂盒优选包括酶联免疫吸附法检测试剂盒、化学发光法检测试剂盒、免疫层析法检测试剂盒、免疫渗滤法检测试剂盒、蛋白质阵列检测试剂盒、流式细胞计数法检测试剂盒、建立于磁珠上的多重免疫测定法检测试剂盒、蛋白质印迹检测试剂盒、点印迹检测试剂盒、酶联免疫斑点法检测试剂盒或胶体金法检测试剂盒。在本发明中,当所述试剂盒为酶联免疫吸附法检测试剂盒时,所述反应试剂优选包括样本稀释液、酶标液、洗涤液、显色液和终止液。在本发明中,所述样本稀释液优选包括含牛血清白蛋白的磷酸盐缓冲溶液;所述酶标液优选包括辣根过氧化物酶溶液;所述洗涤液优选包括含聚山梨醇酯的磷酸盐缓冲溶液;所述显色液优选包括由过氧化氢和3,3',5,5'-四甲基联苯胺所组成的群组;所述终止液优选包括硫酸。本发明对所述样本稀释液、酶标液、洗涤液、显色液和终止液的来源没有特殊限定,采用本领域技术人员熟知的用于免疫测定的常规市售样本稀释液、酶标液、洗涤液、显色液和终止液即可。具体的,本发明的检测方法可以采用相关技术领域已知的流程进行,例如可将个体的全血或血清样品点于滤纸上,其可经干燥及储存。然后经干燥的全血斑点或血清渍可用于ELISA中以检测及量化该样品中的生物标记量(可参见Aabye等人,PLoS ONE,7(6);e39228,2012年6月)。也可采用将全血或血清的液体样品与检测溶液混合置于孔板架上,通过特异性识别上述生物标记的抗体并检测其吸光值,借以获得该样品中的生物标记量。在本发明一具体实施例,是通过酶联免疫检测仪配合酶标微孔盘,检测其吸光值来达成生物标记量的测定。当本发明所述试剂盒用于检测川崎病时,本发明所述试剂盒检测的生物标记为CCL23和/或CXCL10,所述生物标记的生物标记量优选用于与一对照样品的对照截止量进行比较。当任一所述生物标记的生物标记量高于其所述对照截止量,则所述个体患有川崎病;当两所述生物标记量均低于其所述对照截止量,则所述个体不患有川崎病。在本发明具体实施例中,当所述CCL23生物标记量高于其所述CCL23截止量,则判定CCL23此生物标记为阳性,反之,则判定为阴性;当所述CXCL10生物标记量高于其所述CXCL10对照截止量时,则判定CXCL10此生物标记为阳性,反之则判定为阴性。若两生物标记皆为阳性,所述个体患有川崎病;若至少一所述生物标记为阳性,则该个体患有川崎病;若两所述生物标记均为阴性,则该个体不患有川崎病。当本发明所述试剂盒用于评估疗效时,本发明优选依据治疗剂治疗前与治疗剂治疗后的生物标记量差异,判定所述治疗剂对所述个体有否治疗效果;具体的,优选当所述生物标记量差异为治疗剂治疗前的生物标记量高于治疗剂治疗后的生物标记量30%以上,则判定所述治疗剂对所述个体有治疗效果。在具体实施方案中,所述CXCL10的生物标记量用于样本个体治疗前、治疗后的CXCL10进行比较,所述CCL23的生物标记量用于与样本个体治疗前、治疗后的CCL23进行比较,当所述治疗前CXCL10的生物标记量高于其所述治疗后CXCL10生物标记量的30%,且所述治疗前CCL23的生物标记量高于所述治疗后CCL23生物标记量的30%,则判定所述治疗剂对所述个体有治疗效果;当所述治疗前比治疗后CXCL10的生物标记量低于30%,或所述治疗前比治疗后CCL23的生物标记量低于30%,则判定所述治疗剂对所述个体无治疗效果。
本发明所述试剂盒的其它成分可以为一外包装,用以将本发明的各试剂(包括抗体)保持在一确定的范围内,例如放置在一盒体内。适用于本发明之外包装的材料包含玻璃、塑胶(聚乙烯、聚丙烯、聚碳酸酯及其类似物)、瓶、罐、纸、束袋及其类似物。
本发明试剂盒可更包含操作说明书,其中记载本发明各种配方的添加方式。所述操作说明书可为纸本或可备读取的电子媒介物,例如:电子储存媒体(磁碟、磁带及其类似物)、光学媒体(CD-ROM、DVD)及其类似物。所述媒体可额外或替选地包含网路页面,以提供上述说明资讯
本发明还提供了上述技术方案所述的试剂在制备诊断个体川崎病的试剂盒中的应用。本发明所述应用制备得到的试剂盒能够快速有效诊断区别川崎病个体与非川崎病个体,能够提升川崎病的诊断的准确性及有效性。在本发明中,所述试剂盒诊断的样品优选包括全血、血清、血浆、尿液或唾液,更优选为全血、血清或血浆。本发明可从一个体取得样品,并从中检测目标生物标记的量;本发明所述个体可以是确定患有川崎病或是具有与川崎病相似症状疑似患有川崎病之人类个体。
在本发明具体实施例中,生样品中的目标蛋白分子,亦即作为生物标记的CXCL10与CCL23。在具体实施方式中,本发明将样品置于酶标板(ELISA plate),常见是96孔的微孔盘。对该样品溶液添加特异性结合至CXCL10蛋白与CCL23蛋白的抗体,再加入酶标液进行反应,最后在特定光波长(例如在450nm光波长)下测定反应物吸光值。通过前述流程,可得到个体样品中,CXCL10及CCL23于一特定波长的吸光值,该吸光值大小分别反应样品中CXCL10及CCL23的表达水平,亦即生物标记量。
依据本发明一具体实施方式,可从健康个体取得之对照样品,从中CXCL10及CCL23在相同检测流程及相同光波长下,测得一吸光值。在具体实施方式中,该吸光值可为对照截止量,亦可将该吸光值做为一基础值,从而计算出对照截止量。
在本发明一些较为优选的实施方式中,生物标记量用于与对照截止量进行比较,基本上,若该个体样品的生物标记量高于对照截止量(包含等于对照截止量),则视为该个体患有川崎病。
或者是,可以同时检测样品中的所有生物标记,包含CXCL10及CCL23。CXCL10及CCL23的生物标记量分别与对照组(例如一标准品或是一健康个体之样品)的CXCL10及CCL23对照截止量比较,看是否有高于各自的对照截止量。若个体样品的生物标记量高于各自的对照截止量,则判断该生物标记为阳性;反之则为阴性。
具体来说,若CXCL10的生物标记量高于CXCL10对照截止量,则判定生物标记CXCL10为阳性,反之为阴性。若CCL23的生物标记量高于CCL23对照截止量,则判定生物标记CCL23为阳性;反之为阴性。
当检测样品中的CXCL10生物标记量为阳性时,判断所述个体患有川崎病。
当检测样品中的CCL23的生物标记量为阳性时,亦说明所述个体患有川崎病。
当同时检测样品中CXCL10及CCL23的生物标记量时,若其中一生物标记或是两个生物标记均被判断为阳性,则该个体患有川崎病。若两生物标记量均判断为阴性,则该个体不患有川崎病,是健康个体。
除了前述应用,也可以通过具体的对照截止量的数值来执行本发明诊断个体是否患有川崎病的应用。
在一具体实施方式中,具体对照截止量为标准品中CXCL10蛋白或CCL23蛋白的定量浓度,各介于每毫升100微微克(pg)至每毫升4000微微克之间,例如每毫升100、200、300、400、500、600、700、800、900、1000、1100、1200、1300、1400、1500、1600、1700、1800、1900、2000、2100、2200、2300、2400、2500、2600、2700、2800、2900、3000、3100、3200、3300、3400、3500、3600、3700、3800、3900或4000微微克的CXCL10蛋白或CCL23蛋白。在一较佳实施方式中,对照截止量是浓度为100pg/ml的CXCL10蛋白或CCL23蛋白;在另一实施方式中,对照截止量是浓度为400pg/ml的CXCL10蛋白或CCL23蛋白;在另一实施方式中,对照截止量的CXCL10蛋白或CCL23蛋白的浓度为1000pg/ml;在又另一实施方式中,对照截止量的CXCL10蛋白或CCL23蛋白的浓度为4000pg/ml。可通过判断个体样品中的浓度是否高于前述对照截止量浓度,来判断该个体是否罹患川崎病。在具体实施例中,CXCL10蛋白对照截止量或CCL23蛋白对照截止量分别是400pg/ml,当待测个体样品CXCL10蛋白或CCL23蛋白高于400pg/ml,则该生物标记为阳性,所述个体患有川崎病。
在另一具体实施方式中,CXCL10对照截止量是对照样品中CXCL10蛋白在光波长450nm的吸光值乘以第一系数。CCL23对照截止量是对照样品中CCL23蛋白在光波长450nm的吸光值乘以第二系数。可通过个体待测样品中实测的生物标记量与其对照截止量的比值,来判断该个体是否罹患川崎病。
依据本揭示内容的部份实施方式,所述第一系数介于0.1至0.6之间,例如可以是0.1、0.11、0.12、0.13、0.14、0.15、0.16、0.17、0.18、0.19、0.2、0.21、0.22、0.23、0.24、0.25、0.26、0.27、0.28、0.29、0.3、0.31、0.32、0.33、0.34、0.35、0.36、0.37、0.38、0.39、0.4、0.41、0.42、0.43、0.44、0.45、0.46、0.47、0.48、0.49、0.5、0.51、0.52、0.53、0.54、0.55、0.56、0.57、0.58、0.59或0.6。在某些实施方式中,第一系数可介于0.2至0.4之间;在一较佳实施例中,第一系数为0.33。所述第二系数为0.8至1.2之间,例如可以是0.8、0.81、0.82、0.83、0.84、0.85、0.86、0.87、0.88、0.89、0.9、0.91、0.92、0.93、0.94、0.95、0.96、0.97、0.98、0.99、1.0、1.01、1.02、1.03、1.04、1.05、1.06、1.07、1.08、1.09、1.1、1.11、1.12、1.13、1.14、1.15、1.16、1.17、1.18、1.19或1.2。在某些实施例中,所述第二系数是介于0.9至1.1之间;在一较佳实施例中,第二系数为1.02。
承前所述,当第一系数为0.33时,获得一CXCL10对照截止量。若个体样品中CXCL10在光波长450nm的数值与CXCL10对照截止量的比值是大于等于0.8(亦即,大于0.8或是等于0.8,≧0.8;或0.8以上),则判断该生物标记CXCL10是阳性,且该个体患有川崎病。另一方面,当第二系数为1.02时,获得一CCL23对照截止量。当个体样品中CCL23在光波长450nm的数值与CCL23对照截止量的比值是大于等于0.9(亦即,大于0.9或是等于0.9,≧0.9;或0.9以上),则判断该生物标记CCL23为阳性,且该个体患有川崎病。反之,若CXCL10生物标记量/CXCL10对照截止量的比值小于0.8(亦即,<0.8,不包含0.8)且CCL23生物标记量/CCL23对照截止量的比值小于0.9(亦即,<0.9,不包含0.9),则可判定该个体为非患有川崎病的个体,或是健康个体。
本揭示内容还包含判断待测个体是否患有川崎症,特别是准确鉴别同为发热个体是否为川崎症。依据一具体实施方式,待测个体发热至少3日。当个体体温(例如额温或腋温)达到37.3℃以上,则该个体为发热状态。举例来说,所述个体发热至少3日,至少4日,至少5日,至少6日,至少7日。在较佳实施例中,所述个体发热3至7日。在本发明中,同为发热3日至7日的个体,可依照所量测的CXCL10与CCL23蛋白的表达水平是否高于其各自的对照截止量,据以判定该个体是否为真正的川崎病患者,并排除非川崎病患者。
本发明还提供了上述技术方案所述的试剂在制备预测一治疗剂对川崎病个体的疗效的试剂盒中的应用。在本发明中,所述治疗剂包括免疫球蛋白、解热镇痛剂(例如:阿斯匹灵)、肿瘤坏死因子抗体(例如:英利昔单抗)和糖皮质激素中的一种或两种以上。在本发明中,所述个体优选正在经一治疗剂治疗或是已经经所述治疗剂治疗,以及样品是全血、血清、尿液、或唾液样品。本发明优选依据治疗剂治疗前与治疗剂治疗后的生物标记量差异,判定所述治疗剂对所述个体有否治疗效果;具体的,优选当所述生物标记量差异为治疗剂治疗前的生物标记量高于治疗剂治疗后的生物标记量30%以上,则判定所述治疗剂对所述个体有治疗效果。在具体实施方案中,所述CXCL10的生物标记量用于样本个体治疗前、治疗后的CXCL10进行比较,所述CCL23的生物标记量用于与样本个体治疗前、治疗后的CCL23进行比较,当所述治疗前CXCL10的生物标记量高于其所述治疗后CXCL10生物标记量的30%,且所述治疗前CCL23的生物标记量高于所述治疗后CCL23生物标记量的30%,则判定所述治疗剂对所述个体有治疗效果;当所述治疗前比治疗后CXCL10的生物标记量低于30%,或所述治疗前比治疗后CCL23的生物标记量低于30%,则判定所述治疗剂对所述个体无治疗效果。
在一实施方式中,本发明对正在进行或是已经经过治疗剂治疗之个体进行检测。经投予所述治疗剂之后,对所述个体进行检测,若该个体的CXCL10生物标记量且CCL23生物标记量均低于对照截止量(亦即同时低于CXCL10对照截止量也低于CCL23对照截止量),则表示该治疗剂对于该个体的川崎病可产生治疗效果。若该个体的CXCL10生物标记量或是CCL23生物标记量任一高于其对照截止量,则表示该治疗剂对于该个体的川崎病无产生治疗效果。
下面结合具体实施例对本发明所述的一种用于检测川崎病及疗效评估的试剂、试剂盒及应用做进一步详细的介绍,本发明的技术方案包括但不限于以下实施例。实施方式中涵盖了多个具体实施例的特征以及用以建构与操作这些具体实施例的方法步骤与其顺序。然而,亦可利用其他具体实施例来达成相同或均等的功能与步骤顺序。在对实例描述前,有必要提供一些备注说明:采用不同厂家、不同批次的试剂会造成实验结果的些微差异,属于正常现象。在进行小规模实验时,为保证平行实验间的重复性,建议配置试剂后,充分混匀并分装,以保证每次实验试剂的均一性。
定义
为了便于说明,此处统整性地说明本权利要求书及说明书所记载的特定用语。除非本文另有定义,本文所有的技术及科学术语与本领域技术人员熟知的术语的意思相同。在不和上下文冲突的情形下,本说明书所用的单数名词“一”涵盖该名词的复数型;而所用的复数名词时亦涵盖该名词的单数型。
本文中“生物标记”的用语是指,存在于个体的生物样品中,并可从中获取以检测该个体或患者的表现型(例如,可检测该个体的病理学状态或可能对特定治疗剂的响应性)的指标。生物标记包括但不限于DNA、RNA、蛋白质、碳水化合物,蛋白聚糖、糖脂分子以及其组合。在本发明中,生物标记物是从个体样品中取得的人γ干扰素诱导蛋白-10(CXCL10)以及骨髓造血祖细胞抑制因子(Myeloid Progenitor Inhibitory Factor-1,CCL23)。
所述生物“样品”或“样本”,可交替使用,并且是指,获自或来自目标个体的组合物,所述组合物含有待被表征和/或鉴定的细胞和/或其它生物分子实体(例如根据身体的、生物化学的、化学的和/或生理学的特征)。所述样品预期或已知含有待表征的细胞和/或分子实体。依据本发明一实施方案,样品是提取自于疑似罹患或患有川崎病人类个体的体液检体,所述体液检体包含但不限于:全血样品、血浆样品、血清样品、尿液样品、唾液样品及粘液样品。在本发明中,所述样品是全血、血清、尿液或唾液。
本文中“对照截止量”(cutoff control)一词是指,相对于待测个体,基于作为对照组的个体或群体中该相同生物标记的一确定代表量或数值。在本发明中,生物标记的对照截止量可基于非川崎病个体或健康个体体内的生物标记量来确定。非川崎病个体可包括具有与川崎病相似的临床特征及/或实验室参数重叠的疾病或症状(例如,持续发烧、皮疹、幼年型类风湿性关节炎、特定病毒及细菌感染)。透过统计分析获得的非川崎病个体或健康个体的生物标记定量数值具有非川崎病个体的普遍代表性意义,借此可用以区分川崎病患者与非川崎病个体(尤其具有临床上类似川崎病的症状的非川崎病个体)。前述可作为区分个体是否患病的生物标记定量数值称为一对照截止量。举例来说,在本发明,对照截止量是指健康个体的CXCL10及/或CCL23的蛋白表达水平,可用来作为与待测样品比较的基准点,借以区分个体是否罹患川崎病。
本文中“吸光值”(absorbance)是指光线通过待测样品(在本发明中为溶液形式)前的入射光强度与该光线通过所述待测样品后的透射光强度比值,并以10为底的对数(即Al=log(I0/I)),其中I0为入射光强度,I为透射光强度。一般而言,若某一化学物质可吸收某一固定波长的光(如H2O2可吸收波长240nm的光),则该化学物质在溶液中的浓度,会与溶液对该波长的吸光值成一正比关系,因此可通过测得该溶液的吸光值来推知化学物质在溶液中的浓度。在本发明中,含有生物标记物的酶联反应待测溶液可吸收450nm波长的光,故而将待测溶液与标准品或对照品于同一光波长下检测,在固定溶液体积下,依其正比关系可推知待测溶液中生物标记物的浓度(含量)是否高于标准品/对照品。
实施例1
CXCL10与CCL23的ELISA酶标板配制
在本实施例中,依照以下表1至表3所列的特定组分及条件配制ELISA反应所需的前导反应溶液。即将CXCL10包被液、CCL23包被液分别加入不同96孔板中;倒掉包被液,每孔中加入封稳液封稳;封稳时间为室温反应2小时,并风干5小时后,连同干燥剂放入铝箔袋中冷藏保存。
表1 CXCL10包被液组份配制表
注:所述CXCL10单株抗体的市售信息为:厂家:invitrogen,品名:CXCL10Polyclonal Antibody,产品编号:PA5-95302或厂家:Abcam,品名:Anti-IP10 antibody[6D4],产品编号:ab8098。
表2 CCL23包被液组份配制表
注:所述CCL23单株抗体的市售信息为:厂家:Sino biological,品名:CCL23/MIP3Antibody,Rabbit MAb,产品编号:10655-R004或厂家:BioLegend,品名:Purified anti-CCL23(MPIF-1)Antibody,产品编号:684002。
表3封稳液组份配制表
实施例2
ELISA操作与结果分析
接着,依照以下步骤进行ELISA操作。
1、试剂准备:将试剂盒(含实施例1制备的酶标板)各组份取出,室温下(18~25℃)平衡约30分钟,酶标板开封后取所需量,其余应及时封存,并保存于2~8℃。
在本实施例中,总检测样品数为8例,并依照表4列出的试剂组分配制表制备样品。若检测样品数为16例,则将表4列出的组分各增加两倍。以此类推。
表4试剂组份配制表
其中,10倍样本稀释液包含PBS缓冲液及BSA;酶标液为辣根过氧化物酶;10倍洗涤液包含PBS缓冲液以及吐温20;显色液A为含有过氧化氢的溶液、显色液B为含有3,3',5,5'-四甲基联苯胺的溶液。所述CCL23抗体的市售信息为:厂家:Sino biological,品名:CCL23antibody,Mouse MAb,产品编号:10655-MM03;所述CXCL10单株抗体的市售信息为:厂家:Sino biological,品名:CXCL10 antibody,Mouse MAb产品编号:10768-MM07。
2、对照样品稀释:从健康个体以及市售厂商取得CXCL10对照样品和CCL23对照样品。采用表4列出配制好的样本稀释工作液分别对CXCL10对照样品和CCL23对照样品进行梯度稀释。具体从对CXCL10对照样品和CCL23对照样品分别取4μL加入36μL的对照样品稀释工作液中,完成10倍CXCL10对照稀释样品或10倍CCL23对照稀释样品,再从其各取32μL加至288μL的对照样品稀释工作液中,完成100倍CXCL10对照稀释样品工作液及100倍CCL23对照稀释样品工作液。未使用完的100倍CXCL10或CCL23对照稀释样品工作液可于2~8℃保存14天。配制好的对照稀释样品工作液中的CXCL10及CCL23蛋白浓度均分别介于100至4000pg/ml之间。具体为400、800、1200、1600、2000、2400、2800、3200、3600或4000pg/ml。
3、待测样品制备:接着进行待测样品的制备。首先,从待测个体或是疑似患病个体取得之样品需进行4倍稀释。将60μL的全血、血浆或血清样品与180μL样本稀释工作液混合均匀,稀释后样本总体积240μL,仅限当日使用。
4、加样:将相同数目的CXCL10酶标96孔微孔盘与CCL23酶标96孔微孔盘至于板架上,设置对照孔和样品孔,每孔加入100μL的100倍CXCL10或CCL23对照样品稀释工作液(步骤2制备)、阴性对照(即表4的样本稀释工作液)、空白对照(不加样本、抗体工作液、酶标工作液,其余步骤相同)与稀释后待测样品(步骤3制备)。
5、孵育:裁剪适当大小的封板膜封板,室温下孵育60分钟。
6、洗板:甩掉反应液,每孔加300μL的洗涤工作液洗板,共洗板3次,无需浸泡时间,在吸水纸上扣干后使用。
7、一抗:在微孔中各加入100μL的CXCL10抗体工作液或100μL的CCL23抗体工作液,仅空白对照不加。
8、孵育:封板膜覆盖微孔板,室温下孵育60分钟。
9、洗板:重复实验步骤6。
10、加酶:在每个微孔中各加入100μL的酶标工作液,空白对照不加。
11、孵育:封板膜覆盖微孔板,室温下孵育20分钟。
12、洗板:重复实验步骤6。
13、显色:每孔中加入100μL的显色工作液,室温下孵育15分钟。
14、终止:每孔中加入50μL的终止液(2N硫酸),并确认混合均匀蓝色中间产物均转为黄色终产物。
15、测量:在10分钟内,使用酶标仪读取波长为O.D.450nm的吸光值。
16、质量控制:检测对照样品、阴性对照及空白对照在光波长450nm的吸光值是否均符合下表(表5)标准。若是,表示此次检测数据有效,否则应进行重复检测。
表5质量控制标准对照表
17、结果判读:
对照截止量(C.O.)计算公式如下:
C.O.CXCL10=CXCL10对照样品O.D.450nm吸光值×0.33
C.O.CCL23=CCL23对照O.D.450nm吸光值×1.02
18、结果判定:
将待测样品中,CXCL10与CCL23酶标板的O.D.450nm吸光值(S),分别除以上述步骤17计算而得的C.O.CXCL10与C.O CCL23,借以判读待测样品所反应的个体是否罹患川崎病。
表6生物标记判断
根据表6,若待测样品CXCL10生物标记量与CXCL10对照截止量的比值大于等于0.8,判定为阳性;若CCL23生物标记量与CCL23对照截止量的比值大于等于0.9,亦判定此项指标为阳性。经检测后,若CXCL10或CCL23检测至少一项判定为阳性,判定该个体为川崎病阳性病患。若CXCL10或CCL23检测两项均为阴性,方判定该个体为川崎病阴性个体。
检测物中的生物标记浓度越高,其OD值也会越高,同样地,OD值的比值也会提高。透过大量临床检体验证,计算得到截止量,可区分阳性与阴性病患的OD值的比值以及其对应的生物标记浓度,再依据此截止量对应的浓度,设计阴、阳性等参考品验证试剂盒检测性能,确保检测的有效性。
实施例3
试剂性能验证
本实施例取得现有的10份阳性参考品及4份阴性参考品,加以对本发明检测试剂及前述判断方式进行双重验证。结果显示,本发明的检测试剂及其判定准则均符合参考品的阳性及阴性要求(表7及表8)。
表7-1阳性参考品CXCL10及CCL23浓度及判定结果
表7-2阳性参考品判定结果
表8-1阴性参考品CXCL10及CCL23浓度及判定结果
表8-2阴性参考品判定结果
根据检测结果,通过比较市售参考品与对照稀释样品中的CXCL10浓度及CCL23浓度,可判定生物标记CXCL10或CCL23分别为阴性或阳性。当一生物标记(例如CXCL10)在若市售参考品的浓度高于其在对照稀释样品中的浓度,则该生物标记为阳性;反之则为阴性。若其中一生物标记(亦即CXCL10或CCL23),被判断为阳性;或是两个生物标记(CXCL10和CCL23)均为阳性,则验证该市售参考品是来自川崎病个体。反之,若市售参考品中两生物标记的浓度皆同时低于对照稀释样品中的相同生物标记的浓度,亦即两生物标记同时为阴性时,检证市售参考品非来自川崎病个体。
另,检测2份重复性参考品(KD-LPC01、KD-LPC02)各10重复,结果显示,通过本发明实施例1的试剂检测,批内精密度均符合O.D.值的变异系数(CV,%)<10%要求(表9)。
表9-1重复性参考品CXCL10及CCL23浓度及判定结果
表9-2重复性参考品判定结果
接着检测本发明实施例1及2的试剂检测流程是否符合检测限参考品标准。检测4份检测限参考品(KD-LOD01、KD-LOD02、KD-LOD03、KD-LOD04),如表10所示,结果均符合判定为阳性或阴性要求。
表10-1检测限参考品的CXCL10及CCL23浓度及其判定结果
注:每个参考品每次检测结果只会有阳性或阴性一个判定结果,表10-2为某次测试的结果,该次结果KD-LOD03为KD阳性,而在另一次检测KD-LOD03结果检测为KD阴性,所以10-1表格中KD-LOD03写的是阳性、阴性。
表10-2检测限参考品的CXCL10及CCL23浓度及其判定结果
实施例4
本发明试剂盒用于判断个体是否罹患川崎病
收集201例临床川崎病筛查的EDTA抗凝血清样本,以临床诊断结果作为参比方法,验证结果之间的一致性。其中,阳性组样品是发热3~7天(亦即,体温为37.3℃以上连续数日)且临床诊断具有3至5项川崎病临床表征的个体血清样本;阴性组是发热3~7天且临床诊断具有1~3项川崎病临床表征的个体血清样本。按照实施例2的步骤,进行样本稀释,进行酶联反应相关操作,并进行结果分析。结果如下表11及表12,以及图1及图2。结果显示,阴性血清80例,阳性血清121例,灵敏度90.1%,专一性92.5%。
表11 201例样本检测结果
表12 201例ROC分析结果
实施例5
本发明试剂盒检测川崎病病程
收集22例川崎病个体分别在治疗前及经静脉注射免疫球蛋白(intravenousimmunoglobulin,IVTG)治疗一周之后的血清样本,并透过本发明试剂盒对样品中的生物标记进行检测,以验证是否能反应经免疫球蛋白治疗的效果。按照实施例2的步骤,进行样本稀释,进行酶联反应相关操作,并进行结果分析。表13呈现结果。
表13经IVTG治疗前后,对生物标记检测之灵敏度
结果显示,在22个病例中,有20例是治疗之前检测为阳性的个体,在IVIG治疗下,检测结果由阳性转回阴性;仅有2个个体因有免疫球蛋白丙球耐药型,须经第二次以上的治疗,故而无显着变化。显见本试剂盒可透过两种生物标记组合预测免疫球蛋白治疗效果,作为检测手段可提升检测的灵敏度。
综合前述实验结果,通过可诊断个体川崎病的检测试剂盒及其应用,本发明可增加川崎病检测灵敏度及准确度。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 一种用于检测川崎病及疗效评估的试剂、试剂盒及应用
- 一种用于评估肿瘤免疫治疗效果的分子检测方法及引物组合物及试剂盒