一种银柴胡样品的真伪鉴别方法
文献发布时间:2024-04-18 19:58:26
技术领域
本发明属于药物检测技术领域,具体涉及一种银柴胡样品的真伪鉴别方法。
背景技术
《中国药典》(2020年版)一部收载的银柴胡为石竹科(Caryophyllaceae)繁缕属植物银柴胡Stellaria dichotoma L.var.lanceolata Bge.的干燥根,别名银胡、山菜根、沙参儿等。主要分布于宁夏、内蒙古、甘肃、陕西等地。味甘,性微寒,归肝、胃经,具有清虚热、除疳热的功效。主要用于治疗阴虚发热、骨蒸劳热、小儿疳热等症。现代研究表明,银柴胡中含有甾醇类、环肽类、生物碱类、黄酮类等化学成分,具有良好的解热抗炎、抗过敏、抗癌、抗动脉粥样硬化等药理作用,临床应用广泛。
近年来,因价格昂贵,市场上常会出现用丝石竹(Gypsophila oldhamiana Miq.)、灯心蚤缀(Arenaria juncea Bieb.)等石竹科植物的根混充银柴胡的情况,严重影响药材市场和临床用药效果。出于确保市场、临床用药的安全性和有效性的考虑,如何快速、准确地鉴别银柴胡的真伪已成为消费者、生产者和质检管理机构的迫切需求。
目前,关于银柴胡与其混伪品的鉴别主要有性状、显微、紫外光谱及薄层色谱等方面,并且现有技术多是对银柴胡药材或饮片进行鉴别,而实际中,为了使用方便,往往将银柴胡药材制成标准汤剂或配方颗粒进行使用,而标准汤剂或配方颗粒为银柴胡药材水提后的有效成分,要想鉴别标准汤剂或配方颗粒的真伪或对其质量进行把控,也需要对其进行鉴别检测,但实际上,现有的适用于银柴胡药材的检测方法对于标准汤剂或配方颗粒的检测则不适用。虽然现有的用于银柴胡与其混伪品两者之间的鉴别方法可以在一定程度上对其混伪品进行区分,但是均存在一些不足之处:如传统的性状及显微鉴别依赖经验、主观性大,尤其是对于市场上两者混合品或粉碎状态无法鉴别;紫外光谱在鉴定中药材质量与含量时,需要使用溶液提取药材,因此会导致其紫外光谱吸收峰值变宽抑或是重叠,若中药材的相似度高,则紫外光谱的吸收峰值较相似,会出现鉴别不准确的现象;薄层色谱法存在结果重复性差、稳定性差、鉴别速度不及高效液相色谱快、展开剂毒性大等缺陷。
另外,《中国药典》(2020年版)一部银柴胡项下,暂无含量测定项。现有文献大多对银柴胡中黄酮类、甾醇类成分进行含量测定,专属性较差。银柴胡胺B,为银柴胡中专属的一种卡巴林生物碱类成分(SUN B.,Structures of newβ-carboline-type alkaloids withantiallergic effects from Stellaria dichotoma[J],J.Nat.Prod.,2004,67:1464-1469),研究表明该类型生物碱具有明显的抗氧化、抗应激、抗炎症等多种药理活性,对人体具有明显的保护作用(李静,敖亮.,银柴胡的生物碱成分及其抗炎活性研究[J],中草药,2018,49(22):5259-5263)。迄今为止,仅有一篇文献(CN 113759011 B)公开了一种银柴胡及其制剂特征图谱的建立方法,但适用于不同形态银柴胡样品(例如药材、饮片、标准汤剂、配方颗粒等)中银柴胡胺B的含量测定,并以此为基础来鉴别银柴胡样品真伪的方法尚未见文献报道。
发明内容
发明要解决的问题
本发明的目的是为克服上述现有技术的不足,提供一种利用超高效液相色谱鉴别银柴胡样品真伪的方法。本方法以银柴胡胺B为指标,采用超高效液相色谱法检测银柴胡和丝石竹等银柴胡混伪品,通过判断银柴胡胺B色谱峰响应信号的有无,来鉴别银柴胡样品的真伪。
用于解决问题的方案
本发明提供了一种银柴胡样品的真伪鉴别方法,包括下列步骤:
1)对照品溶液的制备;
2)供试品溶液的制备;和
3)采用超高效液相色谱法的检测和鉴别;
其中,
所述银柴胡样品为银柴胡的药材、饮片、标准汤剂和/或配方颗粒;
所述银柴胡的伪品为丝石竹和/或灯心蚤缀,优选丝石竹;
步骤1)中所述对照品为银柴胡胺B;
步骤3)中所述超高效液相色谱法的色谱条件包括:
固定相为十八烷基硅烷键合硅胶;
流动相为乙腈-酸水溶液二元体系,优选乙腈-三氟乙酸水溶液二元体系;
洗脱方式采用等度洗脱。
优选地,步骤1)中所述对照品溶液采用下列方法制备:取银柴胡胺B适量,精密称定,精密加溶剂溶解,制成浓度为5~50μg/mL,优选10μg/mL的溶液,即得所述对照品溶液。
更优选地,步骤1)中所述溶剂选自醇和醇的水溶液。
进一步优选地,所述醇为甲醇。
进一步优选地,所述醇的水溶液中醇的体积百分比为70%以上。
优选地,步骤2)中所述供试品溶液采用下列方法制备:取作为供试品的银柴胡样品适量,任选粉碎后精密称定,精密加提取溶剂提取,用提取溶剂补足减失重量,取滤液,即得所述供试品溶液。
更优选地,步骤2)中所述提取溶剂选自水、醇和醇的水溶液。
进一步优选地,所述醇为甲醇。
进一步优选地,所述醇的水溶液中醇的体积百分比为30%-70%,优选70%。
优选地,步骤2)中所述提取的方式选自超声提取、振摇提取和回流提取,优选超声提取。
优选地,步骤2)中所述提取的时间为15-60min,优选15-30min,更优选30min。
优选地,步骤2)中所述银柴胡样品与所述提取溶剂的用量比为1g:30-200mL,优选1g:50-100mL,更优选1g:100mL。
优选地,步骤3)中所述超高效液相色谱法的色谱条件包括:
色谱柱为ZORBAX Eclipse Plus C18、CORTECS UPLC T3或ZORBAX SB-AQ;
流动相为乙腈-0.1%v/v三氟乙酸二元体系,优选体积比为10:90至15:85的乙腈-0.1%v/v三氟乙酸二元体系;更优选体积比为11:89的乙腈-0.1%v/v三氟乙酸二元体系。
更优选地,步骤3)中所述超高效液相色谱法的色谱条件还包括:
检测波长为270-280nm,优选275nm;
流速为0.25mL/min~0.40mL/min,优选0.25mL/min~0.35mL/min,更优选0.3mL/min;
柱温为25℃~40℃,优选25℃~35℃,更优选30℃。
优选地,步骤3)中所述检测包括定性检测。
优选地,步骤3)中所述鉴别的标准如下:若所述供试品溶液的超高效液相色谱图中存在与所述对照品溶液的超高效液相色谱图中银柴胡胺B色谱峰相应的色谱峰,则判断所述银柴胡样品为正品,否则判断为伪品。
更优选地,步骤3)中所述检测还包括定量检测;其中,所述定量检测的方法如下:基于所述对照品溶液的超高效液相色谱图中银柴胡胺B的色谱峰,确定所述供试品溶液的超高效液相色谱图中与之对应的色谱峰,并利用其峰面积,按照峰面积归一化法,计算所述银柴胡样品中银柴胡胺B的含量。
发明的效果
本发明提供了一种银柴胡样品的真伪鉴别方法,该方法可快速、高效地鉴别银柴胡和丝石竹等银柴胡混伪品,是对中药经典方法的创新,较传统性状鉴别客观、精准,较显微、理化及薄层鉴定方法快速、简便、节省成本。
本发明建立的银柴胡胺B含量检测方法,分析快速、准确,阴性样品没有干扰,有较强的专属性,良好的重现性和稳定性,所得到的色谱图色谱峰峰型较好,可准确地检测银柴胡样品中的银柴胡胺B含量。
本发明既包含定性鉴别方法又包含目标成分的含量测定方法,可为不同形态银柴胡样品(例如药材、饮片、标准汤剂、配方颗粒等)生产过程中的内在质量监测、控制与质量评价提供依据。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为银柴胡胺B对照品在210~400nm波长下的UV图谱;
图2为不同配比条件下银柴胡配方颗粒供试品的UPLC图谱;
图3为不同提取溶剂条件下银柴胡配方颗粒供试品的UPLC图谱;
图4为不同提取方式条件下银柴胡配方颗粒供试品的UPLC图谱;
图5为不同提取时间条件下银柴胡配方颗粒供试品的UPLC图谱;
图6为不同提取溶剂体积条件下银柴胡配方颗粒供试品的UPLC图谱;
图7为银柴胡胺B对照品的线性关系图;
图8为银柴胡配方颗粒供试品的专属性试验;
图9为银柴胡配方颗粒供试品的延迟性试验;
图10为银柴胡配方颗粒供试品在不同流速考察条件下的UPLC图谱;
图11为银柴胡配方颗粒供试品在不同柱温考察条件下的UPLC图谱;
图12为银柴胡配方颗粒供试品在不同色谱柱考察条件下的UPLC图谱;
图13为不同批次银柴胡标准汤剂中银柴胡胺B含量测定;
图14为不同批次银柴胡饮片中银柴胡胺B含量测定;
图15为银柴胡和丝石竹的鉴别结果。
具体实施方式
为了更好地说明本发明,在下文的具体实施方式中给出了众多的具体细节。本领域技术人员应当理解,没有某些具体细节,本发明同样可以实施。在另外一些实例中,对于本领域技术人员熟知的方法、手段、器材和步骤未作详细描述,以便于凸显本发明的主旨。
除非另有说明,本文中所使用的术语“供试品”是指用作检测或鉴定的实验样品。
除非另有说明,本文中所使用的术语“对照品”是指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质。
除非另有说明,本文中所使用的术语“精密称定”是指称取重量应准确至所取重量的千分之一,术语“称定”是指称取重量应准确至所取重量的百分之一,术语“精密量取”是指量取体积应准确至所取体积的千分之一,术语“精密吸取”是指通过微量进样器来准确量取样品的操作方式。
除非另有说明,本文中所使用的术语“填充剂”是指在色谱分析法中用作固定相的物质,术语“固定相”是指在色谱分析法中位置固定不动、对样品产生吸附效果的一相,术语“流动相”是指在色谱分析法中可以自由流动、对样品产生解吸效果的一相。
除非另有说明,本文中所使用的术语“等度洗脱”是指在样品的色谱分析周期中,流动相的组成、比例和流速恒定不变的洗脱方式。
除非另有说明,本文中所使用的术语“超高效液相色谱”(或称“UPLC”)是指在高效液相色谱(HPLC)的基础上开发一种全新技术,具有填料颗粒小、检测速度快、分析通量大、灵敏度高等特点。
实施例
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例1:银柴胡配方颗粒中银柴胡胺B含量检测方法的构建
1.仪器、试剂与样品
(1)仪器:Waters-ACQUIYT-UPLC-H-Class超高效液相色谱系统;Waters-Quaternary-Solvent-Manager四元泵;Sample-manager-FTN自动进样器;Waters-UPLC-PDA检测器;Empower-3色谱工作站;Agilent Technologies 1290Infinity超高效液相色谱仪;1290DAD二极管阵列检测器;1290MCT柱温箱;1290Vialsampler自动进样器;1290Fiexiblepump四元泵;OpenLAB CDS2.3色谱工作站;KQ-250E超声清洗机(昆山超声仪器有限公司);ME204E/02型电子分析天平(梅特勒-托利多仪器(上海)有限公司);GKC114型控温水浴锅(南通华泰实验仪器有限公司);HY-4振荡器(金坛市科兴仪器厂);PO Lab plus-L型纯水系统(Sartorius公司);TGL-16C型离心机(上海安亭科学仪器厂)。
(2)试剂:三氟乙酸(色谱纯,Thermo Fisher公司),乙腈(色谱纯,Thermo Fisher公司);水为超纯水;其它试剂均为分析纯。
(3)样品:对照品:银柴胡胺B(CAS:755036-41-0;批号:PS012961),购自于成都普思生物技术股份有限公司,供含量测定用,纯度以98%计,使用前无须处理;供试品:银柴胡配方颗粒、标准汤剂、药材、饮片以及丝石竹药材均由江阴天江药业有限公司提供。
2.色谱条件的确定
2.1检测波长的确定
采集银柴胡胺B对照品在210~400nm的紫外吸收光谱,结果如图1所示,在紫外吸收光谱中,银柴胡胺B对照品在275nm左右具有最大吸收,因此确定检测波长为275nm,结果见图1。
2.2流动相的选择
本发明根据现有实验条件对流动相的配比进行优化(条件1:乙腈:0.2%磷酸=10:90;条件2:乙腈:0.2%磷酸=11:89;条件3:乙腈:水=11:89;条件4:乙腈:0.2%磷酸=12:88;条件5:乙腈:0.1%三氟乙酸=11:89;条件6:乙腈:0.2%磷酸=13:87;条件7:乙腈:0.2%磷酸=15:85;条件8:乙腈:0.1%乙酸=11:89),具体见图2。由图2可知,在条件3和条件8的等度洗脱条件下,目标成分并不能获得可接受的分离度,而另外六种等度洗脱条件下,均可以清晰地分辨目标成分,但考虑到目标成分含量测定的准确性,最终选择条件5作为流动相,即体积比为11:89的乙腈-0.1%v/v三氟乙酸,采用该流动相检测基线平稳,目标成分分离度良好,检测时间15min内可完成。
色谱条件的确定:以十八烷基硅烷键合硅胶为固定相(柱长为100mm,内径为2.1mm,粒径为1.8μm);以体积比为11:89的乙腈-0.1%v/v三氟乙酸为流动相;洗脱方式采用等度洗脱;流速为0.3mL/min;柱温为30℃;检测波长为275nm。理论板数按银柴胡胺B峰计算应不低于5000。
3.对照品溶液的制备
取银柴胡胺B适量,精密称定,精密加甲醇溶解,制成浓度为10μg/mL的溶液,即得对照品溶液。
4.供试品溶液的制备
取银柴胡配方颗粒适量,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50mL,密塞,超声处理(功率250W,频率40kHz)30min,放冷,摇匀,滤过,取续滤液,即得供试品溶液。
4.1提取溶剂考察
取本品适量,研细,取约0.5g,平行5组,每组2份,精密称定,置具塞锥形瓶中,精密加入水、30%甲醇、50%甲醇、70%甲醇、甲醇各25mL,密塞,称定重量,分别进行超声处理(功率250W,频率40kHz)30min,放冷,再称定重量,用相应溶剂补足减失的重量,摇匀,滤过,取续滤液,即得。分别精密吸取各供试液1μL,注入液相色谱仪,按上述色谱条件测定,计算银柴胡胺B的含量,结果见表1、图3。
表1.不同提取溶剂的比较
结论:由结果可知,以甲醇、水为提取溶剂时含量相对较低,以30%甲醇、50%甲醇、70%甲醇提取时测得银柴胡胺B含量相近,考虑到70%甲醇较易滤过,最终选择70%甲醇为提取溶剂。
4.2提取方式考察
取本品适量,研细,取约0.5g,平行3组,每组2份,精密称定,置具塞锥形瓶中,精密加入70%甲醇25mL,密塞,称定重量,分别超声处理(功率250W,频率40kHz)、加热回流、振摇提取30min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。分别精密吸取各供试液1μL,注入液相色谱仪,按上述色谱条件测定,计算银柴胡胺B的含量,结果见表2、图4。
表2.不同提取方式的比较
结论:由结果可知,不同提取方式测得银柴胡胺B的含量相近,考虑到超声处理较为简便,选择提取方式为超声处理。
4.3提取时间考察
取本品适量,研细,取约0.5g,平行4组,每组2份,精密称定,置具塞锥形瓶中,精密加入70%甲醇50mL,密塞,称定重量,分别超声处理(功率250W,频率40kHz)15min、30min、45min,60min,取出,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。分别精密吸取各供试液1μL,注入液相色谱仪,按上述色谱条件测定,计算银柴胡胺B的含量,结果见表3、图5。
表3.不同提取时间的比较
结论:由结果可知,不同提取时间提取时测得银柴胡胺B的含量相近,说明提取时间为15min时已基本提取完全,为确保提取充分,确定提取时间为30min。
4.4提取溶剂体积考察
取本品适量,研细,取约0.5g,平行3组,每组2份,精密称定,置具塞锥形瓶中,精密加入70%甲醇15mL、25mL、50mL、100mL,密塞,称定重量,分别超声处理(功率250W,频率40kHz)30min,取出,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。分别精密吸取各供试液1μL,注入液相色谱仪,按上述色谱条件测定,计算银柴胡胺B的含量,结果见表4、图6。
表4.不同提取溶剂体积的比较
结论:由结果可知,提取溶剂体积为15mL、25mL、50mL、100mL时测得银柴胡胺B的含量相近,说明提取溶剂量为25mL时已基本提取完全,为确保提取充分,选择提取溶剂量为50mL。
综合以上结果,本发明在下述实施例2中采用的色谱条件为:以十八烷基硅烷键合硅胶为固定相;以乙腈-0.1%三氟乙酸溶液(体积比11:89)为流动相;洗脱方式为等度洗脱;流速为0.30mL/min;柱温为30℃;检测波长为275nm。理论板数按银柴胡胺B峰计算应不低于5000。
供试品溶液的制备方法为:取本品适量,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50mL,密塞,称定重量,超声处理(功率250W,频率40kHz)30min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
实施例2:银柴胡配方颗粒中银柴胡胺B含量检测方法的方法学验证
1.线性
精密吸取对照品溶液(银柴胡胺B浓度为29.63μg/mL)0.1μL、0.2μL、0.4μL、0.8μL、1.0μL、1.4μL、1.8μL、2.0μL注入液相色谱仪,按上述色谱条件测定,以峰面积积分值为纵坐标,进样量(μg)为横坐标,绘制标准曲线,求得回归方程为:y=25576373.757x-18357.0409,结果见表5,图7。
表5.银柴胡胺B对照品峰面积积分值与进样量关系
结果表明:银柴胡胺B的进样量在0.0030μg~0.0593μg范围内,与峰面积值呈良好的线性关系。
2.精密度
2.1仪器精密度
取银柴胡配方颗粒按实施例1供试品溶液制备方法制成供试液,精密吸取1μL,注入液相色谱仪,按上述色谱条件测定,连续进样6次,记录峰面积测量值,计算相对标准偏差,结果见表6。
表6.仪器精密度试验
结果表明:仪器的精密度试验良好。
2.2中间精密度
取银柴胡配方颗粒,分别由两个实验员按实施例1供试品溶液制备方法制备3份供试品溶液,分别于不同时间、在不同仪器上分别进样1μL,测定银柴胡胺B峰面积值,并计算其含量及RSD,结果见表7。
表7.中间精密度试验
结果:中间精密度试验测得银柴胡胺B含量的RSD为3.86%,结果良好。
2.3重复性
取银柴胡配方颗粒适量,研细,取约0.5g,精密称定,平行6份,分别按实施例1供试品溶液制备方法制成供试品溶液,进样1μL,测定银柴胡胺B峰面积值,计算其含量及RSD,结果见表8。
表8.样品的重复性试验结果
结果:重复性试验测得银柴胡胺B含量的RSD为1.14%,结果良好。
3.准确度(加样回收试验)
取已知含量的样品(银柴胡胺B含量为:0.93mg/g)适量,研细,取约0.25g,精密称定,加入银柴胡胺B(0.02189320mg/mL)对照品溶液5mL、10mL、15mL各3份,按实施例1供试品溶液制备方法制成加样回收供试品溶液,按实施例1的色谱条件,分别进样1μL,按照下列公式计算回收率及RSD,结果见表9。
表9.回收率试验结果表
结果:银柴胡胺B回收率在85%~110%之间,准确度试验良好。
4.专属性
银柴胡配方颗粒中所添加的辅料为麦芽糊精和二氧化硅及硬脂酸镁。本次试验考察缺银柴胡的阴性样品对银柴胡配方颗粒中银柴胡胺B含量测定的影响。取缺银柴胡的阴性样品按实施例1确定的供试品制备方法制备阴性样品溶液,结果见图8。
结果:阴性色谱图中在与对照品相应的保留时间没有色谱峰,说明辅料及溶剂对银柴胡胺B的测定无干扰,以本法测定银柴胡配方颗粒中银柴胡胺B的含量具有专属性。
5.延迟性
取银柴胡标配方颗粒,按实施例1确定的供试品溶液的制备方法制备供试品溶液,按实施例1得到的色谱条件进样分析,在相同的色谱条件上,保持乙腈比例最高时的洗脱梯度,洗脱时间延长一倍,记录色谱图,结果见图9。
结果:原梯度洗脱结束后无明显色谱峰流出,表明该色谱条件基本满足了信息量最大的原则。
6.耐用性
6.1稳定性试验
取本品按实施例1中供试品溶液的制备方法制备供试品溶液,分别于0h、4h、8h、12h、16h、20h、24h时按实施例1方法进行测定,记录银柴胡胺B峰面积的变化情况,计算其RSD,结果见表10。
表10.稳定性试验测定结果
结果:由以上数据可知,24小时内银柴胡胺B峰的峰面积RSD值为1.08%,符合系统适用性试验要求。
6.2不同流速考察
取本品按实施例1中供试品溶液的制备方法制备供试品溶液,考察不同流速下(0.25mL/min、0.30mL/min、0.35mL/min)色谱峰分离效果。实验结果见表11、图10。
表11.不同流速的考察结果
结果:不同流速下,测得银柴胡胺B含量相近,峰分离度较好,说明流速在0.25mL/min~0.35mL/min范围内波动时,耐用性良好。
6.3不同柱温考察
取本品,按实施例1中供试品溶液的制备方法制备供试品溶液,考察不同柱温下(25℃、30℃、35℃)色谱峰分离效果。实验结果见表12、图11。
表12.不同柱温的考察结果
结果:不同柱温下,色谱峰分离效果均较好,测得银柴胡胺B含量相近,说明柱温在25℃~35℃范围内波动时,耐用性良好。
6.4不同色谱柱考察
取本品按实施例1中供试品溶液的制备方法制备供试品溶液,分别考察不同色谱柱(ZORBAX Eclipse Plus C18(Agilent,2.1×100mm,1.8μm)、CORTECS UPLC T3(Waters,2.1×100mm,1.6μm)及ZORBAX SB-AQ(Agilent,2.1×100mm,1.8μm))对银柴胡配方颗粒的分离效果,实验结果见表13、图12。
表13.不同色谱柱的考察结果
结果:三种色谱柱的分离均较好,保留时间适中,说明色谱柱对样品的测定结果影响较小,此方法具有普遍适应性。
实施例3:银柴胡配方颗粒中银柴胡胺B含量测定
取3批银柴胡配方颗粒,按实施例1中确定的供试品溶液的制备方法制备样品,按实施例1制备对照品溶液,按实施例1中确定的色谱条件进样分析,进样1μL,记录其峰面积,计算银柴胡胺B的含量,具体结果见表14。
表14.不同批次银柴胡配方颗粒中银柴胡胺B含量测定
实施例4:银柴胡标准汤剂中银柴胡胺B含量测定
供试品溶液制备方法:取银柴胡标准汤剂约0.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50mL,密塞,称定重量,超声处理(功率250W,频率40kHz)30min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
按实施例1制备对照品溶液,按实施例1中确定的色谱条件进样分析,进样1μL,记录其峰面积,计算银柴胡胺B的含量,具体结果见表15、图13。
表15.不同批次银柴胡标准汤剂中银柴胡胺B含量测定
实施例5:银柴胡饮片中银柴胡胺B含量测定
供试品溶液制备方法:取银柴胡饮片约1.0g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50mL,密塞,称定重量,超声处理(功率250W,频率40kHz)30min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
按实施例1制备对照品溶液,按实施例1中确定的色谱条件进样分析,进样1μL,记录其峰面积,计算银柴胡胺B的含量,具体结果见表16、图14。
表16.不同批次银柴胡饮片中银柴胡胺B含量测定
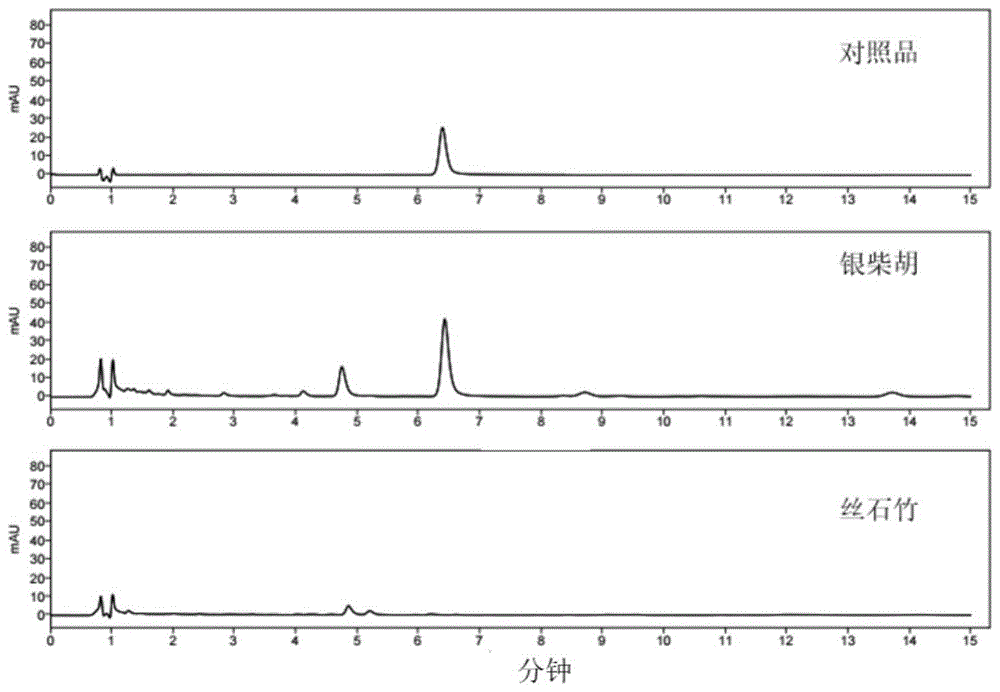
实施例6:银柴胡与丝石竹的鉴别
供试品溶液制备方法:取银柴胡药材约1.0g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50mL,密塞,称定重量,超声处理(功率250W,频率40kHz)30min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。丝石竹药材的供试品溶液制备方法:操作同银柴胡药材。
按实施例1制备对照品溶液,按实施例1中确定的色谱条件进样分析,进样1μL,检测银柴胡胺B色谱峰的有无,结果见图15。
结果:由液相色谱图可知,银柴胡正品药材供试品在与对照品银柴胡胺B相应的保留时间处有明显的色谱峰响应,而丝石竹药材供试品在与对照品银柴胡胺B相应的保留时间处无色谱峰响应。采用本发明建立的银柴胡胺B含量检测方法可将银柴胡正伪品进行区分,进而规范银柴胡使用,达到正本清源的目的。
以上实施例仅用于阐明本发明的若干实施方案,其描述较为具体和详细,但并不能因此而理解为对本发明的范围产生任何限制。应当明确的是,对于本领域技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围之内。
- 手写签名图像真伪鉴别模型建立方法及真伪鉴别方法
- 手写签名图像真伪鉴别模型建立方法及真伪鉴别方法