包含硫代磷酸酯及硼烷磷酸酯的嵌合型核酸低聚物及其制造方法
文献发布时间:2024-04-18 20:01:30
技术领域
本发明涉及核酸低聚物及其制造方法。
背景技术
与靶核酸具有互补碱基序列的反义分子能够与靶核酸形成互补双链而抑制由靶核酸生成蛋白质。在选择疾病相关基因作为靶核酸时,反义分子直接作用于疾病相关基因,因此在基因治疗中作为有效的药物而受到瞩目。
从高效抑制靶蛋白的生成的观点出发,对于反义分子(核酸低聚物),主要要求具有细胞膜穿透性、核酸酶抗性、体内(例如pH7.4的环境下)的化学稳定性、以及仅与特定碱基序列形成稳定的双链的性质。作为反义分子,已知例如使用硫代磷酸酯化合物得到的核酸低聚物(以下称为“硫代磷酸酯型核酸低聚物”)(非专利文献1)及使用硼烷磷酸酯化合物得到的核酸低聚物(以下称为“硼烷磷酸酯型核酸低聚物”)(非专利文献2)。
现有技术文献
非专利文献
非专利文献1:Nucleic Acids Research,2010,Vol.38,No.20,pp.7100-7111
非专利文献2:Chem.Rev.,2007,Vol.107,pp.4746-4796
发明内容
发明要解决的问题
本发明的课题在于,提供新的核酸低聚物及其制造方法。
用于解决问题的方案
作为用于解决上述课题的具体手段,包括以下实施方式。
<1>一种核酸低聚物,其包含下述通式(1)所示的核苷酸单元及下述通式(2)所示的核苷酸单元,并且包含或不包含下述通式(3)所示的核苷酸单元。
(通式(1)、(2)及(3)中,R
<2>根据<1>所述的核酸低聚物,其中,R
(通式(4)中,R
<3>一种<1>或<2>所述的核酸低聚物的制造方法,其包括:
缩合工序,使选自由下述通式(5)所示的化合物及下述通式(6)所示的化合物组成的组中的核苷酸单体、或选自由下述通式(5)所示的化合物、下述通式(6)所示的化合物及下述通式(7)所示的化合物组成的组中的核苷酸单体逐次缩合,得到包含下述通式(8)所示的核苷酸单元及下述通式(9)所示的核苷酸单元、并且包含或不包含下述通式(10)所示的核苷酸单元的核酸低聚物前体;和
氧化工序,利用氧化剂对上述核酸低聚物前体进行氧化,得到<1>所述的核酸低聚物。
(通式(5)、(6)及(7)中,R
(通式(8)、(9)及(10)中,R
<4>根据<3>所述的制造方法,其中,至少上述缩合工序通过使用固相载体的反应来进行。
发明的效果
根据本发明,可以提供新的核酸低聚物及其制造方法。
附图说明
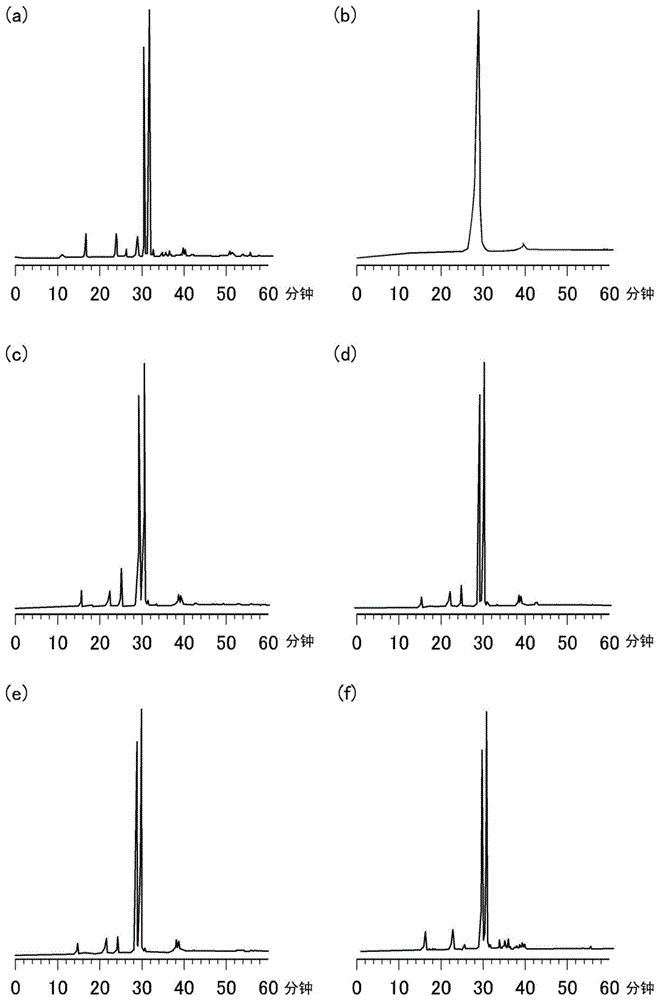
图1为示出比较合成例或合成例中进行的反相HPLC的谱图的图。(a)比较合成例1、(b)比较合成例2、(c)合成例2、(d)合成例3、(e)合成例6、(f)合成例7。
图2为示出对实施例1的核酸低聚物进行的反相HPLC的谱图的图。(a)分离纯化前、(b)分离纯化后。
图3为示出对实施例2的核酸低聚物进行的反相HPLC的谱图的图。(a)分离纯化前、(b)分离纯化后。
图4为示出对实施例3的核酸低聚物进行的反相HPLC的谱图的图。(a)分离纯化前、(b)分离纯化后。
图5为示出对实施例4的核酸低聚物进行的反相HPLC的谱图的图。(a)分离纯化前、(b)分离纯化后。
图6为示出合成例12中进行的反相HPLC的谱图的图。
图7为示出对实施例5的核酸低聚物进行的反相UHPLC的谱图的图。
图8为示出对实施例6的核酸低聚物进行的反相HPLC的谱图的图。(a)分离纯化前、(b)分离纯化后。
具体实施方式
以下对本发明的实施方式进行说明。需要说明的是,本发明不限于以下实施方式。在本说明书中,“工序”这一词语不仅包括独立的工序,即使在无法明确区别于其它工序时只要能够实现该工序的预期目的则也包含于本术语中。另外,在本说明书中,“~”表示包含记载于其前后的数值分别作为最小值和最大值的范围。进而,在本说明书中,关于组合物中各成分的量,在组合物中存在多种对应于各成分的物质的情况下,只要没有特别声明则是指组合物中存在的该多种物质的合计量。
<核酸低聚物>
本实施方式所涉及的核酸低聚物包含下述通式(1)所示的核苷酸单元及下述通式(2)所示的核苷酸单元,并且包含或不包含下述通式(3)所示的核苷酸单元。
通式(1)、(2)及(3)中,R
包含通式(1)所示的核苷酸单元的核酸低聚物虽然具有与靶RNA的亲和性、RNaseH诱导活性及分解酶抗性优异、血中滞留性特别优异的倾向,但是毒性容易变高。包含通式(2)所示的核苷酸单元的核酸低聚物虽然有与靶RNA的亲和性及RNase H诱导活性优异、分解酶抗性特别优异的倾向且毒性不易变高,但是未确认到血中滞留性优异的倾向。包含通式(3)所示的核苷酸单元的核酸低聚物虽然有与靶RNA的亲和性及RNase H诱导活性特别优异的倾向且毒性不易变高,但是分解酶抗性及血中滞留性容易变低。本实施方式所涉及的核酸低聚物由于包含通式(1)所示的核苷酸单元及通式(2)所示的核苷酸单元或者包含通式(1)所示的核苷酸单元、通式(2)所示的核苷酸单元及通式(3)所示的核苷酸单元,可期待使与靶RNA的亲和性、RNase H诱导活性、分解酶抗性及血中滞留性维持较高且将毒性抑制为较低。
作为本实施方式所涉及的核酸低聚物的长度(碱基长度),只要该核酸低聚物具有2个以上碱基则没有特别限制,可以根据靶核酸的种类、长度等来适当选择期望的长度。该核酸低聚物的长度没有特别限定,从应用于反义药物的观点出发,优选8~50个碱基,更优选10~30个碱基,进一步更优选10~21个碱基。
核酸低聚物的碱基序列可基于靶核酸的碱基序列来设定。从双链形成能力的观点出发,核酸低聚物的碱基序列以成为与靶核酸的碱基序列互补的碱基序列的方式来适当选择,根据情况也可以为包含1个以上不同的碱基的碱基序列。另外,核酸低聚物中,通式(1)所示的核苷酸单元、通式(2)所示的核苷酸单元及通式(3)所示的核苷酸单元的比例以使与靶RNA的亲和性、RNase H诱导活性、分解酶抗性及血中滞留性维持较高且将毒性抑制为较低的方式来适当选择。
作为R
作为R
作为R
作为R
作为R
作为R
作为R
作为R
R
从双链形成能力的观点出发,优选R
通式(4)中,R
作为R
R
(式中,Bs如前所述。)
Bs表示选自由腺嘌呤、鸟嘌呤、胞嘧啶、5-甲基胞嘧啶、胸腺嘧啶及尿嘧啶组成的组中的核酸碱基,该核酸碱基任选具有核酸碱基保护基。核酸碱基具有保护基时,保护基的种类没有特别限定。作为保护基,可列举例如苄基、苯甲酰基、4-甲氧基苯甲酰基、乙酰基、丙酰基、丁酰基、异丁酰基、苯基乙酰基、苯氧基乙酰基、氯乙酰基、4-叔丁基苯氧基乙酰基、4-异丙基苯氧基乙酰基、(二甲基氨基)亚甲基等。
X
本实施方式所涉及的核酸低聚物可以为具有除通式(1)所示的核苷酸单元、通式(2)所示的核苷酸单元及通式(3)所示的核苷酸单元以外的核苷酸单元的核酸低聚物。该核酸低聚物中,通式(1)所示的核苷酸单元、通式(2)所示的核苷酸单元及通式(3)所示的核苷酸单元的合计的比例没有特别限定,从双链形成能力的观点出发,相对于核酸低聚物中的全部核苷酸单元优选为40~100摩尔%,更优选为50~100摩尔%,进一步更优选为60~100摩尔%,特别优选为70~100摩尔%。
本实施方式所涉及的核酸低聚物中,5’末端可以为羟基或羟基保护基,3’末端可以为羟基或羟基保护基。作为羟基保护基,可列举例如乙酰基、苯基乙酰基、苯氧基乙酰基、氯乙酰基、特戊酰基、苄基、4-甲氧基苄基、苯甲酰基、4-甲氧基苯甲酰基、三苯基甲基、4,4’-二甲氧基三苯甲基(DMTr)、4-甲氧基三苯甲基(MMTr)、9-苯基呫吨基、叔丁氧基羰基、三甲基甲硅烷基、叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅烷基、氰基甲氧基甲基、2-(氰基乙氧基)乙基、氰基乙氧基甲基等。
<核酸低聚物的制造方法>
本实施方式所涉及的核酸低聚物的制造方法包括:
缩合工序,使选自由下述通式(5)所示的化合物及下述通式(6)所示的化合物组成的组中的核苷酸单体、或选自由下述通式(5)所示的化合物、下述通式(6)所示的化合物及下述通式(7)所示的化合物组成的组中的核苷酸单体逐次缩合,得到包含下述通式(8)所示的核苷酸单元及下述通式(9)所示的核苷酸单元、并且包含或不包含下述通式(10)所示的核苷酸单元的核酸低聚物前体;和
氧化工序,利用氧化剂对上述核酸低聚物前体进行氧化,得到本实施方式所涉及的核酸低聚物。
通式(5)、(6)及(7)中,R
通式(8)、(9)及(10)中,R
上述缩合工序中,使选自由通式(5)所示的化合物及通式(6)所示的化合物组成的组中的核苷酸单体、或选自由通式(5)所示的化合物、通式(6)所示的化合物及通式(7)所示的化合物组成的组中的核苷酸单体逐次缩合,得到包含通式(8)所示的核苷酸单元及通式(9)所示的核苷酸单元且包含或不包含通式(10)所示的核苷酸单元的核酸低聚物前体。此时,核酸低聚物的核苷结构中的核糖结构的5’位的碳原子上所键合的羟基与单体的磷酸部进行缩合。
上述缩合工序中可以使用缩合剂。作为缩合剂,可列举例如N,N-双(2-氧代-3-噁唑烷基)次膦酰氯、3-硝基-1,2,4-三唑-1-基-三(吡咯烷-1-基)鏻·六氟磷酸盐(PyNTP)或1,3-二甲基-2-(3-硝基-1,2,4-三唑-1-基)-2-吡咯烷-1-基-1,3,2-二氮杂鏻·六氟磷酸盐(MNTP)等。这些缩合剂不易与固相载体上的羟基发生副反应,适合于合成长链的核酸低聚物。通式(5)所示的化合物在分子内具有作为亲核性原子的氧原子和硫原子。氧原子与缩合剂反应则进行所期望的反应,而硫原子与缩合剂反应则发生副反应。从抑制副反应的观点出发,在使通式(5)所示的化合物缩合时,优选使用PyNTP作为缩合剂。在使通式(6)所示的化合物或通式(7)所示的化合物缩合时,从使反应快速完成的观点出发,优选使用MNTP作为缩合剂。
上述缩合工序可以在进一步存在碱性化合物的条件下进行。作为碱性化合物,可列举例如1,8-双(二甲基氨基)萘(DMAN)、2,2,6,6-四甲基哌啶(TMP)、二异丙基乙基胺(DIPEA)、2,6-二甲基吡啶、2,4,6-三甲基吡啶、吡啶、喹啉等。其中,从抑制副反应的观点出发,在使以通式(5)表示且Bs为腺嘌呤、胞嘧啶或5-甲基胞嘧啶的化合物缩合时,优选喹啉,在使通式(6)所示的化合物或通式(7)所示的化合物缩合时,优选2,6-二甲基吡啶。需要说明的是,从抑制副反应的观点出发,在使以通式(5)表示且Bs为鸟嘌呤、胸腺嘧啶或尿嘧啶的化合物缩合时,优选在不存在碱性化合物的条件下进行上述缩合工序。
上述缩合工序中的缩合反应可以在例如冰冷却下(0℃)以上且室温(25℃)以下、优选20℃以上且25℃以下的范围以例如1分钟以上且数小时以下、优选3分钟以上且1小时以下的范围来进行。
作为反应溶剂,可列举例如非活性溶剂等。作为非活性溶剂,可列举例如乙腈等。
本实施方式所涉及的核酸低聚物的制造方法中,优选至少上述缩合工序通过使用固相载体的反应来进行(固相法)。具体而言,既可以通过使用固相载体的反应来进行上述缩合工序及上述氧化工序这两者,也可以通过使用固相载体的反应来进行上述缩合工序、并通过其以外的方法、例如液相中的反应来进行上述氧化工序。通过固相反应制造核酸低聚物时,核酸低聚物的3’末端的包含碱基的单体例如可以借助键合在核糖结构的3’位的碳原子上的氧原子与固相载体结合。此时,单体与固相载体之间可根据需要存在连接体(linker)。
上述固相载体的种类没有特别限定,可列举例如可控微孔玻璃(controlled poreglass;CPG)、草酰化-可控微孔玻璃、TentaGel支撑体-氨基聚乙二醇衍生物化支撑体、高交联氨基甲基聚苯乙烯、Pros-聚苯乙烯/二乙烯基苯的共聚物等。上述固相载体与单体的连接中,可以利用固相载体上的氨基。作为上述固相载体上的氨基,可列举3-氨基丙基、长链烷基氨基(LCAA)等。上述固相载体与单体之间,可以存在连接体。作为上述连接体,可列举琥珀酰基、草酰基等。
上述氧化工序中,利用氧化剂对缩合工序中得到的上述核酸低聚物前体进行氧化,得到本实施方式所涉及的核酸低聚物。作为氧化剂,可列举例如(+)-樟脑磺酰哑嗪(CSO)、(+)-(8,8-二氯樟脑磺酰)-哑嗪(DCSO)、叔丁基过氧化氢、甲乙酮过氧化物、正电性卤素(positive halogen)试剂(四氯碳、四溴碳、碘、N-氯琥珀酰亚胺、N-溴琥珀酰亚胺、N-碘琥珀酰亚胺等)与水的组合等。氧化工序中,作为氧化剂,可以在碱性化合物存在下进行。作为碱性化合物,可列举例如三乙胺、二异丙基乙基胺等。
本实施方式所涉及的核酸低聚物的制造方法中的缩合工序包括:核酸低聚物的核苷酸结构中的核糖结构的5’位的碳原子所键合的羟基与单体的磷酸部进行缩合的反应。因此,5’位的碳原子所键合的羟基具有保护基时,例如包括使保护基与脱保护试剂反应而使保护基脱离的工序(第1保护基脱离工序)。只要核酸低聚物的制造中的缩合反应可充分进行,则任何时候进行第1保护基脱离工序均可,优选在该缩合反应之前进行。上述第1保护基脱离工序中可以使用脱保护剂。作为第1保护基脱离工序中可使用的脱保护剂,可列举例如卤代烷基羧酸等。作为卤代烷基羧酸,可列举例如三氟乙酸、三氯乙酸、二氯乙酸等。使用三苯基甲基、4,4’-二甲氧基三苯甲基(DMTr)、4-甲氧基三苯甲基(MMTr)等三苯甲基系保护基作为与5’位的碳原子键合的羟基所具有的保护基时,为了避免第1保护基脱离工序中产生的三苯甲基阳离子与硼烷基的副反应,期望添加阳离子捕捉剂。作为阳离子捕捉剂,可列举例如三乙基硅烷。具有保护基的核酸低聚物优选与2~100当量的脱保护剂进行反应。
另外,核酸低聚物的制造方法中使用的单体中的碱基具有保护基时,上述制造方法可以还包括使碱基的保护基脱离的工序(第2保护基脱离工序)。在哪个阶段进行第2保护基脱离工序并没有特别限定,只要最终得到的核酸低聚物中的碱基不具有保护基即可。第2保护基脱离工序中,例如用氨水对单体或核酸低聚物进行处理。作为第2保护基脱离工序中使用的氨水,可列举例如25质量%氨水或25质量%氨水-乙醇混合溶液(3:1、v/v)。
得到的核酸低聚物例如可以通过反相高效液相色谱(反相HPLC)、离子交换HPLC、柱色谱、重结晶等公知的纯化方法来进行纯化。
实施例
以下通过实施例更具体地说明本发明,但是本发明不受这些实施例限制。
各种分析设备使用了以下所示的机型。
1
31
需要说明的是,1H-NMR中使用四甲基硅烷(TMS)作为内标,
HRMS(ESI-TOF):Sciex X500R QTOF
离子交换HPLC:GE Healthcare AKTA purifier 10
S
作为柱色谱中的填充硅胶,使用KANTO CHEMICAL的Silica Gel 60N。
反相HPLC中使用的柱(分析):watersμ-BOUNDASPHERE,C18 5μm,
离子交换HPLC中使用的柱(分析·制备):GE Healthcare MiniQTM 4.6/50PE,3μm,4.6mm×50mm)
反相UHPLC:waters ACQUITY UPLC H-Class Bio
PDA:waters ACQUITY UPLC PDA eλDetector
LRMS(ESI):waters ACQUITY QDa Detector
反相UHPLC中使用的柱(分析):waters ACQUITY UPLC Oligonucleotide BEH C18Column,
需要说明的是,只要没有特别声明则“%”为质量基准。实施例中,DMTr表示4,4’-二甲氧基三苯甲基,其它缩写与上述说明中含义相同。
<单体的合成1>
/>
(H-膦酸酯单体及H-硼烷膦酸酯单体的合成)
H-膦酸酯单体3a、3c、3g及3t以及H-硼烷膦酸酯单体4a、4c、4g及4t通过已知方法来合成(J.Org.Chem.,2019,Vol.84,pp.15032-15041;J.Org.Chem.,2014,Vol.79,pp.3465-3472)。
(H-硫代膦酸酯单体的合成)
参考已知方法(J.Org.Chem.,1990,Vol.55,pp.3503-3506),按照下述方案合成了H-硫代膦酸酯单体。
将上述方案左端记载的核苷衍生物1a、1c、1g或1t(A:0.90g,1.5mmol;C:0.96g,1.5mmol;G:0.97g,1.5mmol;或T:0.96g,1.5mmol)与三乙基次磷酸铵0.19mL(1.0mmol)在氩气气氛下用吡啶(3ml×3次)进行共沸干燥,溶解于吡啶(5mL)。对于得到的吡啶溶液,边在冰浴下(0℃)搅拌边加入特戊酰氯0.17mL(1.5mmol),搅拌15分钟。之后,将反应温度设为室温(25℃),加入硫48mg(1.5mmol)并进一步搅拌1小时。将该溶液用氯仿30mL稀释后,添加1MTEAB缓冲溶液(pH7.0、30mL×3次)进行清洗。将水层用氯仿(30mL×3次)进行反萃取。用无水硫酸钠除去有机层中的水分。接着,馏去有机层的溶剂,将残渣用硅胶柱色谱(展开溶剂A:乙酸乙酯-甲醇-三乙胺(100:0:1-100:4:1,v/v/v)、之后氯仿-甲醇-三乙胺(100:2:1,v/v/v);C:乙酸乙酯-甲醇-三乙胺(100:0:1-100:4:1,v/v/v)、之后氯仿-甲醇-三乙胺(100:4:1,v/v/v);G:乙酸乙酯-甲醇-三乙胺(100:2:1,v/v/v)、之后氯仿-甲醇-三乙胺(100:3:1-100:5:1,v/v/v);T:乙酸乙酯-甲醇-三乙胺(100:0:1-100:1:1,v/v/v)、之后氯仿-甲醇-三乙胺(100:0:1-100:3:1,v/v/v))进行分离纯化,利用0.2M DBU二碳酸盐进行盐交换,将有机层用无水硫酸钠干燥,将溶剂减压馏去,由此得到目标化合物(H-硫代膦酸酯单体5a、5c、5g或5t)。收率A:0.88g、0.95mmol、95%;C:0.89g、0.99mmol、99%;G:0.58g、0.64mmol、64%;T:0.64g、0.65mmol、65%。
H-硫代膦酸酯单体5a
1
31
H-硫代膦酸酯单体5c
1
31
H-硫代膦酸酯单体5g
1
31
H-硫代膦酸酯单体5t
1
31
(2’-OMe修饰H-硼烷膦酸酯单体的合成)
按照下述方案合成了2’-OMe修饰H-硼烷膦酸酯单体。
将上述方案左端记载的核苷衍生物2a、2c、2g或2t(A:0.69g,1.0mmol;C:0.68g,1.0mmol;G:0.70g,1.0mmol;或U:0.67g,1mmol)和吡啶鎓H-硼烷膦酸酯(0.28g,2.0mmol)在氩气气氛下用吡啶(3mL×3次)进行共沸干燥,溶解于吡啶25mL。对于得到的吡啶溶液,边在冰浴下搅拌边加入BopCl(0.51g,2.0mmol),之后将反应温度设为室温并搅拌1小时。将该溶液用氯仿30mL稀释后,将有机层用1.0M TEAB缓冲溶液(pH7.0)清洗(30mL×3次)。将水层用氯仿进行反萃取(30mL×3次)。合并有机层,用无水硫酸钠除去水分后,将有机层的溶剂减压馏去,将得到的残渣用硅胶柱色谱进行分离纯化(展开溶剂A:乙酸乙酯-甲醇-三乙胺(100:0:1-100:4:1,v/v/v)、之后氯仿-甲醇-三乙胺(100:4:1,v/v/v);C:乙酸乙酯-甲醇-三乙胺(100:0:1-100:4:1,v/v/v)、之后氯仿-甲醇-三乙胺(100:4:1,v/v/v);G:乙酸乙酯-甲醇-三乙胺(100:2:1,v/v/v)、之后氯仿-甲醇-三乙胺(100:3:1-100:5:1,v/v/v);U:乙酸乙酯-甲醇-三乙胺(100:0:1,v/v/v)、之后氯仿-甲醇-三乙胺(100:0:1-100:1:1,v/v/v)),利用1.0M TEAB缓冲溶液(pH7.0)和氯仿进行萃取操作(30mL×3次)。将有机层中所含的水分用无水硫酸钠除去,将溶剂减压馏去,由此得到目标物(2’-OMe修饰H-硼烷膦酸酯单体6a、6c、6g或6u)。收率A:0.81g、0.95mmol、95%;C:0.75g、0.90mmol、90%;G:0.78g、0.92mmol、92%;U:0.62g、0.74mmol、74%
2’-OMe修饰H-硼烷膦酸酯单体6a
1
31
2’-OMe修饰H-硼烷膦酸酯单体6c
1
31
2’-OMe修饰H-硼烷膦酸酯单体6g
1
31
2’-OMe修饰H-硼烷膦酸酯单体6u
1
31
<基于固相法的核酸低聚物的合成1>
以下的内部核苷酸键中,用带下划线的PS表示硫代磷酸酯键,用带下划线的PB表示硼烷磷酸酯键,用带下划线的PO表示磷酸二酯键。
(T
[比较合成例1]
对于借助琥珀酰连接体负载于固相载体的5’-二甲氧基三苯甲基-N
[比较合成例2]比较合成例1的对照实验
不加入H-硫代膦酸酯单体5t而是加入(DMP)
[合成例1]
作为缩合剂,使用BOMP(23.3mg、100当量、50μmol)来代替(DMP)
[合成例2]
作为缩合剂,使用MNTP(22.3mg,100当量,50μmol)代替BOMP,除此以外进行与合成例1同样的操作。将结果示于表1及图1的(c)。
[合成例3]
作为缩合剂,使用PyNTP(25.0mg,100当量,50μmol)代替BOMP,除此以外进行与合成例1同样的操作。将结果示于表1及图1的(d)。
[合成例4]
作为缩合反应时添加的碱,使用吡啶(0.6M,120μmol)代替2,6-二甲基吡啶,除此以外进行与合成例3同样的操作。将结果示于表1。
[合成例5]
作为缩合反应时添加的碱,使用喹啉(0.6M,120μmol)代替2,6-二甲基吡啶,除此以外进行与合成例3同样的操作。将结果示于表1。
[合成例6]
作为缩合反应时添加的碱,使用喹啉(1.8M,355μmol)代替2,6-二甲基吡啶,除此以外进行与合成例3同样的操作。将结果示于表1及图1的(e)。
[合成例7]
作为缩合反应时的反应溶剂,仅使用未加入碱的乙腈,除此以外进行与合成例3同样的操作。将结果示于表1及图1的(f)。
【表1】
表1中的化学选择性由HPLC结果中的、作为副产物的T
图1示出比较合成例1及2以及合成例2、3、6及7的反相HPLC分析结果。结果是,与比较合成例1相比,合成例6及7的目标物的收率高。需要说明的是,仅比较合成例1中在洗脱时间28分钟附加确认到无法鉴定的峰。因此在比较合成例2中进行了在缩合反应时未加入单体的对照实验,结果通过反相HPLC在洗脱时间28分钟观察到峰。由该结果可认为,比较合成例1中在洗脱时间28分钟附加观察到的峰为源自羟基与缩合剂反应而得的副产物。上述合成例及比较合成例中合成的是二聚体,但是在合成低聚物时如果发生这样的副反应,则反应效率大幅下降这一点令人担心,因此可确认合成例的方法适合于低聚物的合成。
(N
[合成例8]
对于借助琥珀酰连接体负载于固相载体的5’-二甲氧基三苯甲基-N
[合成例9]
作为反应溶剂,仅使用未加入碱的乙腈,除此以外进行与合成例8同样的操作。将结果示于表2。
[合成例10]
合成例10仅实施使用H-硫代膦酸酯单体5g的情况。将该单体的量变更为36.2mg(80当量、40μmol),将PyNTP的量变更为50.0mg(200当量、100μmol),除此以外进行与合成例9同样的操作。将结果示于表2。
【表2】
表2中的化学选择性由HPLC结果中的、作为副产物的dN
(N
[合成例11]
以下,N
对于借助琥珀酰连接体负载于固相载体的5’-二甲氧基三苯甲基-N
【表3】
表3中的HPLC收率由HPLC结果中的面积比N
[实施例1]PB/PS/PO嵌合型核酸低聚物(十二聚体)的合成
对于借助琥珀酰连接体负载于固相载体的5’-二甲氧基三苯甲基-N
(i)工序:将根据序列而选择的单体、MNTP或PyNTP及根据需要的2,6-二甲基吡啶或喹啉在乙腈(0.2mL)中混合,反应3分钟。3分钟后,将反应溶液除去,将反应后的固相载体用乙腈清洗4次,用二氯甲烷清洗4次。将使用各种单体的缩合条件示于以下。
【表4】
(ii)工序:使上述(i)工序后的固相载体与3%二氯乙酸/二氯甲烷-三乙基硅烷(1:1、v/v)溶液(1mL)反应15秒。将同样的操作重复进行4次后,用二氯甲烷(1mL)清洗4次,用乙腈(1mL)清洗4次,将固相载体真空干燥。
对于链长延伸后的固相载体,加入0.1M三乙胺/四氯化碳-2,6-二甲基吡啶-H
之后,对于清洗后的固相载体,使25%氨水-乙醇(3:1、v/v)在50℃下反应20小时,由此进行核酸碱基部的脱保护和从固相载体的核酸低聚物切割。滤出固相载体并回收滤液后,进行溶剂的减压馏去。
将包含得到的PB/PS/PO嵌合型核酸低聚物(十二聚体)的溶液用25%NH
收率6%。HRMS(ESI-TOF):对于[M-6H]
得到的PB/PS/PO嵌合型核酸低聚物(十二聚体)为具有d(C
[实施例2]PB/PS/PO嵌合型核酸低聚物(含有apoB序列的十二聚体)的合成
选择编码载脂蛋白B(apoB)(载脂蛋白B-100:Nature,2006,Vol.441,pp111-114)的mRNA的一部分(3’-rCGU AAC CAU AAG-5’:互补链RNA、序列号2)作为靶核酸。合成具有与RNA的碱基序列互补的碱基序列的PB/PS/PO嵌合型核酸低聚物。
对于借助琥珀酰连接体负载于固相载体的5’-O-DMTr-N
收率19%。HRMS(ESI-TOF):对于[M-6H]
得到的PB/PS/PO嵌合型核酸低聚物(十二聚体)为具有d(G
[实施例3]PB/PS/PO-gapmer(十二聚体)的合成
与实施例2同样地,以形成与靶核酸(载脂蛋白B)的碱基序列互补的序列的方式,在实施例2所使用的单体的基础上,还使用具有N
将包含得到的PB/PS/PO-gapmer(十二聚体)的溶液用25%NH
收率13%。HRMS(ESI-TOF):对于[M-6H]
得到的PB/PS/PO-gapmer(十二聚体)为具有G
[实施例4]PB/PS嵌合型核酸低聚物(十二聚体)的合成
与实施例2同样地,以成为与靶核酸(载脂蛋白B)的碱基序列互补的序列的方式,依次重复进行具有保护基的羟基的脱保护反应、缩合反应直至得到十二聚体后,进行氧化反应及核酸碱基的脱保护反应、从固相载体的切割,得到PB/PS嵌合型核酸低聚物(十二聚体)。
将含有得到的PB/PS嵌合型核酸低聚物(十二聚体)的溶液用25%NH
收率5%。HRMS(ESI-TOF):对于[M-6H]
得到的PB/PS嵌合型核酸低聚物(十二聚体)为具有d(G
<单体的合成2>
(2’-O-4’-C-Locked-H-硼烷膦酸酯单体7a或7c的合成)
以下,也将2’-O-4’-C-Locked-H-硼烷膦酸酯单体称为锁核酸(以下称为“LNA”。)修饰H-硼烷膦酸酯单体。
将上述方案左端记载的核苷衍生物(A:0.69g,1.0mmol;或
LNA修饰H-硼烷膦酸酯单体7a
1
31
LNA修饰H-硼烷膦酸酯单体7c
1
31
(2’-O-4’-C-Locked-H-硼烷膦酸酯单体7g的合成)
将2’-O-4’-C-Locked-鸟苷(0.443g,1.5mmol)在氩气气氛下溶解于吡啶5mL,依次加入4-二甲基氨基吡啶(0.189g、1.5mmol)、异丁酸酐(1.25mL、7.5mmol),在110℃下反应9小时。向混合物中加入氯仿(20mL),用1M盐酸(20mL)清洗,将水层用氯仿(20mL)反萃取。将合并后的有机层用饱和碳酸氢钠水溶液(40mL)清洗2次,将水层用氯仿(40mL)反萃取。将合并后的有机层用硫酸钠干燥后进行过滤,将溶剂减压馏去。不进行此外的纯化操作而直接用于下一个反应中。将混合物在氩气气氛下溶解于吡啶(4mL),依次加入二异丙基乙基胺(0.4mL、2.25mmol)、二苯基氨基甲酰氯(0.69g、3.0mmol),在室温下搅拌1小时。加入甲醇(5mL)使反应停止,加入氯仿(20mL),用饱和碳酸氢钠水溶液(20mL)清洗3次。将合并后的水层用氯仿(20mL)反萃取。将合并后的有机层用硫酸钠干燥后过滤,将溶剂减压馏去。将得到的残渣用硅胶柱色谱(展开溶剂:乙酸乙酯-己烷(40:60-50:50、v/v))纯化,由此得到2’-O-4’-C-Locked-3’,5’-O-二异丁酰基-2-N-异丁酰基-6-O-二苯基氨基甲酰基-鸟苷(0.859g、1.23mmol、82%)。
将得到的2’-O-4’-C-Locked-3’,5’-O-二异丁酰基-2-N-异丁酰基-6-O-二苯基氨基甲酰基-鸟苷全部溶解于吡啶-甲醇混合溶剂(1:1、v/v、21mL),在0℃下将甲醇钠(26.4mg、0.488mmol)分阶段地分成4次加入,搅拌14小时。再升温到室温并搅拌4.5小时后,加入阳离子交换树脂而使反应停止,通过过滤除去阳离子交换树脂后,将溶剂减压馏去。将得到的残渣用硅胶柱色谱(展开溶剂:氯仿-甲醇(100:0-100:4、v/v))纯化,由此得到2’-O-4’-C-Locked-2-N-异丁酰基-6-O-二苯基氨基甲酰基-鸟苷(0.401g、0.715mmol、58%)。
将得到的2’-O-4’-C-Locked-2-N-异丁酰基-6-O-二苯基氨基甲酰基-鸟苷全部在氩气气氛下溶解于吡啶(7mL),加入4,4’-二甲氧基三苯甲基氯(0.266g、0.79mmol),在室温下搅拌1.5小时后,进一步加入4,4’-二甲氧基三苯甲基氯(61mg、0.18mmol),搅拌40分钟。加入甲醇(5mL)而使反应停止,加入氯仿(20mL),用饱和碳酸氢钠水溶液(20mL)清洗3次。将合并后的水层用氯仿(20mL)反萃取。将合并后的有机层用硫酸钠干燥后过滤,将溶剂减压馏去。将残渣用硅胶柱色谱(展开溶剂:氯仿-甲醇-吡啶(100:0:0.5-100:1.5:0.5、v/v/v))纯化,得到2’-O-4’-C-Locked-5’-O-(4,4’-二甲氧基三苯甲基)-2-N-异丁酰基-6-O-二苯基氨基甲酰基-鸟苷(0.617g、0.451mmol、63%)。
将上述方案左端记载的LNA修饰鸟苷衍生物(0.39g,0.45mmol)和吡啶鎓H-硼烷膦酸酯(0.19g,0.90mmol)在氩气气氛下用吡啶(3mL×3次)进行共沸干燥,溶解于吡啶(5mL)。对于得到的吡啶溶液,边在冰浴下搅拌边加入BopCl(0.23g,0.90mmol),之后将反应温度设为室温并搅拌1小时。将该溶液用氯仿(30mL)稀释后,将有机层用1.0M TEAB缓冲溶液(pH7.0)清洗(30mL×3次)。将水层用氯仿反萃取(30mL×3次)。合并有机层,用无水硫酸镁除去水分后,将有机层的溶剂减压馏去,将得到的残渣用硅胶柱色谱分离纯化(展开溶剂:乙酸乙酯-三乙胺(100:0.5,v/v)、之后氯仿-甲醇-三乙胺(98:2:0.5,v/v/v)),将溶剂减压馏去,由此得到LNA修饰H-硼烷膦酸酯单体7g(0.35g,0.92mmol,92%)。
LNA修饰H-硼烷膦酸酯单体7g
1
31
(2’-O-4’-C-Locked-H-硼烷膦酸酯单体7t的合成)
2’-O-4’-C-Locked-H-硼烷膦酸酯单体7t通过文献(The Journal of OrganicChemistry、2014年、第79卷、3465-3472页)记载的合成方法来合成。
(2’-O-MOE修饰H-硼烷膦酸酯单体的合成)
按照下述方案合成2’-O-MOE修饰H-硼烷膦酸酯单体。
将上述方案左端记载的鸟苷衍生物8g(0.722g,1.0mmol)在氩气气氛下用吡啶(3mL×3次)共沸干燥,与吡啶鎓H-硼烷膦酸酯(0.30g,2.0mmol)一起溶解于吡啶25mL。对于得到的吡啶溶液,边在冰浴下搅拌边加入BopCl(0.51g,2.0mmol),之后将反应温度设为室温并搅拌6小时。将该溶液用氯仿30mL稀释后,将有机层用1.0M TEAB缓冲溶液(pH7.0)清洗(30mL×3次)。将水层用氯仿反萃取(30mL×3次)。合并有机层,用无水硫酸钠除去水分后,将有机层的溶剂减压馏去,将得到的残渣用硅胶柱色谱分离纯化(展开溶剂:乙酸乙酯-甲醇-三乙胺(100:0:1,v/v/v)、之后氯仿-甲醇-三乙胺(100:10:1,v/v/v)),将溶剂减压馏去,由此得到2’-O-MOE修饰H-硼烷膦酸酯单体9g。收量0.57g、0.60mmol,收率60%
2’-O-MOE修饰H-硼烷膦酸酯单体9g
1
31
<基于固相法的核酸低聚物的合成2>
(N
[合成例12]
以下,N
对于借助琥珀酰连接体负载于固相载体的5’-二甲氧基三苯甲基-N
【表5】
表5中的HPLC收率由HPLC结果中的面积比G
[实施例5]PB/PS-LNA-gapmer(十二聚体)的合成
与实施例2同样地合成作为与靶核酸(载脂蛋白B)的碱基序列互补的序列的核酸低聚物。作为固相载体,使用具有5’-二甲氧基三苯甲基的UnyLinker(UniversalUnyLinker Support
(A)工序:向填充有固相载体的固相合成用柱中通入4次3%三氯乙酸/二氯甲烷溶液并反应40秒(10秒/次),由此进行脱三苯甲基化。反应结束后,用乙腈清洗固相载体,接着用氩气进行通气干燥。然后,向柱中通入4次以0.1mol/L溶解有碱基部具有N
(B)工序:之后,对于第二个碱基及此后的碱基,通过与实施例2同样的方法依次重复进行具有保护基的羟基的脱保护反应、缩合反应,直至得到十二聚体为止。其中,作为本序列中使用LNA修饰H-硼烷膦酸酯单体的缩合用的缩合剂,使用MNTP,作为使用2’-脱氧核糖核酸型的H-硼烷膦酸酯单体的缩合用的缩合剂,使用PyNTP(将各种单体的缩合条件示于表6)。最后进行氧化反应,得到包含PB/PS-LNA-gapmer(十二聚体)的固相载体。
【表6】
对于得到的包含PB/PS-LNA-gapmer(十二聚体)的固相载体,在25℃(室温)使28%NH
纯度18.7%(洗脱时间:11.3分钟)。LRMS(ESI-MS):对于[M-4H]4-,计算值977.71;实测值977.89.对于[M-5H]5-,计算值781.97;实测值781.87
得到的PB/PS-LNA-gapmer(十二聚体)为具有
[实施例6]PB/PS/PO-mixmer(十二聚体)的合成
与实施例2同样地,以成为与靶核酸(载脂蛋白B)的碱基序列互补的序列的方式,在实施例2所使用的单体的基础上,还使用具有N
之后,用反相HPLC进行分离纯化。利用反相HPLC的分析及分离纯化在60℃下使用10~45%甲醇的0.4M六氟异丙醇-0.008M三乙胺缓冲溶液作为流动相以流速0.5mL/分钟进行20分钟。将实施例6中分离纯化前及分离纯化后进行的反相HPLC的谱图分别示于图8的(a)及图8的(b)。
收率20%。HRMS(ESI-TOF):对于[M-4H]
得到的PB/PS/PO-mixmer(十二聚体)为具有G
【表7】
/>
序列表
<110>学校法人东京理科大学(TOKYO UNIVERSITY OF SCIENCE FOUNDATION)
<120>包含硫代磷酸酯及硼烷磷酸酯的嵌合型核酸低聚物及其制造方法
<130>RKF-070PCT
<150>JP 2021-092873
<151>2021-06-02
<160>7
<210>1
<211>12
<212>DNA
<213>人工序列(Artificial Sequence)
<220>
<223>示意性序列
<400>1
cagtcagtca gt 12
<210>2
<211>12
<212>RNA
<213>智人(Homo sapiens)
<400>2
gaauaccaau gc 12
<210>3
<211>12
<212>DNA
<213>人工序列(Artificial Sequence)
<220>
<223>apoB (PB/PS/PO 嵌合体)反义低聚物
<400>3
gcattggtat tc 12
<210>4
<211>12
<212>DNA
<213>人工序列(Artificial Sequence)
<220>
<223>apoB (PB/PS/PO-gapmer)反义低聚物
<220>
<223>DNA/RNA嵌合体
<400>4
gcattggtau uc 12
<210>5
<211>12
<212>DNA
<213>人工序列(Artificial Sequence)
<220>
<223>apoB (PB/PS嵌合体)反义低聚物
<400>5
gcattggtat tc 12
<210>6
<211>12
<212>DNA
<213>人工序列(Artificial Sequence)
<220>
<223>apoB (PB/PS-LNA-gapmer)反义低聚物
<220>
<223>DNA/RNA嵌合体
<400>6
gcattggtat tc 12
<210>7
<211>12
<212>DNA
<213>人工序列(Artificial Sequence)
<220>
<223>apoB (PB/PS/PO-mixmer)反义低聚物
<220>
<223>DNA/RNA嵌合体
<400>7
gcattggtat tc 12
- 一种光纤束中心线调节装置及松套管生产装置
- 一种有限旋转角度线束保持装置
- 一种应用于泵浦合束器中心支路的回返光处理方法及装置
- 一种CBCT等中心治疗设备用滤波限束装置
- 一种CBCT等中心治疗设备用滤波限束装置