一种对特征丰度数据和样本表型数据进行关联分析的方法
文献发布时间:2023-06-19 11:26:00
技术领域
本发明涉及生物学领域,具体涉及一种对特征丰度数据和样本表型数据进行关联分析的方法。
背景技术
随着生物技术、计算机技术以及高通量技术的发展,各个领域积累了大量样本表型与其特征丰度相互关联的文献和数据。面对海量数据,如何对其进行系统分析和深入挖掘成为了生命科学研究领域的研究热点。其中深入挖掘复杂表型的关联特征成为在相关研究中的一项重要挑战,对于实际问题的研究具有重要的指导意义。
各组学数据在与表型数据进行关联分析的时候,主要面临以下几个问题:第一、表型数据的数据量一般不会给分析过程带来麻烦,但是有时特征数据的量级却是很大,比如,微生物中的基因数量就是人的基因数量的100倍不止。第二、每个样本的特征本身就存在很大的差异性,给分析建模带来巨大的挑战,例如,人的个体都具有相同的基因,但是他们所携带的微生物的种类和基因却是千差万别的。第三、特征数据的定量存在困难,人的基因表达量很容易计算,而大部分微生物组数据只能通过相对丰度进行量化。第四、特征本身就有很大的波动性,例如人的基因组是不会改变的,当然,除了癌症等特殊情况除外,而个体所携带的微生物组却在无时无刻发生着变化。
关于表型数据和特征数据的关联分析,其中较为典型的是应用宏基因组测试来研究微生物与疾病之间的关联,被称之为宏基因组关联分析(MWAS)。现有的MWAS研究,是将宏基因组数据中特征基因的相对丰度与感兴趣的疾病建立关联,常见的做法是对基因进行聚类,可以降低数据的维度,将物种分辨率提高至菌株水平。通过MWAS,使得人类微生物组与复杂疾病,如二型糖尿病、肥胖症、肝硬化、结直肠癌和类风湿型关节炎等之间高分辨率的关联研究成为可能。
MWAS研究发现,健康人群和二型糖尿病(T2D)患者在肠道菌群结构和功能上有着较大的差异。健康人肠道菌群中富含促进有益代谢产物(如短链脂肪酸、维生素)生成的菌群。短链脂肪酸(SCFA)经肠道上皮细胞吸收,与受体结合诱导Treg细胞的分化,进而抑制炎症反应,促进组织损伤修复,有利于维持肠道的完整性和能量平衡。SCFA也可以促进胰高血糖素肽的分泌,从而调节血糖平衡并控制食物摄取。T2D患者中这些细菌和功能的丰富程度明显少于正常人。
结直肠癌(CRC)是常见的恶行肿瘤,通常由结直肠腺瘤缓慢发展而成,具有高度破坏性和侵袭性,在中老年中由较高发病率。MWAS研究发现,一些厌氧口腔菌与CRC相关联,如梭杆菌和微小微单胞菌。与CRC相关的功能改变包括:氨基酸发酵或胆汁酸代谢产生致癌物质,产生短链脂肪酸功能下降。
类风湿性关节炎(RA)是一个主要影响关节的长期持续性疾病。MWAS分析发现,人体口腔菌群和肠道菌群与RA疾病具有相关性。RA患者口腔和肠道菌群中一些共有的微生物物种会同时增加,如唾液乳杆菌。此外RA患者口腔和肠道样本检测到的菌群变化具有相关性,如肠道中的克雷白氏肺炎杆菌丰度和口腔中的乳球菌丰度是正相关的;而肠道中天门冬形梭菌与口腔的中间普氏菌丰度负相关。
未来基于MWAS技术对微生物组在相关疾病中作用的研究会越来越深入,科学家们希望能够发展一个微生物全球定位系统来对疾病人群进行分层,指导精准医疗,从而维护人类健康。
发明内容
针对上述问题,本发明的目的是提供一种对特征丰度数据和样本表型数据进行关联分析的方法。
为实现上述目的,本发明采取以下技术方案:一种对特征丰度数据和样本表型数据进行关联分析的方法,其包括如下步骤:
(1)获取特征丰度数据矩阵和样本表型数据矩阵,并同时对这两个数据矩阵进行预处理;
(2)对预处理后的特征丰度数据矩阵和样本表型数据矩阵进行LASSO回归,过滤掉回归过程中,回归系数被LASSO回归模型压缩为0的特征和样本;
(3)计算剩余特征丰度数据矩阵中特征之间以及样本表型数据矩阵中样本之间的相关系数,并将相关性未达到预设阈值的特征和样本进行过滤;
(4)对步骤(3)中最终剩余的特征丰度数据矩阵与样本表型数据矩阵进行线性回归分析,最终得到特征丰度数据矩阵中和样本表型数据有关联的特征。
进一步,所述步骤(1)中,获取特征丰度数据矩阵和表型数据矩阵,并同时对这两个数据矩阵进行预处理的方法,包括以下步骤:
(1.1)获取特征丰度数据矩阵和样本表型数据矩阵,并同时对这两个数据矩阵进行标准化处理,得到标准化特征丰度数据矩阵和标准化样本表型数据。
(1.2)对标准化特征丰度数据矩阵中“零值”到达预设比例的特征以及标准化样本表型数据矩阵中含有异常值的样本进行剔除。
(1.3)计算剔除后标准化特征丰度数据矩阵的条件数,判断特征之间的多重共线性的严重程度,并使用方差膨胀系数过滤多重共线性超过预设阈值的特征。
进一步,所述步骤(1.1)中,对特征丰度数据矩阵和样本表型数据矩阵进行标准化处理的方法包括:将数据转换为有无类型、最大值标准化、总和标准化、最小最大标准化、模标准化、hellinger转化和z值标准化。
进一步,所述步骤(1.2)中,对标准化特征丰度数据中“零值”到达预设比例的特征进行剔除时,若标准化特征丰度矩阵中多于20%样本中特征取值为“零值”,则进行去除,否则保留不变。
进一步,所述步骤(1.2)中,对于标准化样本表型数据矩阵中的异常值样本进行处理时,处理过程为:
首先,对标准化样本表型数据矩阵进行检测,得到异常值样本数据;
然后,对异常值样本数据进行处理,处理时包括直接剔除或通过拟合插值的方法,为异常值重新赋值。
进一步,所述步骤(1.3)中,计算特征丰度数据矩阵的条件数,判断特征之间的多重共线性的严重程度,然后使用方差膨胀系数过滤多重共线性超过预设阈值的特征的方法,包括以下步骤:
(1.3.1)计算剔除零值后标准化特征丰度数据矩阵的条件数,并基于计算得到的条件数对特征之间的多重共线性的严重程度进行判断,将该严重程度分为不存在、中等程度以及严重程度三类;
(1.3.2)采用方差膨胀系数对步骤(1.3.1)中多重共线性为严重程度的特征进行过滤,得到过滤后的特征丰度数据矩阵。
进一步,所述步骤(1.3.1)中,计算特征丰度数据矩阵的条件数,并基于计算得到的条件数对特征之间的多重共线性的严重程度进行判断,将该严重程度分为不存在、中等程度以及严重程度三类的方法,包括以下步骤:
首先,计算特征丰度数据矩阵的条件数,所述特征丰度数据矩阵的条件数是指数据矩阵的kappa值,计算方法是将数据矩阵的相关系数矩阵与其转置矩阵做矩阵乘法运算,得到的新矩阵的最大特征值与最小特征值的比值就是数据矩阵的kappa值;
其次,根据计算得到的kappa值将特征对特征之间的多重共线性的严重程度进行判断:
当kappa值小于100,特征之间不存在多重共线性问题;
当kappa值介于100和1000之间时,特征之间存在中等程度的多重共线性问题;
当kappa值大于1000的时候,特征之间存在严重的多重共线性问题。
进一步,所述步骤(1.3.2)中,采用方差膨胀系数对步骤(1.3.1)中多重共线性为严重程度的特征进行过滤时:
首先,计算所有自变量的方差膨胀系数,删除那些方差膨胀系数大于10的自变量;
然后,再次计算所有剩余自变量的方差膨胀系数,再删除掉那些方差膨胀系数大于10的自变量,直到所有自变量的方差膨胀系数都在10以下。
进一步,所述步骤(3)中,所述相关系数包括pearson相关系数、spearman相关系数和kendall相关系数中的至少一种。
进一步,所述步骤(4)中,特征丰度数据矩阵中和样本表型数据有关联的特征的方法,包括以下步骤:
(4.1)采用线性回归方法对步骤(3)中最终剩余的特征丰度数据矩阵与特定表型数据进行线性回归分析,得到各特征的线性回归系数;
(4.2)对每个特征的线性回归系数进行T检验,线性回归系数不为0的特征结合相关系数结果得到最终和表型有关联的特征。
本发明由于采取以上技术方案,其具有以下优点:1、本发明通过对特征丰度数据矩阵和样本表型数据矩阵进行LASSO回归,同时通过计算pearson相关系数、spearman相关系数和kendall相关系数,在进行关联分析之前,进行了特征筛选,降低了特征丰度数据的维度,缩短了分析时间,适用于数据量较大的情况,经过多种统计模型的层层筛选,在一定程度上降低了结果的假阳性。2、本发明通过对达到一定相关性的特征与表型做一般线性回归分析,结合回归系数的T检验结果来最终确定与特征表型相关联的特征,有效提高了准确性和效率。因此,本发明可以广泛应用于生物数据分析领域。
附图说明
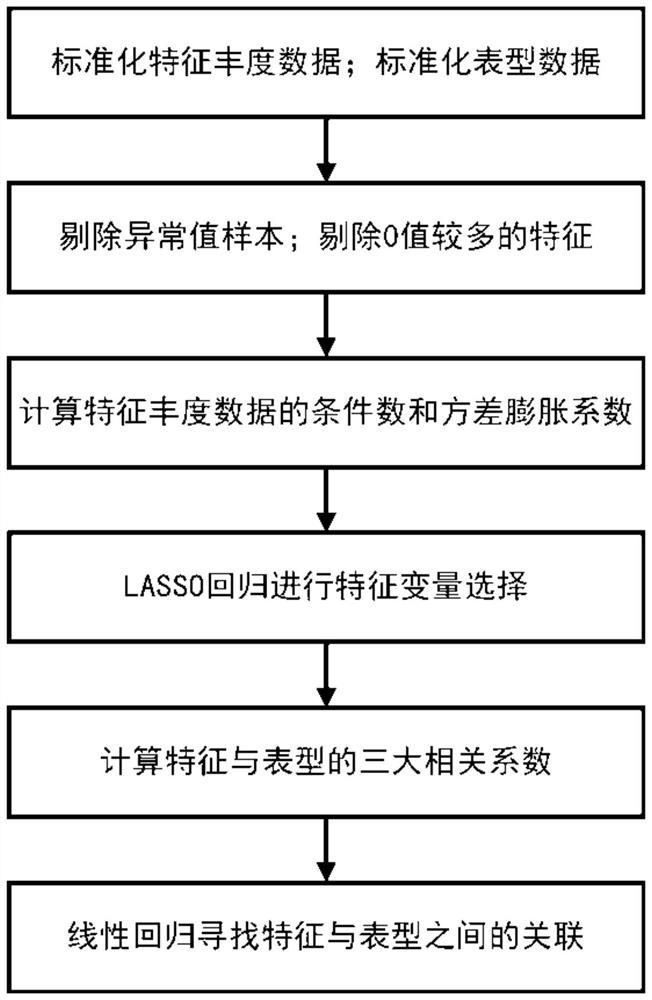
图1是本发明实施例提供的一种对特征丰度数据和样本表型数据进行关联分析的方法流程图。
具体实施方式
下面将对本发明的具体实施方式进行详细说明。需要理解的是以下过程的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。
本发明在对高通量测序技术产生的丰度数据和采集的样本表型数据的研究基础上,提供了一种通过组合多种统计模型来找出与特定表型数据具有关联关系的特征的方法。具体的,本发明提供的一种对特征丰度数据和样本表型数据进行关联分析的方法,其包括如下步骤:
(1)获取特征丰度数据矩阵和样本表型数据矩阵,并同时对这两个数据矩阵进行预处理;
(2)对预处理后的特征丰度数据矩阵和样本表型数据矩阵进行LASSO回归,过滤掉回归过程中,回归系数被LASSO回归模型压缩为0的特征和样本;
(3)计算剩余特征丰度数据矩阵中特征之间以及样本表型数据矩阵中样本之间的pearson相关系数、spearman相关系数和kendall相关系数,并将相关性未达到预设阈值的特征和样本进行过滤;
(4)对步骤(3)中最终剩余的特征丰度数据矩阵与样本表型数据矩阵进行线性回归分析,最终得到特征丰度数据矩阵中和样本表型数据有关联的特征。
上述步骤(1)中,获取数据并进行预处理的方法,包括以下步骤:
(1.1)获取特征丰度数据矩阵和样本表型数据矩阵,并同时对这两个数据矩阵进行标准化处理,得到标准化特征丰度数据矩阵和标准化样本表型数据。
(1.2)对标准化特征丰度数据矩阵中“零值”到达预设比例的特征以及标准化样本表型数据矩阵中含有异常值的样本进行剔除。
(1.3)计算剔除后标准化特征丰度数据矩阵的条件数,判断特征之间的多重共线性的严重程度,并使用方差膨胀系数过滤多重共线性超过预设阈值的特征。
上述步骤(1.1)中,获取的特征丰度数据矩阵中,行为样本,列为特征;获取的样本表型数据矩阵中,同样行为样本,列为表型。需要注意的是,由于本发明中采用的统计模型多为回归模型,所以如果要进行标准化处理,建议同时对特征丰度数据矩阵和样本表型数据矩阵进行标准化处理。可以使用的标准化方法包括:将数据转换为有无类型、最大值标准化、总和标准化、最小最大标准化、模标准化、hellinger标准化和z值标准化等。
数据标准化方法各有各的特征,将数据转换为有无类型,也就是数据当中只包含0和1两个数字,这种标准化方法一般用于研究非加权下的菌落结构;最大值标准化方法,是将数据除以该行或者该列的最大值,若数据非负,则最大值标准化后数据全部位于0到1之间;总和标准化方法,是将数据除以该行或者该列的总和,也即求相对丰度,总和标准化后数据全部位于0到1之间;最大最小标准化方法,将数据减去该行或者该列的最小值,并且比上最大值与最小值之差,最大最小标准后的数据全部位于0到1之间;模标准化方法,将数据除以每行或者每列的平方和的平方根,模标准化后每行、列的平方和为1,也即向量的模为1,就是在笛卡尔坐标系中到原点的欧式距离为1,样本分布在一个圆弧上,彼此之间的距离为弦长,因此这种标准化方法也叫做弦转化,弦转化后的数据使用欧式距离函数计算就可以得到弦距离矩阵;hellinger标准化方法,就是总和标准化数据的平方根,hellinger转换后的数据使用欧式距离函数计算将得到hellinger距离矩阵;z值标准化方法,最常用的标准化方法之一,将数据减去均值后比上标准差,z值标准化后的数据均值为0,方差为1,服从正态总体的数据标准化后服从标准正态分布,z值标准化可以去除不同环境因子量纲的影响。
上述步骤(1.2)中,对标准化特征丰度数据矩阵和标准化样本表型数据矩阵中的数据进行剔除时,包括两个部分,一是去掉标准化特征丰度数据矩阵中含有较多“零值”的特征,因为这些特征对于建模分析没有实质性的帮助;二是去掉标准化样本表型数据矩阵中具有异常值的样本数据,这些数据出现异常,可能是在数据收集过程中发生了记录错误,这一类数据除了剔除掉异常样本之外,还可以按照处理缺失值的办法处理这一类数据,因为有时样本数量有限,应该尽可能保留每一个样本。
关于标准化特征丰度数据矩阵,具体的,对于“零值”的处理具有一定的难度,因为无法分辨“零值”产生的原因,是测序深度不够,该特征没有被检测出来还是就是丰度信息就是为零,并且零值太多在计算相关性的时候会带来很高的假阳性,毕竟两个向量都包含很多“零值”,这本身就是一种相似,所以,当“零值”较多的时候,为了提高模型结果的准确性,应该剔除掉这些特征,本发明建议根据标准化特征丰度数据矩阵的稀疏程度确定预设比例,若标准化特征丰度矩阵数据不是很稀疏,则保留取值都不是“零值”的特征;若标准化特征丰度矩阵数据很稀疏,则去掉那些在多于20%样本中取值为“零值”的特征,这个阈值可以根据矩阵的稀疏程度来作出适当的调整。
关于标准化样本表型数据矩阵而言,对于异常值样本来说,处理过程又可以分为两步,第一步就是检测方法,就是找到含有异常值的样本,第二步就是处理办法,就是将异常值变得不异常。
对于异常值的判断,研究者可以针对具体问题,具体分析。具体的,可以预先估算每一个特征的一个合理的范围,落在这个范围之外的就是异常样本了,还可以借助一些统计学的方法,比如,降维处理,将所有样本可视化在一个人类可视的超平面上,这样异常样本可以一目了然,再有,根据正态分布的理论,均值周围三倍方差之外的样本出现的概率是极小的,这样的样本也可以当中异常样本来处理。
对于检测出来的异常样本,可以直接剔除,如果样本数量足够支持这样做的时候,当不支持的时候,可以考虑按照处理缺失值的方法来处理这些异常值,比如通过拟合插值的方法,重新给异常值的样本赋值一个合理的值,又或者所有特征取值的均值或者中位数来代替异常值等等。
上述步骤(1.3)中,采用方差膨胀系数对标准化特征丰度数据矩阵的特征值进行过滤的方法,包括以下步骤:
(1.3.1)计算剔除零值后标准化特征丰度数据矩阵的条件数,并基于计算得到的条件数对特征之间的多重共线性的严重程度进行判断,将该严重程度分为不存在、中等程度以及严重程度三类。
其中,数据矩阵的条件数具体是指数据矩阵的kappa值,具体计算方法是数据矩阵的相关系数矩阵与其转置矩阵做矩阵乘法运算,得到的新矩阵的最大特征值与最小特征值的比值就是数据矩阵的kappa值。一般来说,当kappa值小于100,特征之间不存在多重共线性问题;当kappa值介于100和1000之间时,特征之间存在中等程度的多重共线性问题;当kappa值大于1000的时候,特征之间存在严重的多重共线性问题。
(1.3.2)采用方差膨胀系数对步骤(1.3.1)中多重共线性为严重程度的特征进行过滤,得到过滤后的特征丰度数据矩阵。
方差膨胀系数是衡量多元线性回归模型中多重共线性严重程度的一种度量,它表示回归系数估计量的方差与假设自变量间不线性相关时方差相比的比值,方差膨胀系数越大,表示自变量越有共线性问题。方差膨胀系数通常以10作为判断边界,当方差膨胀系数小于10的时候,不存在多重共线性;当方差膨胀系数介于10到100之间的时候,存在较强的多重共线性;当方差膨胀系数大于100的时候,存在严重的多重共线性。
采用方差膨胀系数对步骤(1.3.1)中多重共线性为严重程度的特征进行过滤时,可以在第一轮计算所有自变量的方差膨胀系数,删除那些方差膨胀系数大于10的自变量,然后再次计算所有剩余自变量的方差膨胀系数,再删除掉那些方差膨胀系数大于10的自变量,以此类推,直到所有自变量的方差膨胀系数都在10以下。
上述步骤(2)中,LASSO回归的特点是在拟合广义线性模型的同时进行变量筛选和复杂度调整,不仅如此,不论因变量是连续的还是离散的,都可以用LASSO回归建模然后预测,这里的变量筛选是指不把所有变量都放入模型中进行拟合,而是有选择的把变量放入模型从而得到更好的性能参数,LASSO回归复杂度调整的程度是由一个参数λ来控制的,λ越大对变量较多的线性模型的惩罚力度就越大,从而最终获得一个变量较少的模型,降低模型复杂度,避免了过度拟合的问题。
上述步骤(3)中,pearson、spearman、kendall三个相关系数反映的都是两个变量之间变化趋势的方向及程度,其值的范围都是-1到1,0表示两个变量不相关,正值表示正相关,负值表示负相关,值的绝对值越大表示相关性就越大。
Pearson相关性系数是协方差于标准差的比值,所以它对数据是有比较高的要求的,第一、数据通常假设是成对的来自于正态分布的总体,之所以假设正态分布,是因为通过数据计算pearson相关系数之后,通常还会用T检验的方法来进行相关系数检验,而T检验是基于数据呈正态分布假设的。第二、数据之间的差距不能太大,或者说pearson相关系数受异常值的影响比较大,异常值的存在会大大干扰计算结果的。
Spearman相关性系数,通常叫做Spearman秩相关系数,“秩”,可以理解成就是一种顺序或者排序,那么它就是根据原始数据的排序位置进行求解,这种做法弥补了pearson相关系数的那些限制,也就是说,计算过程不用管两个变量具体的值差了多少,只需要计算一下它们每个值所处的排列位置,就可以求相关性系数,而且,即使在变量值没有变化的情况下,也不会出现pearson系数那样分母为0而无法计算的情况,另外,即使出现异常值,由于异常值的秩通常不会有明显的变化,所以异常值对spearman相关系数的影响也非常小,也正是因为这样,在生物实验数据分析中,尤其是在分析多组学数据中说明不同组学数据之间相关性时,使用的频率很高。
Kendall相关性系数是针对有序分类变量所提出来的一种等级相关系数,两个变量按照特定属性排序,同序对和异序对之差与总对数的比值定义为kendall相关系数,它的取值范围在-1到1之间,当系数取值为1时,表示两个变量拥有一致的等级相关性;当系数取值为-1时,表示两个变量拥有完全相反的等级相关性;当系数取值为0时,表示两个变量是相互独立的。
上述步骤(4)中,得到最终和表型有关联的特征的方法,包括以下步骤:
(4.1)采用线性回归方法对步骤(3)中最终剩余的特征丰度数据矩阵与特定表型数据进行线性回归分析,得到各特征的线性回归系数;
(4.2)对每个特征的线性回归系数进行T检验,线性回归系数不为0的特征结合相关系数结果得到最终和表型有关联的特征。
线性回归是利用数理统计中的回归分析,来确定两种或两种以上变量间相互依赖的定量关系的一种统计方法。通过计算,每个特征变量都会得到一个回归系数,通常称为直线的斜率,其意义在于,当特征变量每变动一个单位时,表型变量平均变动的数量。建立回归方程后,只要回归系数不为0,就建立了方程,这里使用T检验对回归系数是否为0进行推断,来完成对回归系数的假设检验。
下面过程中所使用的软件如无特殊说明,均为常规软件。
下面过程中展现了本发明的一般计算过程,具体包括如下步骤:
S01:对测序数据进行标准化流程分析,即对物种或者功能进行定量分析得到要做关联分析的特征丰度矩阵,对样本信息进行收集、评估、过滤、筛选、整理,最终得到表型数据矩阵,选择一种标准化方式同时对两个数据矩阵进行标准化处理,或者根据实际问题需要,不进行标准化处理,或者数据已经是标准化之后的数据,进入后续分析步骤,这里使用R软件(version 4.0.0)对数据进行预处理,可以调用scale函数对两个数据矩阵进行标准化处理;
S02:首先对特征丰度矩阵进行进一步处理,对于特征丰度数据矩阵来说,“零值”就是数字0,逐个检查每个特征,如果某个特征当中数字0所占的比例超过该特征数据的20%,则删除该特征,然后检查下一个特征,直到所有特征符合分析要求。接下来处理表型数据,仍然使用R软件,调用prcomp函数对表型数据矩阵进行PCA分析,使用ggplot2对结果可视化,将观察到的远离数据聚集中心的样本删除;
S03:调用kappa函数计算特征丰度数据矩阵的条件数,调用car包当中的vif函数计算每一个特征的方差膨胀系数,然后删除掉方差膨胀系数大于10的那些特征,之后使用剩余的特征从新计算每个特征的方差膨胀系数,不断重复这个过程,直到所有特征的方差膨胀系数小于10后结束该计算过程;
S04:调用glmnet包里的glmnet函数来进行LASSO回归分析,glmnet函数有一个参数alpha,当alpha=0时,glmnet函数执行岭回归,当alpha=1时,glmnet函数执行LASSO回归,这里设置alpha=1,glmnet函数会自动计算一个最优的惩罚项系数λ,在λ等于这个最优值的情况下,将那些回归系数为0的特征删除,只保留回归系数不等于0的那些特征,筛选过程可以调用coef函数来进行;
S05:调用psych包当中的corr.test函数进行相关系数的计算,通过将该函数的参数method分别设置为pearson、spearman、kendall,来分别计算pearson相关系数、spearman相关系数、kendall相关系数,同时得到每个相关系数统计显著性的p值,这里只保留相关系数大于0.7,同时p值小于0.05的那些特征,可以保留三种相关系数同时满足以上条件的,也可以保留只要有一种相关系数满足以上条件的,根据剩余的特征数量决定;
S06:调用lm函数进行线性回归分析,使用summary函数对lm函数返回的对象进行解析,可以得到每一个特征的回归系数,并且对每个回归系数的统计显著性做出评价,最终回归系数有统计意义,即p值小于0.05的特征就是被筛选出来的与表型相关联的特征,关联性大小可以由回归系数的大小,相关系数的大小,以及它们的统计显著性来衡量。
上述各实施例仅用于说明本发明,其中各部件的结构、连接方式和制作工艺等都是可以有所变化的,凡是在本发明技术方案的基础上进行的等同变换和改进,均不应排除在本发明的保护范围之外。
- 一种对特征丰度数据和样本表型数据进行关联分析的方法
- 一种大样本高通量生物数据关联分析方法