用于借助于循环肿瘤DNA的个人化检测的癌症检测和监测的方法
文献发布时间:2023-06-19 09:33:52
相关申请的引证
本申请案要求以下的优先权:2018年4月14日提交的美国临时申请案第62/657,727号;2018年5月9日提交的美国临时申请案第62/669,330号;2018年7月3日提交的美国临时申请案第62/693,843号;2018年8月6日提交的美国临时申请案第62/715,143号;2018年10月16日提交的美国临时申请案第62/746,210号;2018年12月11日提交的美国临时申请案第62/777,973号;和2019年2月12日提交的美国临时申请案第62/804,566号。以上列举的这些申请案各自以全文引用的方式并入本文中。
背景技术
癌症的早期复发或转移的检测传统上依赖于成像和组织活检。肿瘤组织的活检是侵袭性的并且具有可能有助于转移或手术并发症的风险,而基于成像的检测对检测早期阶段的复发或转移的敏感性不足。需要更好的且侵袭性更低的用于检测癌症的复发或转移的方法。
发明内容
本文中所描述的本发明的一个方面涉及用于监测和检测癌症(例如乳癌、膀胱癌或结肠直肠癌)的早期复发或转移的方法,其包含通过对核酸进行多重扩增反应来产生扩增子集合,所述核酸是从来自已接受癌症(例如乳癌、膀胱癌或结肠直肠癌)治疗的患者的血液或尿液样品或其一部分分离,其中扩增子集合中的每个扩增子跨越与癌症(例如乳癌、膀胱癌或结肠直肠癌)相关联的患者特异性单核苷酸变异体基因座集合中的至少一个单核苷酸变异体基因座;和确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含患者特异性单核苷酸变异体基因座,其中一种或多种(或两种或更多种,或三种或更多种,或四种或更多种,或五种或更多种,或六种或更多种,或七种或更多种,或八种或更多种,或九种或更多种,或十种或更多种)患者特异性单核苷酸变异体的检测指示癌症(例如乳癌、膀胱癌或结肠直肠癌)的早期复发或转移。
除乳癌、膀胱癌和结肠直肠癌以外,本文中所描述的方法还可以用于监测和检测其它类型的癌症的早期复发或转移,如:急性淋巴母细胞性白血病;急性骨髓性白血病;肾上腺皮质癌;AIDS相关癌症;AIDS相关淋巴瘤;肛门癌;阑尾癌;星形细胞瘤;非典型畸胎样/横纹肌样肿瘤;基底细胞癌;脑干神经胶质瘤;脑肿瘤(包括脑干神经胶质瘤、中枢神经系统非典型畸胎样/横纹肌样肿瘤、中枢神经系统胚胎肿瘤、星形细胞瘤、颅咽管瘤、室管膜母细胞瘤、室管膜瘤、神经管胚细胞瘤、髓上皮瘤、中度分化型松果体实质性肿瘤、幕上原始神经外胚层肿瘤和成松果体细胞瘤);支气管肿瘤;伯基特淋巴瘤(Burkitt lymphoma);原发位点未知的癌症;类癌肿瘤;原发位点未知的癌瘤;中枢神经系统非典型畸胎样/横纹肌样肿瘤;中枢神经系统胚胎肿瘤;子宫颈癌;儿童癌症;脊索瘤;慢性淋巴细胞性白血病;慢性骨髓性白血病;慢性骨髓增生性病症;结肠癌;颅咽管瘤;皮肤T细胞淋巴瘤;内分泌胰岛细胞瘤;子宫内膜癌;室管膜母细胞瘤;室管膜瘤;食道癌;鼻腔神经胶质瘤;尤文氏肉瘤(Ewingsarcoma);颅外生殖细胞肿瘤;性腺外生殖细胞肿瘤;肝外胆管癌;胆囊癌;胃部(胃)癌症;胃肠道类癌肿瘤;胃肠道基质细胞肿瘤;胃肠道基质瘤(GIST);妊娠期滋养细胞肿瘤;神经胶质瘤;毛状细胞白血病;头颈癌;心脏癌症;霍奇金氏淋巴瘤(Hodgkin lymphoma);下咽癌症;眼内黑素瘤;胰岛细胞瘤;卡波西肉瘤(Kaposi sarcoma);肾脏癌;兰格汉氏细胞组织细胞增多病(Langerhans cell histiocytosis);喉癌;唇癌;肝癌;恶性纤维组织细胞瘤骨癌;神经管胚细胞瘤;髓上皮瘤;黑素瘤;梅克尔细胞癌(Merkel cell carcinoma);梅克尔细胞皮肤癌瘤(Merkel cell skin carcinoma);间皮瘤;隐性原发性转移性鳞状颈部癌症;口腔癌;多发性内分泌瘤形成综合症;多发性骨髓瘤;多发性骨髓瘤/血浆细胞赘瘤;蕈样真菌病;骨髓发育不良综合症;骨髓增生赘瘤;鼻腔癌;鼻咽癌;神经母细胞瘤;非霍奇金氏淋巴瘤;非黑素瘤型皮肤癌;非小细胞肺癌;口部癌症;口腔癌症;口咽癌;骨肉瘤;其它脑部和脊髓肿瘤;卵巢癌;卵巢上皮癌症;卵巢生殖细胞肿瘤;卵巢低恶性潜能肿瘤;胰脏癌;乳头瘤病;副鼻窦癌;副甲状腺癌;骨盆癌;阴茎癌;咽癌;中度分化型松果体实质性肿瘤;成松果体细胞瘤;垂体肿瘤;血浆细胞赘瘤/多发性骨髓瘤;胸膜肺母细胞瘤;原发性中枢神经系统(CNS)淋巴瘤;原发性肝细胞肝癌;前列腺癌;直肠癌;肾癌;肾细胞(肾脏)癌;肾细胞癌;呼吸道癌症;成视网膜细胞瘤;横纹肌肉瘤;唾液腺癌症;塞氏综合症(Sezary syndrome);小细胞肺癌;小肠癌;软组织肉瘤;鳞状细胞癌;鳞状颈部癌症;胃(胃部)癌症;幕上原始神经外胚层肿瘤;T细胞淋巴瘤;睾丸癌;喉癌;胸腺癌;胸腺瘤;甲状腺癌;移行细胞癌症;肾盂和输尿管的移行细胞癌症;滋养细胞肿瘤;输尿管癌症;尿道癌;子宫癌;子宫瘤;阴道癌;外阴癌;瓦尔登斯特伦巨球蛋白血症(Waldenstrom macroglobulinemia);或威尔姆氏肿瘤(Wilm's tumor)。
在一些实施例中,从患者的肿瘤分离核酸且在确定血液或尿液样品或其一部分的扩增子集合中的每个扩增子的至少一个区段的序列之前,针对患者特异性单核苷酸变异体基因座集合鉴别肿瘤中的体细胞突变,且其中单核苷酸变异体。
在一些实施例中,所述方法包含纵向地从患者收集血液或尿液样品并且进行测序。
在一些实施例中,检测到至少2种或至少5种SNV且存在至少2种或至少5种SNV指示乳癌、膀胱癌或结肠直肠癌的早期复发或转移。
在一些实施例中,乳癌、膀胱癌或结肠直肠癌是1期或2期乳癌、膀胱癌或结肠直肠癌。在一些实施例中,乳癌、膀胱癌或结肠直肠癌是3期或4期乳癌、膀胱癌或结肠直肠癌。
在一些实施例中,在分离血液或尿液样品之前,个体已用手术治疗。
在一些实施例中,在分离血液或尿液样品之前,个体已用化学疗法治疗。
在一些实施例中,在分离血液或尿液样品之前,个体已用辅助疗法或新辅助疗法治疗。
在一些实施例中,在分离血液或尿液样品之前,个体已用放射线疗法治疗。
在一些实施例中,所述方法进一步包含向个体给予化合物,其中已知所述化合物能够尤其有效治疗具有一种或多种所确定的单核苷酸变异体的乳癌、膀胱癌或结肠直肠癌。
在一些实施例中,所述方法进一步包含由序列确定来确定每种单核苷酸变异体的变异体等位基因出现率。
在一些实施例中,乳癌、膀胱癌或结肠直肠癌治疗计划是基于变异体等位基因出现率确定来确认。
在一些实施例中,所述方法进一步包含向个体给予化合物,其中已知所述化合物能够尤其有效治疗具有满足以下条件的单核苷酸变异体中的一种的乳癌、膀胱癌或结肠直肠癌:其可变等位基因出现率大于至少一半的其它所确定的单核苷酸变异体。
在一些实施例中,通过多个单核苷酸变异基因座的高通量DNA测序来确测序列。
在一些实施例中,所述方法进一步包含通过基于一系列扩增子的多个拷贝的序列确定每种SNV基因座的变异体等位基因出现率,检测乳癌、膀胱癌或结肠直肠癌中的克隆单核苷酸变异体,其中与多个单核苷酸变异体基因座的其它单核苷酸变异体相比较高的相对等位基因出现率指示乳癌、膀胱癌或结肠直肠癌中的克隆单核苷酸变异体。
在一些实施例中,所述方法进一步包含向个体给予化合物,所述化合物靶向一种或多种克隆单核苷酸变异体,但不靶向其它单核苷酸变异体。
在一些实施例中,变异体等位基因出现率大于1.0%指示存在克隆单核苷酸变异体。
在一些实施例中,所述方法进一步包含通过以下方式来形成扩增反应混合物:组合聚合酶、核苷酸三磷酸酯、来自由样品产生的核酸库的核酸片段和各自在单核苷酸变异体基因座的150个碱基对内结合的引物的集合或各自跨越包含单核苷酸变异体基因座的具有160个或更少的碱基对的区域的引物的集合,和使扩增反应混合物经历扩增条件以产生扩增子集合。
在一些实施例中,确定样品中是否存在单核苷酸变异体包含至少部分地基于基因座的读段深度来鉴别每个单核苷酸变异基因座集合处的每次等位基因确定的置信度值。
在一些实施例中,如果关于存在单核苷酸变异体的置信度值大于90%,那么作出单核苷酸变异体识别。
在一些实施例中,如果关于存在单核苷酸变异体的置信度值大于95%,那么作出单核苷酸变异体识别。
在一些实施例中,单核苷酸变异基因座集合包含在乳癌、膀胱癌或结肠直肠癌的TCGA和COSMIC数据集中鉴别的所有单核苷酸变异基因座。
在一些实施例中,单核苷酸变异位点集合包含在乳癌、膀胱癌或结肠直肠癌的TCGA和COSMIC数据集中鉴别的所有单核苷酸变异位点。
在一些实施例中,在单核苷酸变异基因座集合的读段深度是至少1,000的情况下进行所述方法。
在一些实施例中,单核苷酸变异体基因座集合包含25到1000个已知与乳癌、膀胱癌或结肠直肠癌相关联的单核苷酸变异基因座。
在一些实施例中,确定单核苷酸变异基因座的多重扩增反应中的每次扩增反应的每个循环的效率和误差率,并且使用所述效率和误差率确定样品中是否存在单一变异体基因座的集合中的单核苷酸变异体。
在一些实施例中,扩增反应是PCR反应且粘接温度比引物集合中至少50%的引物的熔融温度高1到15℃。
在一些实施例中,扩增反应是PCR反应且PCR反应中的粘接步骤的长度是15到120分钟。
在一些实施例中,扩增反应是PCR反应且PCR反应中的粘接步骤的长度是15到120分钟。
在一些实施例中,扩增反应中的引物浓度是1到10nM。
在一些实施例中,引物集合中的引物被设计成最大限度地减少引物二聚体形成。
在一些实施例中,扩增反应是PCR反应,粘接温度比引物集合中至少50%的引物的熔融温度高1到15℃,PCR反应中的粘接步骤的长度在15与120分钟之间,扩增反应中的引物浓度在1与10nM之间,并且引物集合中的引物被设计成最大限度地减少引物二聚体形成。
在一些实施例中,在限制性引物条件下进行多重扩增反应。
本文中所描述的本发明的另一个方面涉及一种组合物,其包含循环肿瘤核酸片段,所述循环肿瘤核酸片段包含通用衔接子,其中所述循环肿瘤核酸是来源于乳癌、膀胱癌或结肠直肠癌。
在一些实施例中,循环肿瘤核酸是来源于患有乳癌、膀胱癌或结肠直肠癌的个体的血液或尿液样品或其一部分。
本文中所描述的本发明的另一个方面涉及一种组合物,其包含固体负载物,所述固体负载物包含核酸的多个克隆群体,其中所述克隆群体包含由循环游离核酸的样品产生的扩增子,其中所述循环肿瘤核酸是来源于乳癌、膀胱癌或结肠直肠癌。
在一些实施例中,循环游离核酸是来源于患有乳癌、膀胱癌或结肠直肠癌的个体的血液或尿液样品或其一部分。
在一些实施例中,不同克隆群体中的核酸片段包含相同通用衔接子。
在一些实施例中,核酸的克隆群体是来源于来自两名或更多名个体的样品集合的核酸片段。
在一些实施例中,核酸片段包含对应于样品集合中的样品的一系列分子条形码中的一个。
本文中所描述的本发明的另一个方面涉及用于监测和检测乳癌、膀胱癌或结肠直肠癌的早期复发或转移的方法,其包含基于在已诊断患有乳癌、膀胱癌或结肠直肠癌的患者的肿瘤样品中鉴别的体细胞突变,选择具有至少8个或16个患者特异性单核苷酸变异体基因座的集合;在患者已用手术、一线化学疗法及/或辅助疗法治疗之后,从患者纵向收集一个或多个血液或尿液样品;通过对从每个血液或尿液样品或其一部分分离的核酸进行多重扩增反应来产生扩增子集合,其中扩增子集合中的每个扩增子跨越与乳癌、膀胱癌或结肠直肠癌相关联的患者特异性单核苷酸变异体基因座集合中的至少一个单核苷酸变异体基因座;以及确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含患者特异性单核苷酸变异体基因座,其中来自血液或尿液样品的一种或多种(或两种或更多种,或三种或更多种,或四种或更多种,或五种或更多种,或六种或更多种,或七种或更多种,或八种或更多种,或九种或更多种,或十种或更多种)患者特异性单核苷酸变异体的检测指示乳癌、膀胱癌或结肠直肠癌的早期复发或转移。
本文中所描述的本发明的另一个方面涉及用于治疗乳癌、膀胱癌或结肠直肠癌的方法,其包含用手术、一线化学疗法及/或辅助疗法治疗已诊断患有乳癌、膀胱癌或结肠直肠癌的患者;从患者纵向收集一个或多个血液或尿液样品;通过对从每个血液或尿液样品或其一部分分离的核酸进行多重扩增反应来产生扩增子集合,其中扩增子集合中的每个扩增子跨越具有至少8个或16个与乳癌、膀胱癌或结肠直肠癌相关联的患者特异性单核苷酸变异体基因座的集合中的至少一个单核苷酸变异体基因座,所述患者特异性单核苷酸变异体基因座是基于在患者的肿瘤样品中鉴别的体细胞突变而选择;确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含患者特异性单核苷酸变异体基因座,其中来自血液或尿液样品的一种或多种(或两种或更多种,或三种或更多种,或四种或更多种,或五种或更多种,或六种或更多种,或七种或更多种,或八种或更多种,或九种或更多种,或十种或更多种)患者特异性单核苷酸变异体的检测指示乳癌、膀胱癌或结肠直肠癌的早期复发或转移;以及向个体给予化合物,其中已知所述化合物可以有效治疗具有一种或多种在血液或尿液样品中检测到的单核苷酸变异体的乳癌、膀胱癌或结肠直肠癌。
本文中所描述的本发明的另一个方面涉及用于监测或预测对乳癌、膀胱癌或结肠直肠癌治疗的反应的方法,其包含从正在经历乳癌、膀胱癌或结肠直肠癌治疗的患者纵向收集一个或多个血液或尿液样品;通过对从每个血液或尿液样品或其一部分分离的核酸进行多重扩增反应来产生扩增子集合,其中扩增子集合中的每个扩增子跨越具有至少8个或16个与乳癌、膀胱癌或结肠直肠癌相关联的患者特异性单核苷酸变异体基因座的集合中的至少一个单核苷酸变异体基因座,所述患者特异性单核苷酸变异体基因座是基于在患者的肿瘤样品中鉴别的体细胞突变而选择;以及确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含患者特异性单核苷酸变异体基因座,其中来自血液或尿液样品的一种或多种(或两种或更多种,或三种或更多种,或四种或更多种,或五种或更多种,或六种或更多种,或七种或更多种,或八种或更多种,或九种或更多种,或十种或更多种)患者特异性单核苷酸变异体的检测指示对乳癌、膀胱癌或结肠直肠癌治疗的不良反应。
在一些实施例中,本文中所描述的方法包含在疗法之前和/或在新辅助疗法期间(例如在第1循环、第2循环、第3循环、第4循环等之后)检测乳癌患者的血浆中的ctDNA。在一些实施例中,治疗计划是基于ctDNA浓度确定(例如存在/不存在)和在新辅助疗法期间的降低率来定义的。
在一些实施例中,本文中所描述的方法包含评估每位癌症患者的ctDNA存在情况和含量(即,以肿瘤中实际存在的突变为目标)。在一些实施例中,本文中所描述的方法包含检测患者肿瘤中实际存在的突变中的2种或更多种、4种或更多种、10种或更多种、16种或更多种、32种或更多种、50种或更多种、64种或更多种或100种或更多种。
根据本发明的一些实施例,至少50%,或至少60%,或至少70%,或至少80%,或至少90%,或约100%的将具有转移性复发(例如在新辅助疗法和手术之后)的患者在基线处具有可检测的ctDNA。
根据本发明的一些实施例,至少50%,或至少60%,或至少70%,或至少80%,或至少90%,或约100%的将具有转移性复发(例如在新辅助疗法和手术之后)的患者在新辅助疗法的第1循环之后具有可检测的ctDNA。
根据本发明的一些实施例,至少50%,或至少60%,或至少70%,或至少80%,或至少90%,或约100%的将具有转移性复发(例如在新辅助疗法和手术之后)的患者在新辅助疗法的第2循环之后具有可检测的ctDNA。
根据本发明的一些实施例,至少50%,或至少60%,或至少70%,或至少80%,或至少90%,或约100%的将具有转移性复发(例如在新辅助疗法和手术之后)的患者在新辅助疗法之后且在手术之前具有可检测的ctDNA。
根据本发明的一些实施例,至少50%,或至少60%,或至少70%,或至少80%,或至少90%,或约100%的将具有转移性复发(例如在新辅助疗法和手术之后)的患者在手术之后具有可检测的ctDNA。
根据本发明的一些实施例,至少50%,或至少60%,或至少70%,或至少80%,或至少90%,或约100%的具有可检测的ctDNA(例如在手术之后)的患者在不进行进一步治疗的情况下将具有转移性复发(例如在新辅助疗法和手术之后)。
根据本发明的一些实施例,如果不给予其它治疗,那么至少50%,或至少60%,或至少70%,或至少80%,或至少90%,或约100%的在基线与第1循环或第2循环等之间具有增加的ctDNA含量的患者将在手术之后具有转移性复发。
在一些实施例中,本文中所描述的方法包含检测癌症的某些亚型,包括乳癌的某些亚型的发生、复发或转移。在一些实施例中,本文中所描述的方法包含检测HR+/HER2-肿瘤,包括HR+/HER2-乳癌(例如激素受体阳性-ERα+和/或PR+)的发生、复发或转移。HR+肿瘤通常具有较低侵润性且具有良好的预后,其中5年存活率超过90%。
在一些实施例中,本文中所描述的方法包含检测HER2+肿瘤,包括HER2+乳癌(人类表皮生长因子受体2阳性)的发生、复发或转移。HER2+肿瘤与HR+/HER2-乳癌相比通常具有更高的侵袭性、更坏的预后且更可能复发和转移。
在一些实施例中,本文中所描述的方法包含检测HR-/HER2-肿瘤,包括HR-/HER2-乳癌(TNBC或三阴性BC)的发生、复发或转移。三阴性乳癌(TNBC)不表达ERα、PR或HER2。这些肿瘤在所有乳癌亚型中倾向于具有最高的侵润性和最差的预后。
在一些实施例中,本文中所描述的方法能够在至少75%、至少80%、至少85%、至少90%或至少95%的具有癌症的早期复发或转移的患者中检测到患者特异性单核苷酸变异体。
在一些实施例中,本文中所描述的方法能够在至少80%、至少85%、至少90%、至少95%或至少98%的具有HER2+乳癌的早期复发或转移的患者中检测到患者特异性单核苷酸变异体。
在一些实施例中,本文中所描述的方法能够在至少80%、至少85%、至少90%、至少95%或至少98%的具有三阴性乳癌的早期复发或转移的患者中检测到患者特异性单核苷酸变异体。
在一些实施例中,本文中所描述的方法能够在至少75%、至少80%、至少85%、至少90%或至少95%的具有HR+/HER2-乳癌的早期复发或转移的患者中检测到患者特异性单核苷酸变异体。
在一些实施例中,本文中所描述的方法能够在可以通过成像来检测到癌症的临床复发或转移之前至少100天、至少150天、至少200天、至少250天或至少300天和/或在CA15-3含量上升之前至少100天、至少150天、至少200天、至少250天或至少300天,在具有癌症的早期复发或转移的患者中检测到患者特异性单核苷酸变异体。
在一些实施例中,本文中所描述的方法能够在可以通过成像来检测到HER2+乳癌的临床复发或转移之前至少100天、至少150天、至少200天、至少250天或至少300天和/或在CA15-3含量上升之前至少100天、至少150天、至少200天、至少250天或至少300天,在具有HER2+乳癌的早期复发或转移的患者中检测到患者特异性单核苷酸变异体。
在一些实施例中,本文中所描述的方法能够在可以通过成像来检测到三阴性乳癌的临床复发或转移之前至少100天、至少150天、至少200天、至少250天或至少300天和/或在CA15-3含量上升之前至少100天、至少150天、至少200天、至少250天或至少300天,在具有三阴性乳癌的早期复发或转移的患者中检测到患者特异性单核苷酸变异体。
在一些实施例中,本文中所描述的方法能够在可以通过成像来检测到HR+/HER2-乳癌的临床复发或转移之前至少100天、至少150天、至少200天、至少250天或至少300天和/或在CA15-3含量上升之前至少100天、至少150天、至少200天、至少250天或至少300天,在具有HR+/HER2-乳癌的早期复发或转移的患者中检测到患者特异性单核苷酸变异体。
在一些实施例中,本文中所描述的方法在至少95%、至少98%、至少99%、至少99.5%、至少99.8%或至少99.9%的不具有癌症的早期复发或转移的患者中未检测到患者特异性单核苷酸变异体。
在一些实施例中,本文中所描述的方法在至少95%、至少98%、至少99%、至少99.5%、至少99.8%或至少99.9%的不具有HER2+乳癌的早期复发或转移的患者中未检测到患者特异性单核苷酸变异体。
在一些实施例中,本文中所描述的方法在至少95%、至少98%、至少99%、至少99.5%、至少99.8%或至少99.9%的不具有三阴性乳癌的早期复发或转移的患者中未检测到患者特异性单核苷酸变异体。
在一些实施例中,本文中所描述的方法在至少95%、至少98%、至少99%、至少99.5%、至少99.8%或至少99.9%的不具有HR+/HER2-乳癌的早期复发或转移的患者中未检测到患者特异性单核苷酸变异体。
在一些实施例中,当在高于预定置信度阈值(例如0.95、0.96、0.97、0.98或0.99)的情况下检测到两种或更多种患者特异性单核苷酸变异体时,本文中所描述的方法在检测癌症的早期复发或转移方面的特异性是至少95%、至少98%、至少99%、至少99.5%、至少99.8%或至少99.9%。
在一些实施例中,当在高于预定置信度阈值(例如0.95、0.96、0.97、0.98或0.99)的情况下检测到两种或更多种患者特异性单核苷酸变异体时,本文中所描述的方法在检测HER2+乳癌的早期复发或转移方面的特异性是至少95%、至少98%、至少99%、至少99.5%、至少99.8%或至少99.9%。
在一些实施例中,当在高于预定置信度阈值(例如0.95、0.96、0.97、0.98或0.99)的情况下检测到两种或更多种患者特异性单核苷酸变异体时,本文中所描述的方法在检测三阴性乳癌的早期复发或转移方面的特异性是至少95%、至少98%、至少99%、至少99.5%、至少99.8%或至少99.9%。
在一些实施例中,当在高于预定置信度阈值(例如0.95、0.96、0.97、0.98或0.99)的情况下检测到两种或更多种患者特异性单核苷酸变异体时,本文中所描述的方法在检测HR+/HER2-乳癌的早期复发或转移方面的特异性是至少95%、至少98%、至少99%、至少99.5%、至少99.8%或至少99.9%。
在一些实施例中,本文中所描述的方法在至少75%、至少80%、至少85%、至少90%或至少95%的具有肌肉侵袭性膀胱癌(MIBC)的早期复发或转移的患者中检测到患者特异性单核苷酸变异体。
在一些实施例中,本文中所描述的方法在可以通过成像来检测到MIBC的临床复发或转移之前至少100天、至少150天、至少200天或至少250天,在具有癌症的早期复发或转移的患者中检测到患者特异性单核苷酸变异体。
在一些实施例中,本文中所描述的方法在至少95%、至少98%、至少99%、至少99.5%、至少99.8%或至少99.9%的不具有MIBC的早期复发或转移的患者中未检测到患者特异性单核苷酸变异体。
在一些实施例中,当在高于预定置信度阈值(例如0.95、0.96、0.97、0.98或0.99)的情况下检测到两种或更多种患者特异性单核苷酸变异体时,本文中所描述的方法在检测MIBC的早期复发或转移方面的特异性是至少95%、至少98%、至少99%、至少99%、至少99.5%、至少99.8%或至少99.9%。
除单核苷酸变异体以外或代替单核苷酸变异体,本文中所描述的方法也可以基于检测其它基因组变异体,如插入缺失、多核苷酸变异体和/或基因融合体。
因此,本文中所描述的本发明的另一方面涉及用于监测和检测乳癌、膀胱癌或结肠直肠癌的早期复发或转移的方法,其包含基于在已诊断患有乳癌、膀胱癌或结肠直肠癌的患者的肿瘤样品中鉴别的体细胞突变,选择多个基因组变异体基因座(例如SNV、插入缺失、多核苷酸变异体和基因融合体);在患者已用手术、一线化学疗法及/或辅助疗法治疗之后,从患者纵向收集一个或多个血液或尿液样品;通过对从每个血液或尿液样品或其一部分分离的核酸进行多重扩增反应来产生扩增子集合,其中扩增子集合中的每个扩增子跨越与乳癌、膀胱癌或结肠直肠癌相关联的患者特异性基因组变异体基因座集合中的至少一个基因组变异体基因座;以及确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含患者特异性基因组变异体基因座,其中来自血液或尿液样品的一种或多种(或两种或更多种,或三种或更多种,或四种或更多种,或五种或更多种,或六种或更多种,或七种或更多种,或八种或更多种,或九种或更多种,或十种或更多种)患者特异性基因组变异体的检测指示乳癌、膀胱癌或结肠直肠癌的早期复发或转移。
本文中所描述的本发明的另一个方面涉及用于治疗乳癌、膀胱癌或结肠直肠癌的方法,其包含用手术、一线化学疗法和/或辅助疗法治疗已诊断患有乳癌、膀胱癌或结肠直肠癌的患者;从患者纵向收集一个或多个血液或尿液样品;通过对从每个血液或尿液样品或其一部分分离的核酸进行多重扩增反应来产生扩增子集合,其中扩增子集合中的每个扩增子跨越具有至少8个或16个与乳癌、膀胱癌或结肠直肠癌相关联的患者特异性基因组变异体基因座的集合中的至少一个基因组变异体基因座(例如SNV、indel、多核苷酸变异体和基因融合体),所述患者特异性基因组变异体基因座是基于在患者的肿瘤样品中鉴别的体细胞突变而选择;确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含患者特异性基因组变异体基因座,其中来自血液或尿液样品的一种或多种(或两种或更多种,或三种或更多种,或四种或更多种,或五种或更多种,或六种或更多种,或七种或更多种,或八种或更多种,或九种或更多种,或十种或更多种)患者特异性基因组变异体的检测指示乳癌、膀胱癌或结肠直肠癌的早期复发或转移;以及向个体给予化合物,其中已知所述化合物可以有效治疗具有一种或多种在血液或尿液样品中检测到的基因组变异体的乳癌、膀胱癌或结肠直肠癌。
本文中所描述的本发明的另一个方面涉及用于监测或预测对乳癌、膀胱癌或结肠直肠癌治疗的反应的方法,其包含从正在经历乳癌、膀胱癌或结肠直肠癌治疗的患者纵向收集一个或多个血液或尿液样品;通过对从每个血液或尿液样品或其一部分分离的核酸进行多重扩增反应来产生扩增子集合,其中扩增子集合中的每个扩增子跨越具有至少8个或16个与乳癌、膀胱癌或结肠直肠癌相关联的患者特异性基因组变异体基因座的集合中的至少一个基因组变异体基因座(例如SNV、插入缺失、多核苷酸变异体和基因融合体),所述患者特异性基因组变异体基因座是基于在患者的肿瘤样品中鉴别的体细胞突变而选择;以及确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含患者特异性基因组变异体基因座,其中来自血液或尿液样品的一种或多种(或两种或更多种,或三种或更多种,或四种或更多种,或五种或更多种,或六种或更多种,或七种或更多种,或八种或更多种,或九种或更多种,或十种或更多种)患者特异性基因组变异体的检测指示对乳癌、膀胱癌或结肠直肠癌治疗的不良反应。
除患者特异性基因组变异体以外或代替患者特异性基因组变异体,本文中所描述的方法也可以基于检测在许多癌症患者中复发的复发性癌症相关突变(例如热点癌症突变、耐药性标记物、癌症基因突变(cancer panel mutation))。
因此,本文中所描述的本发明的另一个方面涉及用于监测和检测乳癌、膀胱癌或结肠直肠癌的早期复发或转移的方法,其包含选择多个复发性癌症相关突变;在患者已用手术、一线化学疗法和/或辅助疗法治疗之后,从患者纵向收集一个或多个血液或尿液样品;通过对从每个血液或尿液样品或其一部分分离的核酸进行多重扩增反应来产生扩增子集合,其中扩增子集合中的每个扩增子跨越与乳癌、膀胱癌或结肠直肠癌相关联的复发性突变的集合中的至少一种;以及确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含复发性癌症相关突变,其中来自血液或尿液样品的一种或多种(或两种或更多种,或三种或更多种,或四种或更多种,或五种或更多种,或六种或更多种,或七种或更多种,或八种或更多种,或九种或更多种,或十种或更多种)复发性癌症相关突变的检测指示乳癌、膀胱癌或结肠直肠癌的早期复发或转移。
本文中所描述的本发明的另一个方面涉及用于治疗乳癌、膀胱癌或结肠直肠癌的方法,其包含用手术、一线化学疗法和/或辅助疗法治疗已诊断患有乳癌、膀胱癌或结肠直肠癌的患者;从患者纵向收集一个或多个血液或尿液样品;通过对从每个血液或尿液样品或其一部分分离的核酸进行多重扩增反应来产生扩增子集合,其中扩增子集合中的每个扩增子跨越具有至少8种或16种与乳癌、膀胱癌或结肠直肠癌相关联的复发性突变的集合中的至少一种复发性癌症相关突变(例如热点癌症突变、耐药性标记物、癌症基因突变);确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含复发性癌症相关突变,其中来自血液或尿液样品的一种或多种(或两种或更多种,或三种或更多种,或四种或更多种,或五种或更多种,或六种或更多种,或七种或更多种,或八种或更多种,或九种或更多种,或十种或更多种)复发性癌症相关突变的检测指示乳癌、膀胱癌或结肠直肠癌的早期复发或转移;以及向个体给予化合物,其中已知所述化合物可以有效治疗具有一种或多种在血液或尿液样品中检测到的复发性癌症相关突变的乳癌、膀胱癌或结肠直肠癌。
本文中所描述的本发明的另一个方面涉及用于监测或预测对乳癌、膀胱癌或结肠直肠癌治疗的反应的方法,其包含从正在经历乳癌、膀胱癌或结肠直肠癌治疗的患者纵向收集一个或多个血液或尿液样品;通过对从每个血液或尿液样品或其一部分分离的核酸进行多重扩增反应来产生扩增子集合,其中扩增子集合中的每个扩增子跨越具有至少8种或16种与乳癌、膀胱癌或结肠直肠癌相关联的复发性突变的集合中的至少一种复发性癌症相关突变(例如热点癌症突变、耐药性标记物、癌症基因突变);以及确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含复发性癌症相关突变,其中来自血液或尿液样品的一种或多种(或两种或更多种,或三种或更多种,或四种或更多种,或五种或更多种,或六种或更多种,或七种或更多种,或八种或更多种,或九种或更多种,或十种或更多种)复发性癌症相关突变的检测指示对乳癌、膀胱癌或结肠直肠癌治疗的不良反应。
除初始鉴别已诊断患有乳癌、膀胱癌或结肠直肠癌的患者的肿瘤样品中的体细胞突变以外或代替初始鉴别已诊断患有乳癌、膀胱癌或结肠直肠癌的患者的肿瘤样品中的体细胞突变,本文中所描述的方法也可以基于鉴别患者的其它生物样品中的体细胞突变,所述其它生物样品如血液、血清、血浆、尿液、毛发、泪液、唾液、皮肤、指甲、粪便、胆汁、淋巴、子宫颈粘液或精液。
因此,本文中所描述的本发明的另一个方面涉及用于监测和检测乳癌、膀胱癌或结肠直肠癌的早期复发或转移的方法,其包含基于在已诊断患有乳癌、膀胱癌或结肠直肠癌的患者的包含癌症相关突变的生物样品(例如血液、血清、血浆、尿液、毛发、泪液、唾液、皮肤、指甲、粪便、胆汁、淋巴、子宫颈粘液或精液)中鉴别的体细胞突变,选择多个基因组变异体基因座(例如SNV、插入缺失、多核苷酸变异体和基因融合体);在患者已用手术、一线化学疗法和/或辅助疗法治疗之后,从患者纵向收集一个或多个血液或尿液样品;通过对从每个血液或尿液样品或其一部分分离的核酸进行多重扩增反应来产生扩增子集合,其中扩增子集合中的每个扩增子跨越与乳癌、膀胱癌或结肠直肠癌相关联的患者特异性基因组变异体基因座的集合中的至少一个基因组变异体基因座;以及确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含患者特异性基因组变异体基因座,其中来自血液或尿液样品的一种或多种(或两种或更多种,或三种或更多种,或四种或更多种,或五种或更多种,或六种或更多种,或七种或更多种,或八种或更多种,或九种或更多种,或十种或更多种)患者特异性基因组变异体的检测指示乳癌、膀胱癌或结肠直肠癌的早期复发或转移。
本文中所描述的本发明的另一个方面涉及用于治疗乳癌、膀胱癌或结肠直肠癌的方法,其包含用手术、一线化学疗法和/或辅助疗法治疗已诊断患有乳癌、膀胱癌或结肠直肠癌的患者;从患者纵向收集一个或多个血液或尿液样品;通过对从每个血液或尿液样品或其一部分分离的核酸进行多重扩增反应来产生扩增子集合,其中扩增子集合中的每个扩增子跨越具有至少8个或16个与乳癌、膀胱癌或结肠直肠癌相关联的患者特异性基因组变异体基因座的集合中的至少一个基因组变异体基因座(例如SNV、插入缺失、多核苷酸变异体和基因融合体),所述患者特异性基因组变异体基因座是基于在包含癌症相关突变的患者的生物样品(例如血液、血清、血浆、尿液、毛发、泪液、唾液、皮肤、指甲、粪便、胆汁、淋巴、子宫颈粘液或精液)中鉴别的体细胞突变而选择;确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含患者特异性基因组变异体基因座,其中来自血液或尿液样品的一种或多种(或两种或更多种,或三种或更多种,或四种或更多种,或五种或更多种,或六种或更多种,或七种或更多种,或八种或更多种,或九种或更多种,或十种或更多种)患者特异性基因组变异体的检测指示乳癌、膀胱癌或结肠直肠癌的早期复发或转移;以及向个体给予化合物,其中已知所述化合物可以有效治疗具有一种或多种在血液或尿液样品中检测到的基因组变异体的乳癌、膀胱癌或结肠直肠癌。
本文中所描述的本发明的另一个方面涉及用于监测或预测对乳癌、膀胱癌或结肠直肠癌治疗的反应的方法,其包含从正在经历乳癌、膀胱癌或结肠直肠癌治疗的患者纵向收集一个或多个血液或尿液样品;通过对从每个血液或尿液样品或其一部分分离的核酸进行多重扩增反应来产生扩增子集合,其中扩增子集合中的每个扩增子跨越具有至少8个或16个与乳癌、膀胱癌或结肠直肠癌相关联的患者特异性基因组变异体基因座的集合中的至少一个基因组变异体基因座(例如SNV、插入缺失、多核苷酸变异体和基因融合体),所述患者特异性基因组变异体基因座是基于在包含癌症相关突变的患者的生物样品(例如血液、血清、血浆、尿液、毛发、泪液、唾液、皮肤、指甲、粪便、胆汁、淋巴、子宫颈粘液或精液)中鉴别的体细胞突变而选择;以及确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含患者特异性基因组变异体基因座,其中来自血液或尿液样品的一种或多种(或两种或更多种,或三种或更多种,或四种或更多种,或五种或更多种,或六种或更多种,或七种或更多种,或八种或更多种,或九种或更多种,或十种或更多种)患者特异性基因组变异体的检测指示对乳癌、膀胱癌或结肠直肠癌治疗的不良反应。
所公开的本发明的其它实施例以及特征和优点将由以下详细描述和权利要求书显而易见。
附图说明
本专利案或申请案文件含有至少一幅彩色图。具有彩色图式的本专利案或专利申请公开案的拷贝将在请求和支付必需费用之后由专利局提供。
将参考附图进一步说明本发明所公开的实施例,其中在若干视图中由类似的数字指代类似的结构。所展示的图式未必按比例绘制,重点实际上主要放在说明本发明所公开的实施例的原理上。
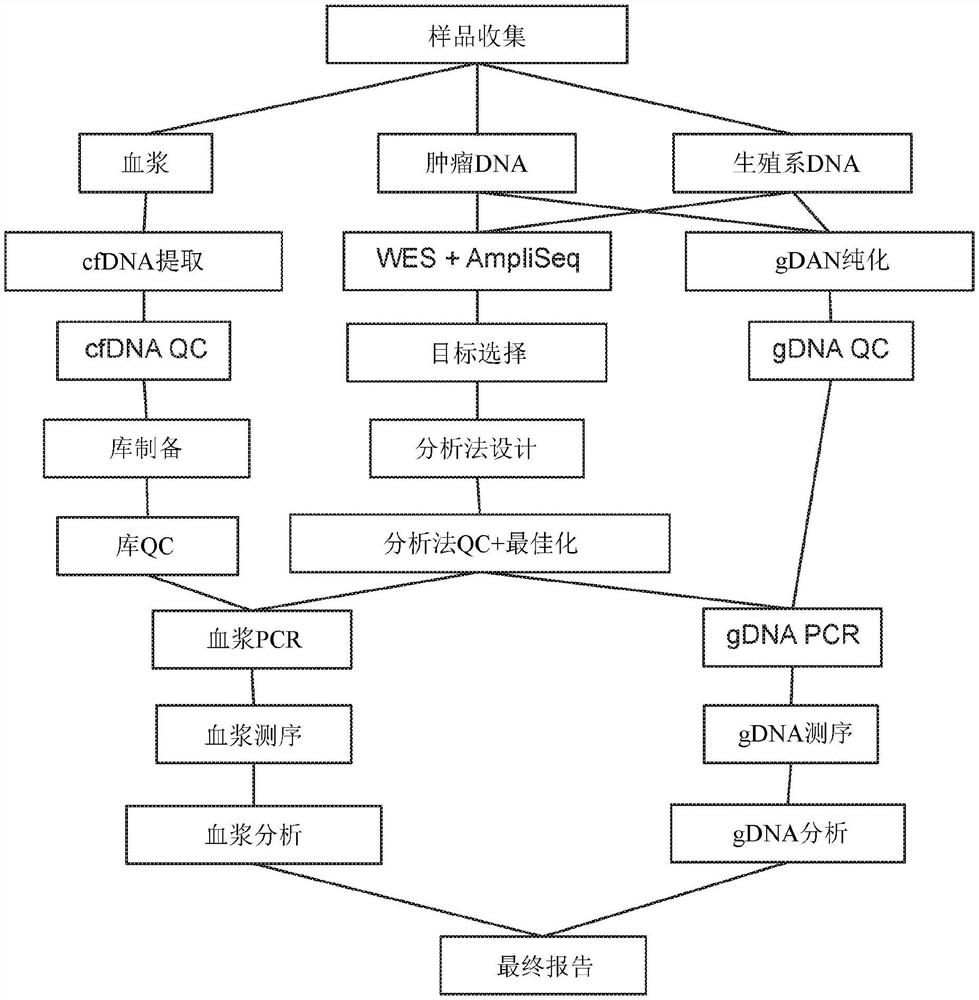
图1是工作流程图。
图2.上部图:每个样品中的SNV数目;下部图:工作分析法,由驱动物类别分类。
图3.所测量的cfDNA浓度。每个数据点指代一个血浆样品。
图4.在预先确定(x轴)的组织VAF测量结果与本文中使用mPCR-NGS(y轴)确定的组织VAF测量结果之间展示良好相关性的样品。每个样品在单独的方框中展示,并且由组织子部分对VAF数据点进行染色。
图5.在预先确定(x轴)的组织VAF测量结果与本文中使用mPCR-NGS(y轴)确定的组织VAF测量结果之间展示不良相关性的样品。每个样品在单独的方框中展示,并且由组织子部分对VAF数据点进行染色。
图6A-B.随所作出的识别而变的读段深度直方图。上部:分析法未检测到预期血浆SNV。下部:分析法检测到预期血浆SNV。
图7.由组织学类型在血浆中检测到的SNV数目。
图8.由肿瘤阶段进行的血浆中的SNV检测(左侧)和样品检测(右侧)。
图9.随肿瘤阶段和SNV克隆性而变的血浆VAF。
图10.在来自每个样品的血浆中检测到的随cfDNA输入量而变的SNV数目。
图11.随平均肿瘤VAF而变的血浆VAF。计算来自每个肿瘤的所分析的所有肿瘤子部分的平均肿瘤VAF。
图12展示每个所检测的SNV的克隆率(红色对比蓝色)和突变型变异体等位基因出现率(MutVAF)。将由每个样品检测到的全部SNV放置于单一栏中且由肿瘤阶段(pTNM阶段)对样品进行分类。包括未检测到SNV的样品。克隆率定义为其中观察到SNV的肿瘤子部分的数目与来自所述肿瘤的所分析的子部分的总数之间的比率。
图13展示每个所检测的SNV的克隆状态(蓝色表示克隆且红色表示亚克隆)和突变型变异体等位基因出现率(MutVAF)。将由每个样品检测到的全部SNV放置于单一栏中且由肿瘤阶段(pTNM阶段)对样品进行分类。包括未检测到SNV的样品。使用来自肿瘤组织的完全外显子组测序数据,通过PyCloneCluster确定克隆状态。
图14展示每个所检测的SNV的克隆状态(蓝色表示克隆且红色表示亚克隆)和突变型变异体等位基因出现率(MutVAF),其中上部图仅展示克隆SNV且下部图仅展示亚克隆SNV。将由每个样品检测到的全部SNV放置于单一栏中且由肿瘤阶段(pTNM阶段)对样品进行分类。包括未检测到SNV的样品。使用来自肿瘤组织的完全外显子组测序数据,通过PyCloneCluster确定克隆状态。
图15展示在血浆中检测到的随组织学类型和肿瘤尺寸而变的SNV数目。通过病理报告来确定组织学类型和肿瘤阶段。根据尺寸来对每个数据点进行染色,其中红色表示最大肿瘤尺寸且蓝色表示最小肿瘤尺寸。
图16是cfDNA分析的表格,其展示所有样品中的DNA浓度、用于库制备的基因组拷贝当量、血浆溶血等级和cDNA分布。
图17是每个样品的在血浆中检测到的SNV的表格。
图18是在血浆中检测到的其它SNV的表格。
图19是所检测的分析法和其在复发时间的血浆样品的背景等位基因部分(LTX103)的实例。
图20A-B:临床和分子方案的示意图。
图21:研究概述。
图22:36个月的监测和血浆收集的患者概述。
图23A-B:由手术后ctDNA状态进行分级的复发风险。
图24A-B:由手术后ctDNA状态进行分级的疗法后复发风险。
图25:辅助疗法在预防复发中的有效性。
图26A-B:基于放射学和ctDNA的释放时间。
图27A-D:复发的早期检测和治疗反应的预测。
图28:临床样品收集的示意图。
图29:血浆测序QC。
图30A-F:早期复发检测。
图31:在诊断时和进行切除术以后的无复发存活率和ctDNA状态。
图32A-B:新辅助治疗反应。
图33:实体瘤患者定制化监测技术(Signatera;RUO)方法。
图34:血浆测序QC。
图35:单一SNV检测的敏感性。
图36:预期输入对比用实体瘤患者定制化监测技术(RUO)观察的VAF。
图37:实例6中的乳癌研究的患者概述。
图38A-H:关于实例6的研究中分析的样品的信息的表格。图38A是表格的第1部分。图38B是表格的延续部分。图38C是表格的延续部分。图38D是表格的延续部分。图38E是表格的延续部分。图38F是表格的延续部分。图38G是表格的延续部分。图38H是表格的延续部分。
图39:实例6的乳癌研究中患者的人口统计学。接收50名患者的WES原始数据(具有35名患者的驱动物变异体)。接受不同数目的时间点(1至8)时的218个血浆样品。接收108个额外提取的DNA样品。还收集复发状态。以6个月时间间隔在辅助疗法后收集血液样品。
图40:实例6中的乳癌研究的WES分析和池设计的概述。池A是基于实体瘤患者定制化监测技术方法。池B含有25名患者且在箱须图中用星号指示。池B中的19名患者具有低肿瘤纯度。6名患者具有额外的早期HER2-肿瘤。池B含有驱动物变异体。
图41:实例6中的乳癌研究的血浆样品。中值血浆体积为4mL。中值DNA输入量为26ng。中值DNA输入量低于CRC和MIBC样品(分别是45ng和66ng)。
图42:测序质量控制,其描绘实例6中的乳癌研究的每种类型的中值方法误差率和中值分析法读段深度。总共处理326个血浆测序样品。估计突变识别FP率为0.28%。
图43:实例6中的乳癌研究的血浆样品。呈现49名患者的319个被测序的样品和214个独特的血浆样品。
图44:来自实例6中的乳癌研究的池A的结果。在49名患者中,11名为基线阳性。3名仅具有一个时间点。其余8名患者始终保持阳性。池B和驱动物产生类似结果。驱动物信息:16个具有驱动物突变的复发样品。11个具有至少一种用驱动物进行的分析法的复发样品。
图45:16名具有所检测的ctDNA的患者的概述。
图46:对应于图38中的患者CD047(TNBC)的数据的图形描述。
图47:对应于图38中的患者CD033(TNBC)的数据的图形描述。
图48:对应于图38中的患者CD037(HER2+)的数据的图形描述。
图49:对应于图38中的患者CD040(HER2+)的数据的图形描述。
图50:对应于图38中的患者CD048(HER2-)的数据的图形描述。
图51:对应于图38中的患者CD005(HER2-)的数据的图形描述。
图52:对应于图38中的患者CD036(HER2-)的数据的图形描述。
图53:对应于图38中的患者CD044(HER2-)的数据的图形描述。
图54:对应于图38中的患者CD049的数据的图形描述。
图55:对应于图38中的患者CD029的数据的图形描述。
图56:对应于图38中的患者CD026的数据的图形描述。
图57:对应于图38中的患者CD017的数据的图形描述。
图58:对应于图38中的患者CD031的数据的图形描述。HW:SHC2、PKD1、COLEC12。
图59:对应于图38中的患者CD025的数据的图形描述。在这名患者中,在2个连续时间点时针对FGF9中的突变观察ctDNA。这名患者近期可能经历复发。
图60:患者募集和临床样品的收集。对于在本研究中监测的49名BC女性,使用Signatera
图61A-C:ctDNA分析的概述和结果。(A)每位患者的(n=49)治疗方案和所分析的连续血浆样品(n=208)的结果的概述。(B)概述表格,其展示每种乳癌亚型之全部患者、复发数目、由ctDNA分析检测的百分比和中值前置时间(天)。(C)使用配对威尔科克森符号秩检验(Wilcoxon signed rank test)(p值<0.001)的通过乳癌亚型HR+、HER2+、TNBC进行染色的分子和临床复发的比较。
图62A-B:连续血浆样品的CtDNA检测预测无复发存活率(A)根据手术后任何随访血浆样品中的ctDNA检测的无复发存活率[HR:35.84(7.9626-161.32],p值<0.001。(B)根据第一手术后血浆样品中的ctDNA检测的无复发存活率[HR:11.784(4.2784-32.457]。数据是来自n=49名患者,其中p值<0.001。
图63:(A-E)五名乳癌患者(每张图一名患者)的多个血浆时间点时的ctDNA的血浆含量。原发性肿瘤和相匹配的正常完全外显子组测序鉴别患者特异性体细胞突变。使用以分析方式验证的Signatera
图64A-C:用于49个患者特异性图的实体瘤患者定制化监测技术变异体选择策略。(上部图)患者的定制图中的肿瘤组织VAF分布。不同色彩表示不同亚型:HER2-(深蓝色)、三阴性(橙色)和HER2+(绿色)。(中部图)患者定制图中的所推断的克隆和亚克隆变异体的数目。49个定制图中的克隆变异体的中值数目是13/16。(下部图)患者的WES数据中的所推断的克隆和亚克隆变异体的数目。
图65:(A-L)12名(11名复发性和1名非复发性)乳癌患者的多个血浆时间点时的ctDNA的血浆含量。原发性肿瘤和相匹配的正常完全外显子组测序鉴别患者特异性体细胞突变。使用以分析方式验证的Signatera
图66:VAF和突变体计数的分布。在ctDNA阳性血浆样品中检测总共251个目标。所检测的目标的VAF在0.01%到64%范围内,其中中值是0.82%。我们使用在每个样品中观察到的突变型VAF和DNA分子的总数来计算患者的血浆样品中的肿瘤分子数目。在251个阳性目标中检测的突变型分子的数目在1到6500个突变型分子范围内,其中中值是39个分子。
图67A-D:实体瘤患者定制化监测技术质量控制方法:在工作流程中的每个步骤中进行质量控制。在总共215个血浆样品中,208个在我们的样品QC过程中合格,且在此处设计的784种独特分析法中,767种在我们的分析法QC中合格(对应于在所有样品中,3328种分析法中的总共3237种合格)。A)每毫升提取的cfDNA。通过Quant-iT高敏感性dsDNA分析法试剂盒对从每个血浆样品提取的cfDNA进行定量。将所定量的cfDNA量<5ng的样品标记为WARNING。每毫升提取的cfDNA在1到21.4ng范围内,其中中值是4.7ng。B)库制备DNA输入量。使用来自每个血浆样品的最多66ng cfDNA作为库制备方案的输入。库DNA输入量在1到66ng范围内,其中中值是25.02。在继续进行下一步骤之前,对被纯化的库进行QC。C)测序覆盖率。分析中排除覆盖率小于5000x的分析法。接着,具有小于8种合格分析法的样品在测序覆盖率QC中不合格。在覆盖率QC中合格的分析法的中值读段深度是110,000x。D)样品和谐性。为了追踪样品完整性,使用SNP追踪器测量患者的样品之间的和谐性。对于每个血浆样品,基因分型和谐性评分是相比于其对应的匹配正常基因分型数据来计算。当至少85%的样品的SNP具有一致基因型时,认为样品是来自相同患者。从ctDNA分析排除六个鉴别为待调换的血浆样品。
图68A-B:分析验证结果。(A)单一目标检测敏感性。使用实体瘤患者定制化监测技术,在约0.03%的加标(spiked-in)肿瘤DNA情况下,获得突变检测的分析敏感性是约60%。(B)当由16种目标变异体的集合检测到至少两种突变时,实体瘤患者定制化监测技术的所估计的样品-含量敏感性。
图69:在筛选和募集之后,通过6个每个月的血液样品对患者进行随访。通过免疫组织化学和荧光原位杂交分析法来确定HER2状态。如果任一种分析法呈阳性,那么认为患者具有HER2阳性癌症。NACT:新辅助化学疗法;ACT:辅助化学疗法。
图70:实例9中的肌肉侵袭性膀胱癌研究的工作流程图。
图71A-G:实例9中的肌肉侵袭性膀胱癌研究的患者概述。图71A展示由WES识别的同义与非同义突变的比率。一名患者的肿瘤发生超突变,其中突变负荷是126个突变/Mb且显示先前已证实与超突变子相关联的POLD1突变(Campbell,B.B.等人,《人类癌症中超突变的综合分析(Comprehensive Analysis of Hypermutation in Human Cancer)》,《细胞(Cell)》171,1042-1056.e10(2017))。图71B展示膀胱癌相关突变标签的相对贡献。图71C展示膀胱癌中频繁突变的基因中的突变(TCGA)(Robertson,A.G.等人,《肌肉侵袭性膀胱癌的综合分子表征(Comprehensive Molecular Characterization of Muscle-InvasiveBladder Cancer)》,《细胞》171,540-556.e25(2017))。图71D展示在68个样品中的超过5%的样品中突变的DNA损伤反应(DDR)相关基因中的有害突变。图71E展示有害DDR突变的总数。图71F展示临床和组织病理学特征。图71G展示概述的ctDNA状态。
图72:概述用于实例9中的肌肉侵袭性膀胱癌研究的临床方案和取样时间表的图。
图73:概述Signatera
图74:对应于实例9中的肌肉侵袭性膀胱癌研究的所有分析样品的ctDNA结果的纵向表示。基于ctDNA状态将患者分成三个组:上部图展示在切除术(CX)之前及之后呈ctDNA阳性的患者;中间图展示仅在CX之前呈ctDNA阳性的患者;下部图展示呈ctDNA阴性的患者。水平线表示每个患者疾病病程且圆形表示ctDNA状态,红色圆形指示具有至少2种阳性分析法的样品。指示每位患者的治疗和成像信息。
图75A-E:实例9中的肌肉侵袭性膀胱癌研究的ctDNA检测的预后值的图形描述。卡普兰-迈耶(Kaplan-Meier)存活率分析展示在化学疗法之前(图75A)、在切除术(CX)之前(图75B)和在切除术(CX)之后(图75C),由ctDNA状态进行分级的无复发存活率(RFS)和总存活率(OS)的概率。图75D展示在化学疗法之前、在切除术之前和在切除术之后,疾病复发与ctDNA状态之间的关联性,以及在切除术之前,疾病复发与淋巴结状态之间的关联性。图75E展示切除术(CX)之前的ctDNA状态与进行切除术(CX)时的病理状态之间的关联性。使用用于连续变量的威尔科克森秩和检验(Wilcoxon rank-sum test)和用于类别变量的费舍尔精确检验(Fisher's Exact test)进行统计显著性的评估。
图76:展示实例9中的肌肉侵袭性膀胱癌研究的单独疾病病程中的ctDNA变化的图。图76展示来自所选择的患者的详细描述的疾病病程、所应用的治疗和相关纵向ctDNA分析的表示。根据图例呈现ctDNA状态、所应用的治疗和成像结果。指示基于ctDNA的复发检测的阳性前置时间。
图77:展示实例9中的肌肉侵袭性膀胱癌研究的分子复发(ctDNA阳性)与临床复发(放射性成像阳性)之间的时间差的图。使用配对威尔科克森秩和检验计算P值。
图78A-H:展示实例9中的肌肉侵袭性膀胱癌研究的化学疗法反应的预测性标记物的图。图78A展示疾病复发与对化学疗法的反应之间的关联性。图78B展示分别通过对化学疗法的反应和ERCC2突变状态进行分级的所有患者的相关标签5贡献。图78C展示对与ERCC2突变状态有关的疗法起反应的一部分患者。图78D是RNA亚型figures_NEW图。图78E展示在整个疾病病程期间呈ctDNA阴性的患者、ctDNA含量降到零的患者和ctDNA含量保持阳性的患者中,ctDNA与对化学疗法的反应之间的关联性。图78F展示在化学疗法之前、期间和之后,所有具有可检测的ctDNA的患者的ctDNA含量。由对化学疗法的反应将患者分组且指示复发状态。
图79A-D:展示在实例9中的肌肉侵袭性膀胱癌研究中,与ERCC2状态相关的每位患者中所鉴别的突变的总数或损伤DNA损伤反应(DDR)突变的数目的图。
图80:描绘在实例9中的肌肉侵袭性膀胱癌研究中,原发性肿瘤与转移性复发之间的基因组异质性的图。比较原发性肿瘤的完全外显子组测序(WES)数据与ctDNA。来自在转移性复发时检测到高ctDNA变异体等位基因出现率(VAF)的血浆样品的数据。研究在血浆或肿瘤外显子组数据中鉴别到突变的基因组位置以用于基础计数。展示在血浆和肿瘤外显子组数据中鉴别的所得等位基因出现率。根据突变识别的统计概率(强度)将各个突变标记颜色。文氏图表示仅在肿瘤、血浆或这两者中鉴别的突变数目。
图81:描绘在来自实例9中的肌肉侵袭性膀胱癌研究的8名患者中,与切除术(CX)相关的不同天数时的变异等位基因出现率(VAF%)的图。
图82:展示在实例9中的肌肉侵袭性膀胱癌研究中,与超深测序相比,来自先前由ddPCR进行分析的10名患者的血浆中的ctDNA含量的图。
图83A-E:展示全部125名患者的临床、组织病理学和分子参数的图。图83A展示与突变标签相关的五种最流行的结肠直肠癌的相对贡献。图83B展示由WES识别的同义与非同义突变的比率。图83C描绘展示结肠直肠癌中频繁突变的基因中的突变(TCGA)的图{《癌症基因组图谱(Cancer Genome Atlas)》,2012第52号}。图83D展示临床和组织病理学特征。图83E展示概述手术前和手术后的ctDNA状态的图。
图84:展示用于解决既定临床问题的患者入选、样品收集和患者子组的定义的图。缩写:ctDNA,循环肿瘤DNA;CT-扫描,计算机断层摄影扫描;手术后,手术后;TTR,复发时间。
图85A-C:展示患者样品的完全外显子组测序的工作流程的质量控制(QC)检验的图。795个血浆样品中的793个(99%)在样品QC过程中合格。用SNP追踪器操作194个样品(来自70名患者)以检验血浆样品与其对应的组织活检之间的和谐性。全部194个血浆样品都在和谐性QC中合格。图85A展示库制备DNA输入量。使用来自每个血浆样品的最多66ng游离DNA(cfDNA)作为库制备方案的输入物。库DNA输入量在1到66ng范围内,其中中值是45.66。在继续进行下一步骤之前,对被纯化的库进行质量控制。一个样品在库制备QC中不合格。图85B展示测序覆盖率。分析中排除覆盖率小于5000x的分析法。接着,具有小于8种合格分析法的样品在测序覆盖率QC中不合格。一个样品在测序覆盖率要求方面不合格。在覆盖率QC中合格的分析法的中值读段深度是105,000x。图85C展示在所有血浆样品中测量的测序误差率。平均转换误差率是5e-5且平均颠换误差率是8e-6。
图86展示每个单独患者的循环肿瘤DNA(ctDNA)结果和动力学。
图87A-F展示手术前(op前)、手术后第30天和在辅助化学疗法(ACT)期间的ctDNA状态。图87A展示ctDNA的手术前检测。图87B展示复发率。图87C展示由手术后第30天的ctDNA状态进行分级的94名I-III期患者的TTR的卡普兰-迈耶估计。图87D展示对ctDNA阳性患者的ACT作用,由复发率和纵向ctDNA状态评估。图87E展示在ACT后第一次访视时,由ctDNA状态进行分级的复发率。图87F展示58名用ACT治疗的患者的TTR的卡普兰-迈耶估计,由ACT后第一次访视时的ctDNA状态进行分级。
图88展示125名I-III期CRC患者中的癌胚抗原(CEA)的手术前检测。
图89展示第30天ctDNA分析中所包括的血浆样品的ctDNA分布结果的示意性概述,由复发状态和疾病阶段进行排序。用(s)标记的患者具有同步CRC。用**标记的血浆仅在第二个池中是阳性(n=1)。
图90A-B展示第30天ctDNA分析中所包括的且接受ACT的血浆样品的子集的ctDNA分布结果的示意性概述,由复发状态和疾病阶段进行排序。用(s)标记的患者具有同步CRC。
图91展示纵向ACT后ctDNA分析中所包括的血浆样品的ctDNA分布结果的示意性概述,由复发状态、手术后ctDNA状态和随访长度进行排序。用(s)标记的患者具有同步CRC(n=2)。用**标记的血浆样品仅在第二个池中是阳性(n=1)。
图92展示纵向ACT后ctDNA分析中所包括的血浆样品的CEA分布结果的示意性概述,由复发状态、手术后ctDNA状态和随访长度进行排序。用(s)标记的患者具有同步CRC(n=2)。用**标记的血浆仅在第二个池中是阳性(n=1)。
图93A-D:展示在确定性治疗之后,ctDNA状态与复发之间的关联性的图。图93A展示由纵向ctDNA状态进行分级的复发率。图93B展示用纵向样品进行的75名患者的TTR的卡普兰-迈耶估计,由纵向ctDNA状态进行分级。图93C展示比较时间与放射性和ctDNA复发的图。图93D展示针对放射性复发,血浆中的ctDNA变异等位基因出现率(VAF)增加。省略在ACT之前和在ACT期间的早期时间点。
图94:来自复发性和非复发性患者的纵向血浆样品的ctDNA分布结果的示意性概述。在监测期间仅具有一个阳性血浆样品的患者视为阳性。
图95:来自复发性和非复发性患者的纵向血清样品的CEA分布结果的示意性概述。在监测期间仅具有一个阳性血浆样品的患者视为阳性。
图96:比较时间与放射性和CEA复发的图。
图97A-C:复发患者中的可操作突变的检测。图97A展示在监测期间检测到可操作突变的ctDNA+复发患者的百分比。第一个ctDNA+样品(左侧柱)和所有ctDNA+血浆样品(右侧柱)。图97B展示血液中识别的可操作变异体。使用实体瘤患者定制化监测技术ctDNA+分析法计算的平均血液VAF与可操作突变的变异等位基因出现率(VAF)之间的相关性,在横轴和纵轴上都使用对数尺度标绘。图97C展示具有可操作突变的两名代表性复发患者的连续ctDNA分布。
图98:当前护理标准与可能的ctDNA指导的手术后患者管理的示意性比较。
图99:展示由辅助化学疗法(ACT)降低的ctDNA的图。
图100A-B:展示在确定性治疗之后,ctDNA状态与复发之间的关联性的图。图100A展示由纵向ctDNA状态进行分级的复发率,和使用纵向样品进行的58名患者的TTR的卡普兰-迈耶估计,由纵向ctDNA分析进行分级。图100B展示由CEA分析进行分级的复发率,和使用纵向样品进行的58名患者的TTR的卡普兰-迈耶估计,由CEA分析进行分级。
以上所标识的图是以代表性且非限制性方式提供。
具体实施方式
本文中所提供的方法和组合物改善癌症(例如乳癌、膀胱癌或结肠直肠癌)的检测、诊断、分期、筛选、治疗和管理。在说明性实施例中,本文中所提供的方法分析循环流体,尤其循环肿瘤DNA中的单核苷酸变异体突变(SNV)。所述方法提供以下优点:在利用肿瘤样品的单一检验中,而非需要多个检验,即可鉴别更多的在肿瘤中发现的突变以及克隆和亚克隆突变(如果完全有效)。方法和组合物本身可以是有帮助的,或其可以在与其它用于癌症(例如乳癌、膀胱癌或结肠直肠癌)的检测、诊断、分期、筛选、治疗和管理的方法一起使用时是有帮助的,例如帮助支持这些其它方法的结果以提供置信度更高和/或决定性的结果。
因此,在一个实施例中,本文中提供一种方法,其使用本文中所提供的ctDNA SNV扩增/测序工作流程,通过确定来自个体,如患有或怀疑患有癌症(例如乳癌、膀胱癌或结肠直肠癌)的个体的ctDNA样品中是否存在单核苷酸变异体来确定癌症(例如乳癌、膀胱癌或结肠直肠癌)中是否存在单核苷酸变异体。
术语“癌症”和“癌性”是指或描述特征通常在于不受调控的细胞生长的动物中的生理学病状。“肿瘤”包含一种或多种癌性细胞。存在若干种主要癌症类型。癌瘤是在皮肤中或在沿内脏排列或覆盖内脏的组织中开始的癌症。肉瘤是在骨骼、软骨、脂肪、肌肉、血管或其它连接性或支持性组织中开始的癌症。白血病是在血液形成组织(如骨髓)中开始的癌症,且引起大量异常的血细胞产生和进入血液。淋巴瘤和多发性骨髓瘤是在免疫系统的细胞中开始的癌症。中枢神经系统癌症是在脑部和脊髓的组织中开始的癌症。
在一些实施例中,癌症包含急性淋巴母细胞性白血病;急性骨髓性白血病;肾上腺皮质癌;AIDS相关癌症;AIDS相关淋巴瘤;肛门癌;阑尾癌;星形细胞瘤;非典型畸胎样/横纹肌样肿瘤;基底细胞癌;膀胱癌;脑干神经胶质瘤;脑肿瘤(包括脑干神经胶质瘤、中枢神经系统非典型畸胎样/横纹肌样肿瘤、中枢神经系统胚胎肿瘤、星形细胞瘤、颅咽管瘤、室管膜母细胞瘤、室管膜瘤、神经管胚细胞瘤、髓上皮瘤、中度分化型松果体实质性肿瘤、幕上原始神经外胚层肿瘤和成松果体细胞瘤);乳癌;支气管肿瘤;伯基特淋巴瘤;原发位点未知的癌症;类癌肿瘤;原发位点未知的癌瘤;中枢神经系统非典型畸胎样/横纹肌样肿瘤;中枢神经系统胚胎肿瘤;子宫颈癌;儿童癌症;脊索瘤;慢性淋巴细胞性白血病;慢性骨髓性白血病;慢性骨髓增生性病症;结肠癌;结肠直肠癌;颅咽管瘤;皮肤T细胞淋巴瘤;内分泌胰岛细胞瘤;子宫内膜癌;室管膜母细胞瘤;室管膜瘤;食道癌;鼻腔神经胶质瘤;尤文氏肉瘤;颅外生殖细胞肿瘤;性腺外生殖细胞肿瘤;肝外胆管癌;胆囊癌;胃部(胃)癌症;胃肠道类癌肿瘤;胃肠道基质细胞肿瘤;胃肠道基质瘤(GIST);妊娠期滋养细胞肿瘤;神经胶质瘤;毛状细胞白血病;头颈癌;心脏癌症;霍奇金氏淋巴瘤;下咽癌症;眼内黑素瘤;胰岛细胞瘤;卡波西肉瘤;肾脏癌;兰格汉氏细胞组织细胞增多病;喉癌;唇癌;肝癌;恶性纤维组织细胞瘤骨癌;神经管胚细胞瘤;髓上皮瘤;黑素瘤;梅克尔细胞癌;梅克尔细胞皮肤癌瘤;间皮瘤;隐性原发性转移性鳞状颈部癌症;口腔癌;多发性内分泌瘤形成综合症;多发性骨髓瘤;多发性骨髓瘤/血浆细胞赘瘤;蕈样真菌病;骨髓发育不良综合症;骨髓增生赘瘤;鼻腔癌;鼻咽癌;神经母细胞瘤;非霍奇金氏淋巴瘤;非黑素瘤型皮肤癌;非小细胞肺癌;口部癌症;口腔癌症;口咽癌;骨肉瘤;其它脑部和脊髓肿瘤;卵巢癌;卵巢上皮癌症;卵巢生殖细胞肿瘤;卵巢低恶性潜能肿瘤;胰脏癌;乳头瘤病;副鼻窦癌;副甲状腺癌;骨盆癌;阴茎癌;咽癌;中度分化型松果体实质性肿瘤;成松果体细胞瘤;垂体肿瘤;血浆细胞赘瘤/多发性骨髓瘤;胸膜肺母细胞瘤;原发性中枢神经系统(CNS)淋巴瘤;原发性肝细胞肝癌;前列腺癌;直肠癌;肾癌;肾细胞(肾脏)癌;肾细胞癌;呼吸道癌症;成视网膜细胞瘤;横纹肌肉瘤;唾液腺癌症;塞氏综合症;小细胞肺癌;小肠癌;软组织肉瘤;鳞状细胞癌;鳞状颈部癌症;胃(胃部)癌症;幕上原始神经外胚层肿瘤;T细胞淋巴瘤;睾丸癌;喉癌;胸腺癌;胸腺瘤;甲状腺癌;移行细胞癌症;肾盂和输尿管的移行细胞癌症;滋养细胞肿瘤;输尿管癌症;尿道癌;子宫癌;子宫瘤;阴道癌;外阴癌;瓦尔登斯特伦巨球蛋白血症;或威尔姆氏肿瘤。
在另一实施例中,本文中提供用于在来自个体,如怀疑患有癌症的个体的血液样品或其一部分中检测癌症(例如乳癌、膀胱癌或结肠直肠癌)的方法,其包括使用本文中所提供的ctDNA SNV扩增/测序工作流程,通过确定ctDNA样品中是否存在单核苷酸变异体来确定样品中是否存在单核苷酸变异体。在样品中,在多个单核苷酸基因座处存在作为范围的下端的1、2、3、4、5、6、7、8、9、10、11、12、13、14或15种SNV和作为范围的上端的2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、40或50种SNV指示存在癌症(例如乳癌、膀胱癌或结肠直肠癌)。
在另一实施例中,本文中提供用于检测个体的肿瘤(例如乳癌、膀胱癌或结肠直肠癌)中的克隆单核苷酸变异体的方法。所述方法包括进行如本文中所提供的ctDNA SNV扩增/测序工作流程,并且基于所述系列的扩增子的多个拷贝的序列来确定每个SNV基因座的变异体等位基因出现率。与多个单核苷酸变异体基因座的其它单核苷酸变异体相比较高的相对等位基因出现率指示肿瘤中的克隆单核苷酸变异体。变异体等位基因出现率是测序技术中众所周知的。对这一实施例的支持提供于例如图12-14中。
在某些实施例中,所述方法进一步包括确定治疗计划、疗法和/或向个体给予靶向一种或多种克隆单核苷酸变异体的化合物。在某些实例中,亚克隆和/或其它克隆SNV不是疗法的目标。特定疗法和相关突变提供于本说明书的其它章节中且是所属领域中已知的。因此,在某些实例中,所述方法进一步包括向个体给予化合物,其中已知所述化合物可以特定地有效治疗具有一种或多种所确定的单核苷酸变异体的癌症(例如乳癌、膀胱癌或结肠直肠癌)。
在这一实施例的某些方面中,变异体等位基因出现率大于0.25%、0.5%、0.75%、1.0%、5%或10%指示存在克隆单核苷酸变异体。这些截止值由表格形式图20A-B中的数据支持。
在这一实施例的某些实例中,癌症是1a、1b或2a期乳癌、膀胱癌或结肠直肠癌。在这一实施例的某些实例中,癌症是1a或1b期乳癌、膀胱癌或结肠直肠癌。在实施例的某些实例中,个体未经历手术。在实施例的某些实例中,个体未经历活检。
在这一实施例的一些实例中,如果其它检验(如直接肿瘤检验)表明检验中的SNV是克隆SNV(即可变等位基因出现率大于至少四分之一、三分之一、二分之一或四分之三的其它所确定的单核苷酸变异体的任何检验中的SNV),那么鉴别或进一步鉴别克隆SNV。
在一些实施例中,可以使用本文中的用于检测ctDNA中的SNV的方法代替来自肿瘤的DNA的直接分析。本文中所提供的结果表明,具有显著更高的成为克隆SNV的可能性的SNV具有较高的VAF(参见例如图12-14)。
在本文中所提供的任何方法实施例的某些实例中,在对来自个体的ctDNA进行靶向扩增之前,提供关于在来自个体的肿瘤中发现的SNV的数据。因此,在这些实施例中,对来自个体的一个或多个肿瘤样品进行SNV扩增/测序反应。在这类方法中,本文中所提供的ctDNA SNV扩增/测序反应仍是有利的,因为其提供克隆和亚克隆突变的液体活检。此外,如本文中所提供,如果在来自个体的ctDNA样品中针对SNV确定高VAF百分比,例如超过1、2、3、4、5、6、7、8、9、10%VAF,那么可以在患有癌症(例如乳癌、膀胱癌或结肠直肠癌)的个体中更明确地鉴别克隆突变。
在某一实施例中,本文中所提供的方法可以用于确定是否从来自患有癌症(例如乳癌、膀胱癌或结肠直肠癌)的个体的循环游离核酸分离和分析ctDNA。首先,确定癌症是否是乳癌、膀胱癌或结肠直肠癌。如果癌症是乳癌、膀胱癌或结肠直肠癌,那么从个体分离循环游离核酸。在一些实例中,所述方法进一步包括确定癌症的阶段。
在一些方法中,本文中提供本发明的组合物和/或固体负载物。一种包含循环肿瘤核酸片段的组合物,所述循环肿瘤核酸片段包含通用衔接子,其中循环肿瘤核酸是来源于乳癌、膀胱癌或结肠直肠癌。
在一些实施例中,本文中提供本发明的组合物,其包括包含通用衔接子的循环肿瘤核酸片段,其中循环肿瘤核酸是来源于患有癌症(例如乳癌、膀胱癌或结肠直肠癌)的个体的血液样品或其一部分。这些方法通常包括形成包括通用衔接子的ctDNA片段。此外,这类方法通常包括形成固体负载物,尤其用于高通量测序的固体负载物,其包括核酸的多个克隆群体,其中所述克隆群体包含由循环游离核酸的样品产生的扩增子,其中ctDNA。在基于本文中所提供的出人意料的结果的说明性实施例中,ctDNA是来源于癌症(例如乳癌、膀胱癌或结肠直肠癌)。
类似地,作为本发明的实施例,本文中提供包含核酸的多个克隆群体的固体负载物,其中克隆群体包含由循环游离核酸的样品产生的核酸片段,所述循环游离核酸是来自患有癌症(例如乳癌、膀胱癌或结肠直肠癌)的个体的血液样品或其一部分。
在某些实施例中,不同克隆群体中的核酸片段包含相同通用衔接子。这类组合物通常在本发明的方法中的高通量测序反应期间形成。
核酸的克隆群体可以来源于来自两名或更多名个体的样品集合的核酸片段。在这些实施例中,核酸片段包含对应于样品集合中的样品的一系列分子条形码中的一个。
详细分析方法在本文中以本文中的分析章节中的SNV方法1和SNV方法2形式提供。本文中所提供的任何方法可以进一步包括本文中所提供的分析步骤。因此,在某些实例中,用于确定样品中是否存在单核苷酸变异体的方法包括鉴别在单核苷酸变异基因座集合中的每一个处进行的每一次等位基因确定的置信度值,其可以至少部分地基于基因座的读段深度。置信界限可以设置成至少75%、80%、85%、90%、95%、96%、96%、98%或99%。置信界限可以针对不同类型的突变而设置成不同水平。
所述方法可以在单核苷酸变异基因座集合的读段深度是至少5、10、15、20、25、50、100、150、200、250、500、1,000、10,000、25,000、50,000、100,000、250,000、500,000或1百万的情况下进行。
在某些实施例中,本文中的任何实施例的方法包括确定效率和/或确定单核苷酸变异基因座的多重扩增反应中的每个扩增反应的每个循环的误差率。接着,效率和误差率可以用于确定样品是否中存在单一变异体基因座集合处的单核苷酸变异体。在某些实施例中,还可以包括分析方法中所提供的SNV方法2中所提供的更详细的分析步骤。
在本文中的任何方法的说明性实施例中,单核苷酸变异基因座集合包括在癌症(例如乳癌、膀胱癌或结肠直肠癌)的TCGA和COSMIC数据集中鉴别的所有单核苷酸变异基因座。
在本文中的任何方法的某些实施例中,单核苷酸变异体基因座集合包括作为范围的下端的2、3、4、5、6、7、8、9、10、15、20、25、30、40、50、75、100、250、500、1000、2500、5000或10,000种和作为范围的下端的5、6、7、8、9、10、15、20、25、30、40、50、75、100、250、500、1000、2500、5000、10,000、20,000和25,000种已知与癌症(例如乳癌、膀胱癌或结肠直肠癌)相关联的单核苷酸变异基因座。
在本文中的任何包括ctDNA SNV扩增/测序工作流程的用于检测SNV的方法中,可以使用改善的多重PCR的扩增参数。举例来说,对于引物集合中的至少10、20、25、30、40、50、06、70、75、80、90、95或100%的引物,其中扩增反应是PCR反应且粘接温度比熔融温度高作为范围的下端的1、2、3、4、5、6、7、8、9或10℃到作为范围的上端的2、3、4、5、6、7、8、9、10、11、12、13、14或15℃。
在某些实施例中,其中扩增反应是PCR反应,PCR反应中的粘接步骤的长度是作为范围的下端的10、15、20、30、45和60分钟到作为范围的上端的15、20、30、45、60、120、180或240分钟。在某些实施例中,扩增(如PCR反应)中的引物浓度在1与10nM之间。此外,在例示性实施例中,引物集合中的引物被设计成最大限度地减少引物二聚体形成。
因此,在本文中任何包括扩增步骤的方法的实例中,扩增反应是PCR反应,粘接温度比引物集合中至少90%的引物的熔融温度高1到10℃,PCR反应中的粘接步骤的长度是15到60分钟,扩增反应中的引物浓度是1到10nM,并且引物集合中的引物被设计成最大限度地减少引物二聚体形成。在本实例的另一个方面中,在限制性引物条件下进行多重扩增反应。
在另一实施例中,本文中提供用于支持个体(如怀疑患有癌症(例如乳癌、膀胱癌或结肠直肠癌)的个体)中的由来自个体的血液样品或其一部分进行的癌症(例如乳癌、膀胱癌或结肠直肠癌)诊断的方法,其包括进行如本文中所提供的ctDNA SNV扩增/测序工作流程,以确定多个单核苷酸变异体基因座中是否存在一种或多种单核苷酸变异体。在这一实施例中,以下元素、陈述、指南或规则适用:如果不存在单核苷酸变异体,那么支持1a、1b或2a期腺癌的诊断;如果存在单核苷酸变异体,那么支持鳞状细胞癌或2b或3a期腺癌的诊断;和/或如果存在十种或更多种单核苷酸变异体,那么支持鳞状细胞癌或2b或3期腺癌的诊断。
这些结果将使用来自个体的肺ADC和SCC样品的ctDNA SNV扩增/测序工作流程的分析确定为用于鉴别在ADC肿瘤,尤其2b和3a期ADC肿瘤且尤其任何阶段的SCC肿瘤中发现的SNV的有价值的方法(参见例如图15和图20A-B)。
在某些实施例中,本文中的用于检测SNV的方法可以用于指导治疗方案。以与ADC和SCC相关联的特异性突变为目标的疗法是可用的且正在研发中(《自然癌症综述(NatureReview Cancer)》,14:535-551(2014))。举例来说,在L858R或T790M处检测到EGFR突变可以为选择疗法提供信息。埃罗替尼(erlotinib)、吉非替尼(gefitinib)、阿法替尼(afatinib)、AZK9291、CO-1686和HM61713是当前在美国或在临床试验中被批准的疗法,其靶向特异性EGFR突变。在另一实例中,KRAS中的G12D、G12C或G12V突变可以用于指导个体使用司美替尼(Selumetinib)加多烯紫杉醇(docetaxel)的组合的疗法。作为另一实例,BRAF中V600E的突变可以用于指导个体使用维罗非尼(Vemurafenib)、达拉非尼(dabrafenib)和曲美替尼(trametinib)的治疗。
在某些说明性实施例中,本发明的方法中分析的样品是血液样品或其一部分。在某些实施例中,本文中所提供的方法被专门调适成用于扩增DNA片段,尤其在循环肿瘤DNA(ctDNA)中发现的肿瘤DNA片段。这类片段的长度通常是约160个核苷酸。
在所属领域中已知,游离核酸(cell-free nucleic acid;cfNA),例如cfDNA,可以通过多种形式的细胞死亡(如细胞凋亡、坏死、自噬和坏死性凋亡)而释放至循环中。cfDNA被片段化且片段的尺寸分布在150-350bp到>10000bp范围内(参见Kalnina等人,《世界胃肠病学杂志(World J Gastroenterol.)》,2015年11月7日;21(41):11636-11653)。举例来说,肝细胞癌(HCC)患者中的血浆DNA片段的尺寸分布在长度是100-220bp的范围内,其中在约166bp处具有计数频率的峰值且在长度是150-180bp的片段中具有最高肿瘤DNA浓度(参见:Jiang等人,《美国国家科学院院刊(Proc Natl Acad Sci USA)》,112:E1317-E1325)。
在说明性实施例中,在通过离心来去除细胞碎片和血小板之后,使用EDTA-2Na试管从血液分离循环肿瘤DNA(ctDNA)。血浆样品可以在-80℃下储存直到使用例如QIAampDNA小型试剂盒(Qiagen,Hilden,Germany)提取DNA(例如Hamakawa等人,《英国癌症杂志(BrJ Cancer.)》2015;112:352-356)。Hamakava等人报道所有样品的所提取的游离DNA的中值浓度是每毫升血浆43.1ng(在9.5-1338ng/ml范围内)且突变体分数范围是0.001-77.8%,其中中值是0.90%。
在某些说明性实施例中,样品是肿瘤。鉴于本文中的教示内容,所属领域中已知用于从肿瘤分离核酸和由这类DNA样品创建核酸库的方法。此外,鉴于本文中的教示内容,所属领域的技术人员将认识到如何由除ctDNA样品以外的其它样品(如其中DNA是自由浮动的其它液体样品)创建适用于本文中的方法的核酸库。
在某些实施例中,本发明的方法通常包括由样品产生和扩增核酸库(即,库制备)的步骤。在库制备步骤期间,来自样品的核酸可以具有附接的接合衔接子,通常称为库标签或接合衔接子标签(LT),其中接合衔接子含有通用引发序列,接着是通用扩增。在一个实施例中,这可以使用被设计成在片段化之后创建测序库的标准方案来进行。在一个实施例中,可以对DNA样品进行钝端化,并且接着可以在3'端添加A。可以添加和接合具有T突出端的Y衔接子。在一些实施例中,可以使用除A或T突出端以外的其它粘性末端。在一些实施例中,可以添加其它衔接子,例如环形接合衔接子。在一些实施例中,衔接子可以具有被设计成用于PCR扩增的标签。
本文中所提供的许多实施例包括检测ctDNA样品中的SNV。在说明性实施例中,这类方法包括扩增步骤和测序步骤(在本文中有时称为“ctDNA SNV扩增/测序工作流程”)。在说明性实例中,ctDNA扩增/测序工作流程可以包括通过对核酸进行多重扩增反应来产生扩增子集合,所述核酸是从来自个体(如怀疑患有癌症(例如乳癌、膀胱癌或结肠直肠癌)的个体)的血液样品或其一部分分离,其中扩增子集合中的每个扩增子跨越单核苷酸变异体基因座集合中的至少一个单核苷酸变异体基因座,如已知与癌症(例如乳癌、膀胱癌或结肠直肠癌)相关联的SNV基因座;和确定扩增子集合中的每个扩增子的至少一个区段的序列,其中所述区段包含单核苷酸变异体基因座。以此方式,这种例示性方法确定样品中是否存在单核苷酸变异体。
更详细地,例示性ctDNA SNV扩增/测序工作流程可以包括通过组合以下来形成扩增反应混合物:聚合酶、核苷酸三磷酸酯、来自由样品产生的核酸库的核酸片段以及引物集合(所述引物各自在单核苷酸变异体基因座的有效距离内结合)或引物对集合(所述引物对各自跨越包括单核苷酸变异体基因座的有效区域)。在例示性实施例中,单核苷酸变异体基因座是一个已知与癌症(例如乳癌、膀胱癌或结肠直肠癌)相关联的基因座。接着,使扩增反应混合物经历扩增条件以产生扩增子集合,所述扩增子集合包含优选已知与癌症(例如乳癌、膀胱癌或结肠直肠癌)相关联的单核苷酸变异体基因座集合中的至少一个单核苷酸变异体基因座;和确定扩增子集合中的每个扩增子的至少一个区段的序列,其中所述区段包含单核苷酸变异体基因座。
引物的有效结合距离可以在SNV基因座的1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、20、25、30、35、40、45、50、75、100、125或150个碱基对内。一对引物跨越的有效范围通常包括SNV且通常是160个或更少的碱基对,并且可以是150、140、130、125、100、75、50或25个或更少的碱基对。在其它实施例中,一对引物跨越的有效范围是来自SNV基因座的作为范围的下端的20、25、30、40、50、60、70、75、100、110、120、125、130、140或150个和作为范围的上端的25、30、40、50、60、70、75、100、110、120、125、130、140或150、160、170、175或200个核苷酸。
关于可以用于ctDNA SNV扩增/测序工作流程中以检测SNV,从而用于本发明的方法中的扩增方法的其它细节提供于本说明书的其它章节中。
SNV识别分析
在进行本文中所提供的方法期间,产生由并排多重PCR产生的扩增子的核酸测序数据。可以使用算法设计工具,其可以用于和/或被调适成用于分析这类数据以确定在某些置信度限制内,目标基因中是否存在突变,如SNV。
测序读段可以使用内部工具进行解复用且使用成对末端合并读段,使用巴罗斯-惠勒比对软件(Burrows-Wheeler alignment software),Bwa mem功能(BWA,巴罗斯-惠勒比对软件(参见Li H.和Durbin R.(2010)《用巴罗斯-惠勒转换进行的快速和精确长读段比对(Fast and accurate long-read alignment with Burrows-Wheeler Transform)》,《Bioinformatics(生物信息学)》,电子版[PMID:20080505])以单端模式映射到hg19基因组。可通过分析全部读段、映射的读段数目、中靶的映射的读段数目和进行计数的读段数目来进行扩增统计QC。
在某些实施例中,任何用于由核酸测序数据检测来检测SNV的分析方法都可以与本发明的包括检测SNV或确定是否存在SNV的步骤的方法一起使用。在某些说明性实施例中,使用利用以下SNV方法1的本发明的方法。在其它的说明性甚至更高的实施例中,本发明的包括检测SNV或确定SNV基因座中是否存在SNV的步骤的方法利用以下SNV方法2。
SNV方法1:在本实施例中,使用正常血浆样品构筑背景误差模型,所述正常血浆样品在同一测序操作中测序以解决操作特异性假象。在某些实施例中,在同一测序操作中分析5、10、15、20、25、30、40、50、100、150、200、250或超过250个正常血浆样品。在某些说明性实施例中,在同一测序操作中分析20、25、40或50个正常血浆样品。去除具有大于截止值的正常中值变异体等位基因出现率的噪声位置。举例来说,在某些实施例中,这一截止值是>0.1%、0.2%、0.25%、0.5%、1%、2%、5%或10%。在某些说明性实施例中,去除具有大于0.5%的正常中值变异体等位基因出现率的噪声位置。从模型迭代地去除异常样品以解决噪声和污染。在某些实施例中,从数据分析去除Z评分大于5、6、7、8、9或10的样品。对于每个基因座的每个碱基取代,计算误差的读段深度加权平均值和标准差。举例来说,可以将针对背景误差模型具有至少5个变异体读段且Z评分是10的肿瘤或无细胞血浆样品的位置识别为候选突变。
SNV方法2:在本实施例中,使用血浆ctDNA数据确定单核苷酸变异体(SNV)。PCR方法模型化为随机方法,使用训练集估计参数且产生用于单独检验集合的最终SNV识别。确定跨越多个PCR循环的误差的传播,并且计算背景误差的平均值和方差且在说明性实施例中,区分背景误差与真实突变。
估计每个碱基的以下参数:
p=效率(在每个循环中复制每个读段的概率)
p
X
因为在PCR方法的过程中复制读段,所以存在更多的误差。因此,由与原始读段的分离程度来确定读段的误差分布。如果一个读段在其被产生之前已经历k次复制,那么我们将其称为第k代。
让我们定义每个碱基的以下变量:
X
Y
X
此外,除正常分子X
鉴于在循环j-1中的第i-1代读段的总数,在样品尺寸是Y
我们还具有
最终,E(X
在某些实施例中,如下进行SNV方法2:
a)使用训练数据集估计PCR效率和每个循环误差率;
b)使用在步骤(a)中估计的效率分布,估计每个碱基处的检验数据集的起始分子的数目;
c)如果需要,那么使用在步骤(b)中估计的分子的起始数目更新检验数据集的效率的估计;
d)使用检验集合数据以及在步骤(a)、(b)和(c)中估计的参数,估计分子总数、背景误差分子和真实突变分子的平均值和方差(对于由初始百分比的真实突变分子组成的搜索空间);
e)针对全部分子中的全部误差分子(背景误差和真实突变)的数目拟合分布,且计算搜索空间中的每个真实突变百分比的似然性;和
f)确定最有可能的真实突变百分比且使用来自步骤(e)的数据计算置信度。
可以使用置信度截止值鉴别SNV基因座处的SNV。举例来说,可以使用90%、95%、96%、97%、98%或99%置信度截止值识别SNV。
例示性SNV方法2算法
通过使用训练集估计每个循环的效率和误差率来开始算法。假设n表示PCR循环的总数。
可以由(1+p
类似地,可以使用每个碱基b处的误差e读段R
接着,对于检验数据,将每个碱基处的初始起始拷贝估计为
因此,我们估计了将用于随机方法中的参数。接着,通过使用这些估计值,可以估计在每个循环中创建的分子的平均值和方差(应注意,对于正常分子、误差分子和突变分子,独立地进行所述估计)。
最终,通过使用概率方法(如最大似然性或类似方法),可以确定最佳地拟合误差、突变和正常分子的分布的最佳f
引物尾部可以改善来自通用标记库的片段化DNA的检测。如果库标签和引物尾部含有同源序列,那么杂交可以得到改善(例如,熔融温度(Tm)降低)并且如果仅一部分引物目标序列在样品DNA片段中,那么可以延长引物。在一些实施例中,可以使用13个或更多的目标特异性碱基对。在一些实施例中,可以使用10到12个目标特异性碱基对。在一些实施例中,可以使用8到9个目标特异性碱基对。在一些实施例中,可以使用6到7个目标特异性碱基对。
在一个实施例中,通过使衔接子接合到样品中的DNA片段的末端或由从样品分离的DNA产生的DNA片段的末端来由以上样品产生库。接着,可以使用PCR来扩增片段,例如根据以下例示性方案:
95℃,2分钟;15×[95℃,20秒,55℃,20秒,68℃,20秒],68℃,2分钟,保持在4℃下。
所属领域中已知许多用于产生核酸库的试剂盒和方法,所述核酸库包括用于后续扩增(例如克隆扩增)和子序列测序的通用引物结合位点。为了有助于衔接子的接合,库制备和扩增可以包括末端修复和腺苷酸化(即,A-加尾)。尤其适用于由小型核酸片段(尤其循环游离DNA)制备库的试剂盒可以适用于实践本文中所提供的方法。举例来说,可以从BiooScientific()获得的NEXTflex Cell Free试剂盒或Natera Library Prep试剂盒(可以从Natera,Inc.San Carlos,CA获得)。然而,这类试剂盒通常将被修改以包括被定制成用于本文中所提供的方法的扩增和测序步骤的衔接子。可以使用可商购的试剂盒,如AGILENTSURESELECT试剂盒(Agilent,CA)中的接合试剂盒来进行衔接子接合。
接着,扩增由从样品(尤其用于本发明的方法的循环游离DNA样品)分离的DNA产生的核酸库的目标区域。使用一系列引物或引物对进行这种扩增,所述一系列引物或引物对可以包括作为范围的下端的5、10、15、20、25、50、100、125、150、250、500、1000、2500、5000、10,000、20,000、25,000或50,000个到作为范围的上端的15、20、25、50、100、125、150、250、500、1000、2500、5000、10,000、20,000、25,000、50,000、60,000、75,000或100,000个引物,其各自结合于一系列引物结合位点中的一个。
可以使用Primer3产生引物设计(Untergrasser A,Cutcutache I,Koressaar T,Ye J,Faircloth BC,Remm M,Rozen SG(2012)《Primer3-新功能和界面(Primer3-newcapabilities and interfaces)》,《核酸研究(Nucleic Acids Research)》40(15):e115和Koressaar T,Remm M(2007)《引物设计程序Primer3的增强和修改(Enhancements andmodifications of primer design program Primer3)》,《生物信息学(Bioinformatics)》23(10):1289-91,可以从primer3.sourceforge.net获得源代码)。可以由BLAST评估引物特异性且添加到现有引物设计流水线准则中:
可以使用来自ncbi-blast-2.2.29+程序包的BLASTn程序确定引物特异性。任务选项“blastn-short”可以用于映射针对hg19人类基因组的引物。如果引物对基因组具有小于100个命中且顶部命中是基因组的目标互补引物结合区且比其它命中高至少两分(评分是由BLASTn程序定义),那么引物设计可以确定为“特异性”。可以进行这一过程以具有针对基因组的独特命中且在整个基因组中不具有许多其它命中。
可以使用BED文件和用于验证的覆盖图,在IGV(James T.Robinson,HelgaThorvaldsdóttir,Wendy Winckler,Mitchell Guttman,Eric S.Lander,Gad Getz,JillP.Mesirov.,《整合基因组学查看器(Integrative Genomics Viewer)》,《自然生物技术(Nature Biotechnology)》29,24-26(2011))和UCSC浏览器(Kent WJ,Sugnet CW,FureyTS,Roskin KM,Pringle TH,Zahler AM,Haussler D.,《UCSC的人类基因组浏览器(Thehuman genome browser at UCSC)》,《基因组研究(Genome Res.)》2002年6月12(6):996-1006)中显示最终所选择的引物。
在某些实施例中,本发明的方法包括形成扩增反应混合物。通常通过组合以下来形成反应混合物:聚合酶、核苷酸三磷酸酯、来自由样品产生的核酸库的核酸片段、对含有SNV的目标区域具有特异性的正向和反向引物的集合。在说明性实施例中,本文中所提供的反应混合物本身形成本发明的独立方面。
适用于本发明的扩增反应混合物包括所属领域中已知用于核酸扩增,尤其用于PCR扩增的组分。举例来说,反应混合物通常包括核苷酸三磷酸酯、聚合酶和镁。适用于本发明的聚合酶可以包括任何可以用于扩增反应中,尤其适用于PCR反应中的聚合酶。在某些实施例中,热起始Taq聚合酶尤其适用。适用于实践本文中所提供的方法的扩增反应混合物,如AmpliTaq Gold主混合物(Life Technologies,Carlsbad,CA),是以可商购的。
用于PCR的扩增(例如温度循环)条件是所属领域中众所周知的。本文中所提供的方法可以包括任何引起目标核酸(如来自库的目标核酸)扩增的PCR循环条件。非限制性例示性循环条件提供于本文中的实例部分中。
在进行PCR时,存在许多有可能进行的工作流程;本文中提供本文中所公开的方法中的一些典型工作流程。本文中概述的步骤并不打算排除其它可能步骤,也不暗示本文中所描述的任何步骤是所述方法恰当地起作用所需的。大量参数变化或其它修改在文献中是已知的,并且可以在不影响本发明的本质的情况下进行。
在本文中所提供的方法的某些实施例中,确定扩增子(如外部引物目标扩增子)的至少一部分序列且在说明性实例中,确定扩增子的全部序列。用于确定扩增子的序列的方法是所属领域中已知的。所属领域中已知的任何测序方法(例如桑格测序(Sangersequencing))都可以用于这类序列确定。在说明性实施例中,可以使用高通量下一代测序技术(在本文中也称为大规模平行测序技术)对由本文中所提供的方法产生的扩增子进行测序,如(但不限于)MYSEQ(ILLUMINA)、HISEQ(ILLUMINA)、ION TORRENT(LIFETECHNOLOGIES)、GENOME ANALYZER ILX(ILLUMINA)、GSFLEX+(ROCHE454)中使用的测序技术。
高通量基因测序器允许使用条形码(即,用独特核酸序列标记的样品),以便鉴别来自个体的特异性样品,由此允许在DNA测序器的单次运行中同时分析多个样品。对库制剂(或其它相关的核制剂)中的基因组的既定区域进行测序的次数(读段的数目)将与相关基因组中序列的拷贝数目(或在含有cDNA的制剂的情况下,表达量)成比例。在这类定量确定中,可以考虑扩增效率的偏差。
目标基因
在例示性实施例中,本发明的目标基因是癌症相关基因且在许多说明性实施例中,是癌症相关基因。癌症相关基因(例如癌症相关基因或膀胱癌相关基因或结肠直肠癌相关基因)是指与癌症(例如乳癌、膀胱癌或结肠直肠癌)风险改变或癌症预后改变相关联的基因。促进癌症的例示性癌症相关基因包括致癌基因;增强细胞增殖、侵袭或转移的基因;抑制细胞凋亡的基因;和促血管生成基因。抑制癌症的癌症相关基因包括(但不限于)肿瘤抑制基因;抑制细胞增殖、侵袭或转移的基因;促进细胞凋亡的基因;和抗血管生成基因。
突变检测方法的实施例由选择成为目标的基因区域开始。使用具有已知突变的区域产生用于mPCR-NGS的引物,以扩增和检测突变。
本文中所提供的方法可以用于检测几乎任何类型的突变,尤其已知与癌症相关联的突变且最具体地说,本文中所提供的方法涉及与癌症,具体地说,乳癌、膀胱癌或结肠直肠癌相关联的突变,尤其SNV。例示性SNV可以在以下基因中的一种或多种中:EGFR、FGFR1、FGFR2、ALK、MET、ROS1、NTRK1、RET、HER2、DDR2、PDGFRA、KRAS、NF1、BRAF、PIK3CA、MEK1、NOTCH1、MLL2、EZH2、TET2、DNMT3A、SOX2、MYC、KEAP1、CDKN2A、NRG1、TP53、LKB1和PTEN,其已在多种肺癌样品中鉴别为发生突变、具有增加的拷贝数目或与其它基因融合和其组合(《非小细胞肺癌:一组异质性疾病(Non-small-cell lung cancers:a heterogeneous set ofdiseases)》,Chen等人,《自然癌症综述(Nat.Rev.Cancer)》,2014年8月,14(8):535-551)。在另一实例中,基因列表是上文所列的列表,其中已报道SNV,如在所列举的Chen等人的参考文献中。
扩增(例如PCR)反应混合物:
在某些实施例中,本发明的方法包括形成扩增反应混合物。通常通过组合以下来形成反应混合物:聚合酶、核苷酸三磷酸酯、来自由样品产生的核酸库的核酸片段、一系列正向目标特异性外部引物和第一链反向外部通用引物。另一说明性实施例是一种反应混合物,其包括代替正向目标特异性外部引物的正向目标特异性内部引物,和代替来自核酸库的核酸片段的来自使用外部引物的第一PCR反应的扩增子。在说明性实施例中,本文中所提供的反应混合物本身形成本发明的独立方面。在说明性实施例中,反应混合物是PCR反应混合物。PCR反应混合物通常包括镁。
在一些实施例中,反应混合物包括乙二胺四乙酸(EDTA)、镁、四甲基氯化铵(TMAC)或其任何组合。在一些实施例中,TMAC的浓度在20与70mM之间且包括端值。不希望受任何具体理论约束,相信TMAC结合于DNA、使双螺旋稳定、提高引物特异性和/或使不同引物的熔融温度一致。在一些实施例中,TMAC提高不同目标的扩增产物的量的均匀性。在一些实施例中,镁(如来自氯化镁的镁)的浓度在1与8mM之间。
用于大量目标的多重PCR的大量引物可以螯合大量镁(引物中2份磷酸盐螯合1份镁)。举例来说,如果使用足够的引物使得来自引物的磷酸盐的浓度是约9mM,那么引物可以使有效镁浓度降低约4.5mM。在一些实施例中,使用EDTA降低可以用作聚合酶的辅因子的镁的量,因为高浓度的镁可以引起PCR误差,如非目标基因座的扩增。在一些实施例中,EDTA的浓度使可用的镁的量降低至1到5mM(如3到5mM)。
在一些实施例中,pH值在7.5与8.5之间,如在7.5与8之间、在8与8.3之间或在8.3与8.5之间,且包括端值。在一些实施例中,Tris是以例如10与100mM之间,如10与25mM之间、25与50mM之间、50与75mM之间或25与75mM之间且包括端值的浓度使用。在一些实施例中,Tris的这些浓度中的任一种是在7.5与8.5之间的pH值下使用。在一些实施例中,使用KCl与(NH
在一些实施例中,使用拥挤试剂,如聚乙二醇(PEG,如PEG 8,000)或甘油。在一些实施例中,PEG(如PEG 8,000)的量在0.1到20%之间,如在0.5到15%、1到10%、2到8%或4到8%之间且包括端值。在一些实施例中,甘油的量在0.1到20%之间,如在0.5到15%、1到10%、2到8%或4到8%之间且包括端值。在一些实施例中,拥挤试剂使得能够使用低聚合酶浓度和/或较短粘接时间。在一些实施例中,拥挤试剂改善DOR的均匀性和/或减少脱扣(未检测到的等位基因)。聚合酶在一些实施例中,使用具有矫正活性(proof-readingactivity)的聚合酶、不具有(或具有可忽略的)矫正活性的聚合酶或具有矫正活性的聚合酶与不具有(或具有可忽略的)矫正活性的聚合酶的混合物。在一些实施例中,使用热起始聚合酶、非热起始聚合酶或热起始聚合酶与非热起始聚合酶的混合物。在一些实施例中,使用HotStarTaq DNA聚合酶(参见例如QIAGEN目录号203203)。在一些实施例中,使用AmpliTaq
在一些实施例中,使用5到600个单位/毫升(每1mL反应体积的单位数)的聚合酶,如5到100、100到200、200到300、300到400、400到500或500到600个单位/毫升且包括端值。
PCR方法
在一些实施例中,使用热起始PCR以减少或防止PCR热循环之前的聚合。例示性热起始PCR方法包括初始抑制DNA聚合酶,或物理分离反应组分反应直到反应混合物达到较高温度。在一些实施例中,使用缓慢释放的镁。DNA聚合酶需要镁离子以具有活性,因此通过结合于化合物来从反应物以化学方式分离镁且仅在高温下释放到溶液中。在一些实施例中,使用抑制剂的非共价结合。在这种方法中,肽、抗体或适配体在低温下非共价结合于酶且抑制其活性。在高温下培育之后,释放抑制剂且开始反应。在一些实施例中,使用低温敏感性Taq聚合酶,如在低温下几乎无活性的被修饰的DNA聚合酶。在一些实施例中,使用化学修饰。在这种方法中,分子共价结合于DNA聚合酶的活性位点中的胺基酸的侧链。通过在高温下培育反应混合物来从酶释放分子。在释放分子之后,酶被活化。
在一些实施例中,针对模板核酸(如RNA或DNA样品)的量在20与5,000ng之间,如在20到200、200到400、400到600、600到1,000、1,000到1,500或2,000到3,000ng之间且包括端值。
在一些实施例中,使用QIAGEN Multiplex PCR试剂盒(QIAGEN目录号206143)。对于100×50μl多重PCR反应,试剂盒包括2x QIAGEN多重PCR主混合物(提供3mM MgCl
在一些实施例中,以20μl最终体积使用1x QIAGEN MM最终浓度(建议浓度)、7.5nM库中的每种引物、50mM TMAC和7μl DNA模板。在一些实施例中,PCR热循环条件包括在95℃下保持10分钟(热起始);20个在96℃下保持30秒的循环;在65℃下保持15分钟;和在72℃下保持30秒;接着在72℃下保持2分钟(最终延伸);和接着保持在4℃下。
在一些实施例中,以20μl总体积使用2x QIAGEN MM最终浓度(两倍建议浓度)、2nM库中的每种引物、70mM TMAC和7μl DNA模板。在一些实施例中,还包括最多4mM EDTA。在一些实施例中,PCR热循环条件包括在95℃下保持10分钟(热起始);25个在96℃下保持30秒的循环;在65℃下保持20、25、30、45、60、120或180分钟;和任选地在72℃下保持30秒;接着在72℃下保持2分钟(最终延伸);和接着保持在4℃下。
另一例示性条件集合包括半嵌套式PCR方法。第一PCR反应使用20μl反应体积以及2x QIAGEN MM最终浓度、1.875nM库中的每种引物(外部正向和反向引物)和DNA模板。热循环参数包括在95℃下保持10分钟;25个在96℃下保持30秒的循环、在65℃下保持1分钟、在58℃下保持6分钟、在60℃下保持8分钟、在65℃下保持4分钟和在72℃下保持30秒;和接着在72℃下保持2分钟,且接着保持在4℃下。接着,使用2μl所得产物(以1:200稀释)作为第二PCR反应的输入物。这一反应使用10μl反应体积以及1x QIAGEN MM最终浓度、20nM每种内部正向引物和1μM反向引物标签。热循环参数包括在95℃下保持10分钟;15个在95℃下保持30秒的循环、在65℃下保持1分钟、在60℃下保持5分钟、在65℃下保持5分钟和在72℃下保持30秒;和接着在72℃下保持2分钟,且接着保持在4℃下。如本文中所讨论,粘接温度可以任选地高于一些或全部引物的熔融温度(参见2015年10月20日提交的美国专利申请案第14/918,544号,其以全文引用的方式并入本文中)。
熔融温度(Tm)是满足以下条件的温度:寡核苷酸(如引物)和其完美互补物的二分之一(50%)的DNA双螺旋解离且变成单链DNA。粘接温度(T
在各种实施例中,粘接温度在1、2、3、4、5、6、7、8、9、10、11、12、13℃与作为范围的上端的2、3、4、5、6、7、8、9、10、11、12、13、或15℃之间,高于至少25、50、60、70、75、80、90、95或100%的非一致引物的熔融温度(如凭经验测量或计算的T
例示性多重PCR方法
在各种实施例中,使用长粘接时间(如本文中所讨论和实例10中所例示)和/或低引物浓度。实际上,在某些实施例中,使用限制性引物浓度和/或条件。在各种实施例中,粘接步骤的长度在作为范围的下端的15、20、25、30、35、40、45或60分钟与作为范围的上端的20、25、30、35、40、45、60、120或180分钟之间。在各种实施例中,粘接步骤的长度(每个PCR循环)在30与180分钟之间。举例来说,粘接步骤可以在30与60分钟之间且每种引物的浓度可以小于20、15、10或5nM。在其它实施例中,引物浓度是作为范围的下端的1、2、3、4、5、6、7、8、9、10、15、20或25nM到作为范围的上端的2、3、4、5、6、7、8、9、10、15、20、25和50。
在高度复用的情况下,溶液可能因为溶液中的大量引物而变得粘稠。如果溶液太粘稠,那么可以将引物浓度降低到仍足以使引物结合模板DNA的量。在各种实施例中,使用1,000到100,000个不同的引物且每种引物的浓度小于20nM,如小于10nM或在1与10nM之间且包括端值。
检测拷贝数目变化(CNV)
除SNV和插入缺失以外,本文中所描述的用于监测和检测早期复发和转移的方法也可以受益于CNV的检测。
在一个方面中,本发明通常至少部分涉及改善的用于确定存在或不存在拷贝数目变化(如染色体区段或整个染色体的缺失或复制)的方法。所述方法尤其适用于检测小型缺失或复制,所述小型缺失或复制由于可以从相关染色体区段获得的数据量较小而难以使用先前方法在高特异性和敏感性下检测。所述方法包括改善的分析方法、改善的生物分析方法以及改善的分析方法和生物分析方法的组合。本发明的方法还可以用于检测仅存在于较小百分比的所检验的细胞或核酸分子中的缺失或复制。这使得能够在疾病发生之前(如在癌变前阶段)或在疾病早期(如在具有缺失或复制的大量病变细胞(如癌细胞)积聚之前)检测到缺失或复制。与疾病或病症相关联的缺失或复制的更精确的检测实现改善的用于诊断、预测、预防、延缓、稳定或治疗疾病或病症的方法。已知若干种缺失或复制与癌症或严重的精神或生理障碍相关联。
在另一方面中,本发明通常至少部分涉及改善的用于检测单核苷酸变异(SNV)的方法。这些改善的方法包括改善的分析方法、改善的生物分析方法以及改善的使用改善的分析方法和生物分析方法的组合的方法。在某些说明性实施例中,使用所述方法检测、诊断、监测癌症或对癌症进行分期,例如在SNV以极低浓度(例如以SNV基因座的正常拷贝总数计小于10%、5%、4%、3%、2.5%、2%、1%、0.5%、0.25%或0.1%)存在的样品中,如在循环游离DNA样品中。也就是说,在某些说明性实施例中,这些方法尤其良好地适用于与所述基因座的正常多态等位基因相比,存在相对较低百分比的突变或变异体的样品。最终,本文中提供组合改善的用于检测拷贝数目变化的方法与改善的用于检测单核苷酸变异的方法的方法。
疾病(如癌症)的成功治疗通常依赖于早期诊断、对疾病的正确分期、选择有效治疗方案和密切监测以防止或检测复发。对于癌症诊断,从组织活检获得的肿瘤材料的组织学评估通常被视为最可靠的方法。然而,基于活检的取样的侵袭性使得其不可用于群体筛选和常规随访。因此,本发明的方法具有以下优点:其能够视需要以非侵袭方式进行,从而具有相对较低成本和快速周转时间。可以由本发明的方法使用的靶向测序与鸟枪法测序相比需要更少的读段,如数百万读段而非4千万读段,从而降低成本。可以使用的多重PCR和下一代测序可以增加输送量和降低成本。
在一些例示性实施例中,ctDNA中AAI模式的分析提供对肿瘤的克隆体系的更详细的洞察,以帮助预测其治疗反应和优化治疗策略。因此,在某些实施例中,选择靶向临床上可操作的CNV和SNV的mmPCR-NGS盘。在某些说明性实施例中,这类盘尤其适用于患有其中CNV占显著比例的突变负荷(如通常在乳癌、卵巢癌和肺癌中)的癌症的患者。
在一些实施例中,使用所述方法检测个体中的缺失、复制或单核苷酸变异体。可以分析来自个体的样品,所述样品含有怀疑具有缺失、复制或单核苷酸变异体的细胞或核酸。在一些实施例中,样品是来自怀疑具有缺失、复制或单核苷酸变异体的组织或器官,如怀疑具有癌性的细胞或块状物。本发明的方法可以用于检测仅存在于混合物中的一个细胞或少量细胞中的缺失、复制或单核苷酸变异体,所述混合物含有具有缺失、复制或单核苷酸变异体的细胞和不具有缺失、复制或单核苷酸变异体的细胞。在一些实施例中,分析来自个体的血液样品中的cfDNA或cfRNA。在一些实施例中,cfDNA或cfRNA是由细胞,如癌细胞分泌。在一些实施例中,cfDNA或cfRNA是由经历坏死或细胞凋亡的细胞,如癌细胞释放。本发明的方法可以用于检测仅存在于较小百分比的cfDNA或cfRNA中的缺失、复制或单核苷酸变异体。在一些实施例中,检验来自胚胎的一种或多种细胞。
除确定存在或不存在拷贝数目变化以外,可以视需要分析一种或多种其它因素。这些因素可以用于提高诊断(如确定存在或不存在癌症或增加的癌症风险、对癌症进行分类或对癌症进行分期)或预后的准确性。这些因素还可以用于选择可能在个体中有效的具体疗法或治疗方案。例示性因素包括存在或不存在多态现象或突变;全部或具体cfDNA、cfRNA、微RNA(miRNA)的含量改变(增加或降低);肿瘤分数改变(增加或降低);甲基化水平改变(增加或降低)、DNA完整性改变(增加或降低)、改变(增加或降低)的或替代性mRNA剪接。
以下章节描述用于使用定相数据(如推断或测量的定相数据)或非定相数据检测缺失或复制的方法;可以检验的样品;用于样品制备、扩增和定量的方法;用于定相基因数据的方法;可以检测的多态现象、突变、核酸变化、mRNA剪接变化和核酸含量变化;具有来自所述方法、其它风险因素和筛选方法的结果的数据库;可以诊断或治疗的癌症;癌症治疗;用于检验治疗的癌症模型;和用于制定和给予治疗的方法。
用于使用定相数据确定倍性的例示性方法
本发明的一些方法是部分地基于发现与使用非定相数据相比,使用定相数据检测CNV可以降低假阴性和假阳性比率。这种改良对于具有少量CNV的样品来说是最大的。因此,与使用非定相数据相比,定相数据增加CNV检测的准确性(如以下方法:计算一个或多个基因座处的等位基因比率或聚集等位基因比率,以得到染色体或染色体区段上的聚集值(如平均值),而不考虑不同基因座处的等位基因比率是否指示相同或不同单倍型似乎以异常量存在)。使用定相数据能够作出所测量的与预期的等位基因比率之间的差异是否是由噪声或存在CNV而引起的更精确的确定。举例来说,如果一个区域中的大部分或所有基因座处的所测量的与预期的等位基因比率之间的差异指示相同单倍型被过度呈现,那么更可能存在CNV。使用单倍型中等位基因之间的关联,使得能够确定所测量的基因数据是否与被过度呈现的相同单倍型(而非随机噪声)一致。相比之下,如果所测量的与预期的等位基因比率之间的差异是仅由于噪声(如实验误差)而引起,那么在一些实施例中,在约一半的时间内,第一单倍型似乎被过度呈现且在约另一半的时间内,第二单倍型似乎被过度呈现。
在一些实施例中,使用定相基因数据确定在个体的基因组中(如在一种或多种细胞的基因组中或在cfDNA或cfRNA中),与第二同源染色体区段相比,是否存在第一同源染色体区段的拷贝数目的过度呈现。例示性过度呈现包括第一同源染色体区段的复制或第二同源染色体区段的缺失。在一些实施例中,不存在过度呈现,因为第一和同源染色体区段是以相等比例存在(如二倍体样品中每个区段的一个拷贝)。在一些实施例中,比较核酸样品中的所计算的等位基因比率与预期的等位基因比率,以确定是否存在过度呈现,如下文中进一步描述。在本说明书中,短语“与第二同源染色体区段相比的第一同源染色体区段”意指染色体区段的第一同系物和染色体区段的第二同系物。
在一些实施例中,所述方法包括获得第一同源染色体区段的定相基因数据,其包含第一同源染色体区段上的多态基因座集合中的每个基因座的第一同源染色体区段上的所述基因座处的等位基因的一致性;获得第二同源染色体区段的定相基因数据,其包含第二同源染色体区段上的多态基因座集合中的每个基因座的第二同源染色体区段上的所述基因座处的等位基因的一致性;和获得所测量的遗传等位基因数据,对于多态基因座集合中的每个基因座处的每个等位基因,所述遗传等位基因数据包含来自个体的一种或多种目标细胞和一种或多种非目标细胞的DNA或RNA的样品中的每种等位基因的量。在一些实施例中,所述方法包括列举指定第一同源染色体区段的过度呈现程度的一种或多种假设的集合;对于每种假设,针对来自一种或多种目标细胞的DNA或RNA与样品中的全部DNA或RNA的一种或多种可能的比率,由所获得的定相基因数据计算样品中的多个基因座的预期基因数据;针对DNA或RNA的每种可能的比率和每种假设,计算(如用计算机计算)所述DNA或RNA的可能的比率和所述假设的样品的所获得的基因数据与样品的所预期的基因数据之间的数据拟合;根据数据拟合将一种或多种假设进行分级;和选择等级最高的假设,由此确定来自个体的一种或多种细胞的基因组中的第一同源染色体区段的拷贝数目的过度呈现程度。
在一些实施例中,所述方法涉及使用本文中所描述的任一种方法或任何已知的方法获得定相基因数据。在一些实施例中,所述方法涉及同时或以任何顺序依序进行(i)获得第一同源染色体区段的定相基因数据,其包含第一同源染色体区段上的多态基因座集合中的每个基因座的第一同源染色体区段上的所述基因座处的等位基因的一致性;(ii)获得第二同源染色体区段的定相基因数据,其包含第二同源染色体区段上的多态基因座集合中的每个基因座的第二同源染色体区段上的所述基因座处的等位基因的一致性;和(iii)获得所测量的遗传等位基因数据,其包含来自个体的一种或多种细胞的DNA样品中的多态基因座集合中的每个基因座处的每种等位基因的量。
在一些实施例中,所述方法涉及计算多态基因座集合中的一种或多种基因座的等位基因比率,所述多态基因座集合在至少一种衍生样品的细胞中是杂合的。在一些实施例中,具体基因座的所计算的等位基因比率是所述基因座的一种等位基因的所测量的数量除以所有等位基因的全部所测量的数量。在一些实施例中,具体基因座的所计算的等位基因比率是所述基因座的一种等位基因(如第一同源染色体区段上的等位基因)的所测量的数量除以一种或多种其它等位基因(如第二同源染色体区段上的等位基因)的所测量的数量。所计算的等位基因比率可以使用本文中所描述的任一种方法或任何标准方法(如本文中所描述的所计算的等位基因比率的任何数学变换)来计算。
在一些实施例中,所述方法涉及如果第一和第二同源染色体区段是以相等比例存在,那么通过比较基因座的一种或多种所计算的等位基因比率与所述基因座的所预期的等位基因比率来确定是否存在第一同源染色体区段的拷贝数目的过度呈现。在一些实施例中,所预期的等位基因比率假设基因座的可能的等位基因在是否存在方面具有相等的似然性。在其中具体基因座的所计算的等位基因比率是所述基因座的一种等位基因的所测量的数量除以所有等位基因的全部所测量的数量的一些实施例中,相应的所预期的等位基因比率是0.5(对于双等位基因座)或1/3(对于三等位基因座)。在一些实施例中,所有基因座的所预期的等位基因比率是相同的,如所有基因座的所预期的等位基因比率都是0.5。在一些实施例中,所预期的等位基因比率假设基因座的可能的等位基因在是否存在方面可以具有不同的似然性,如基于个体所属具体群体(如基于个体的世系的群体)中的每种等位基因的出现率的似然性。这类等位基因出现率是可以公开获得的(参见例如《单倍型图计划(HapMap Project)》;《Perlegen人类单倍型计划(Perlegen Human Haplotype Project)》;网址:ncbi.nlm.nih.gov/projects/SNP/;Sherry ST,Ward MH,Kholodov M等人,《dbSNP:基因变异的NCBI数据库(dbSNP:the NCBI database of genetic variation)》,《核酸研究(Nucleic Acids Res.)》,2001年1月1日;29(1):308-11,其各自以全文引用的方式并入本文中)。在一些实施例中,所预期的等位基因比率是具体个体的所预期的等位基因比率,所述具体个体正在检验指定第一同源染色体区段的过度呈现程度的具体假设。举例来说,可以基于来自个体(如来自不太可能具有缺失或复制的个体的样品,如非癌性样品)的定相或非定相基因数据或来自个体的一位或多位亲属的数据来确定具体个体的所预期的等位基因比率。
在一些实施例中,如果满足以下中的任一项,那么所计算的等位基因比率指示第一同源染色体区段的拷贝数目的过度呈现:(i)由第一同源染色体上的所述基因座处的等位基因的所测量的数量除以所述基因座的所有等位基因的全部所测量的数量而获得的等位基因比率大于所述基因座的所预期的等位基因比率,或(ii)由第二同源染色体上的所述基因座处的等位基因的所测量的数量除以所述基因座的所有等位基因的全部所测量的数量而获得的等位基因比率小于所述基因座的所预期的等位基因比率。在一些实施例中,仅在所计算的等位基因比率显著大于或小于所述基因座的预期比率时才认为其指示过度呈现。在一些实施例中,如果满足以下中的任一项,那么所计算的等位基因比率指示第一同源染色体区段的拷贝数目的过度呈现:(i)由第一同源染色体上的所述基因座处的等位基因的所测量的数量除以所述基因座的所有等位基因的全部所测量的数量而获得的等位基因比率小于或等于所述基因座的所预期的等位基因比率,或(ii)由第二同源染色体上的所述基因座处的等位基因的所测量的数量除以所述基因座的所有等位基因的全部所测量的数量而获得的等位基因比率大于或等于所述基因座的所预期的等位基因比率。在一些实施例中,忽略等于相应的预期比率的所计算的比率(因为其指示不存在过度呈现)。
在各种实施例中,使用以下方法中的一种或多种来比较一个或多个所计算的等位基因比率与相应的所预期的等位基因比率。在一些实施例中,确定具体基因座的所计算的等位基因比率是否高于或低于所预期的等位基因比率,而与差的量值无关。在一些实施例中,确定具体基因座的所计算的等位基因比率与所预期的等位基因比率之间的差的量值,而与所计算的等位基因比率是否高于或低于所预期的等位基因比率无关。在一些实施例中,确定具体基因座的所计算的等位基因比率是否高于或低于所预期的等位基因比率和差的量值。在一些实施例中,确定所计算的等位基因比率的平均值或加权平均值是否高于或低于所预期的等位基因比率的平均值或加权平均值,而与差的量值无关。在一些实施例中,确定所计算的等位基因比率的平均值或加权平均值与所预期的等位基因比率的平均值或加权平均值之间的差的量值,而与所计算的等位基因比率的平均值或加权平均值是否高于或低于所预期的等位基因比率的平均值或加权平均值无关。在一些实施例中,确定所计算的等位基因比率的平均值或加权平均值是否高于或低于所预期的等位基因比率的平均值或加权平均值和差的量值。在一些实施例中,确定所计算的等位基因比率与所预期的等位基因比率之间的差的量值的平均值或加权平均值。
在一些实施例中,使用一种或多种基因座的所计算的等位基因比率和所预期的等位基因比率之间的差的量值,确定第一同源染色体区段的拷贝数目的过度呈现是否是由一种或多种细胞的基因组中的第一同源染色体区段的复制或第二同源染色体区段的缺失而引起。
在一些实施例中,如果满足以下条件中的一种或多种,那么确定存在第一同源染色体区段的拷贝数目的过度呈现。在一些实施例中,指示第一同源染色体区段的拷贝数目的过度呈现的所计算的等位基因比率的数值高于阈值。在一些实施例中,指示不存在第一同源染色体区段的拷贝数目的过度呈现的所计算的等位基因比率的数值低于阈值。在一些实施例中,指示第一同源染色体区段的拷贝数目的过度呈现的所计算的等位基因比率与相应的所预期的等位基因比率之间的差的量值高于阈值。在一些实施例中,对于指示过度呈现的所有所计算的等位基因比率,所计算的等位基因比率与相应的所预期的等位基因比率之间的差的量值的总和高于阈值。在一些实施例中,指示不存在第一同源染色体区段的拷贝数目的过度呈现的所计算的等位基因比率与相应的所预期的等位基因比率之间的差的量值低于阈值。在一些实施例中,由第一同源染色体上的等位基因的所测量的数量除以所述基因座的所有等位基因的全部所测量的数量而获得的所计算的等位基因比率的平均值或加权平均值比所预期的等位基因比率的平均值或加权平均值大至少一倍阈值。在一些实施例中,由第二同源染色体上的等位基因的所测量的数量除以所述基因座的所有等位基因的全部所测量的数量而获得的所计算的等位基因比率的平均值或加权平均值比所预期的等位基因比率的平均值或加权平均值小至少一倍阈值。在一些实施例中,所计算的等位基因比率与预测存在第一同源染色体区段的拷贝数目的过度呈现的等位基因比率之间的数据拟合低于阈值(指示良好数据拟合)。在一些实施例中,所计算的等位基因比率与预测不存在第一同源染色体区段的拷贝数目的过度呈现的等位基因比率之间的数据拟合高于阈值(指示不良数据拟合)。
在一些实施例中,如果满足以下条件中的一种或多种,那么确定不存在第一同源染色体区段的拷贝数目的过度呈现。在一些实施例中,指示第一同源染色体区段的拷贝数目的过度呈现的所计算的等位基因比率的数值低于阈值。在一些实施例中,指示不存在第一同源染色体区段的拷贝数目的过度呈现的所计算的等位基因比率的数值高于阈值。在一些实施例中,指示第一同源染色体区段的拷贝数目的过度呈现的所计算的等位基因比率与相应的所预期的等位基因比率之间的差的量值低于阈值。在一些实施例中,指示不存在第一同源染色体区段的拷贝数目的过度呈现的所计算的等位基因比率与相应的所预期的等位基因比率之间的差的量值高于阈值。在一些实施例中,由第一同源染色体上的等位基因的所测量的数量除以所述基因座的所有等位基因的全部所测量的数量而获得的所计算的等位基因比率的平均值或加权平均值减去所预期的等位基因比率的平均值或加权平均值的结果小于阈值。在一些实施例中,所预期的等位基因比率的平均值或加权平均值减去由第二同源染色体上的等位基因的所测量的数量除以所述基因座的所有等位基因的全部所测量的数量而获得的所计算的等位基因比率的平均值或加权平均值的结果小于阈值。在一些实施例中,所计算的等位基因比率与预测存在第一同源染色体区段的拷贝数目的过度呈现的等位基因比率之间的数据拟合高于阈值。在一些实施例中,所计算的等位基因比率与预测不存在第一同源染色体区段的拷贝数目的过度呈现的等位基因比率之间的数据拟合低于阈值。在一些实施例中,由已知具有相关CNV的样品和/或已知不具有CNV的样品的经验检验确定阈值。
在一些实施例中,确定是否存在第一同源染色体区段的拷贝数目的过度呈现包括列举指定第一同源染色体区段的过度呈现程度的一种或多种假设的集合。例示性假设是不存在过度呈现,因为第一和同源染色体区段是以相比比例存在(如二倍体样品中的每个区段的一个拷贝)。其它例示性假设包括第一同源染色体区段被复制一次或多次(如与第二同源染色体区段的拷贝数目相比,第一同源染色体具有1、2、3、4、5个或更多的额外拷贝)。另一种例示性假设包括第二同源染色体区段的缺失。另一种例示性假设是第一和第二同源染色体区段的缺失。在一些实施例中,针对每种假设,鉴于由所述假设指定的过度呈现程度,估计在至少一种细胞中是杂合的基因座的所预测的等位基因比率。在一些实施例中,通过比较所计算的等位基因比率与所预测的等位基因比率来计算表示假设正确的似然性,且选择具有最大似然性的假设。
在一些实施例中,针对每种假设,使用所预测的等位基因比率计算检验统计值的所预期的分布。在一些实施例中,通过比较使用所计算的等位基因比率计算的检验统计值与使用所预测的等位基因比率计算的检验统计值的所预期的分布来计算表示假设正确的似然性,且选择具有最大似然性的假设。
在一些实施例中,鉴于第一同源染色体区段的定相基因数据、第二同源染色体区段的定相基因数据和由假设指定的过度呈现程度,估计在至少一种细胞中是杂合的基因座的所预测的等位基因比率。在一些实施例中,通过比较所计算的等位基因比率与所预测的等位基因比率来计算表示假设正确的似然性;且选择具有最大似然性的假设。
使用混合样品
应理解,在许多实施例中,样品是混合样品,其具有来自一种或多种目标细胞和一种或多种非目标细胞的DNA或RNA。在一些实施例中,目标细胞是具有CNV(如相关缺失或复制)的细胞,且非目标细胞是不具有相关拷贝数目变化的细胞(如具有相关缺失或复制的细胞与不具有任何所检验的缺失或复制的细胞的混合物)。在一些实施例中,目标细胞是与疾病或病症或增加的疾病或病症风险相关联的细胞(如癌细胞),且非目标细胞是不与疾病或病症或增加的疾病或病症风险相关联的细胞(如非癌性细胞)。在一些实施例中,目标细胞都具有相同的CNV。在一些实施例中,两种或更多种目标细胞具有不同的CNV。在一些实施例中,一种或多种目标细胞具有未在至少一种其它目标细胞中发现的与疾病或病症或增加的疾病或病症风险相关联的CNV、多态现象或突变。在一些这类实施例中,假设来自样品的全部细胞中的与疾病或病症或增加的疾病或病症风险相关联的细胞的分数大于或等于样品中这些CNV、多态现象或突变中的最频繁出现的CNV、多态现象或突变的分数。举例来说,如果6%的细胞具有K-ras突变且8%的细胞具有BRAF突变,那么假设至少8%的细胞是癌性的。
在一些实施例中,计算来自一种或多种目标细胞的DNA(或RNA)与样品中全部DNA(或RNA)的比率。在一些实施例中,列举指定第一同源染色体区段的过度呈现程度的一种或多种假设的集合。在一些实施例中,针对每种假设,鉴于DNA或RNA的所计算的比率和由所述假设指定的过度呈现程度,估计在至少一种细胞中是杂合的基因座的所预测的等位基因比率。在一些实施例中,通过比较所计算的等位基因比率与所预测的等位基因比率来计算表示假设正确的似然性,且选择具有最大似然性的假设。
在一些实施例中,针对每种假设,估计使用所预测的等位基因比率和DNA或RNA的所计算的比率计算的检验统计值的所预期的分布。在一些实施例中,通过比较使用所计算的等位基因比率和DNA或RNA的所计算的比率计算的检验统计值与使用所预测的等位基因比率和DNA或RNA的所计算的比率计算的检验统计值的所预期的分布来确定表示假设正确的似然性,且选择具有最大似然性的假设。
在一些实施例中,所述方法包括列举指定第一同源染色体区段的过度呈现程度的一种或多种假设的集合。在一些实施例中,所述方法包括针对每种假设,估计(i)鉴于由所述假设指定的过度呈现程度,在至少一种细胞中是杂合的基因座的所预测的等位基因比率,或(ii)对于DNA或RNA的一种或多种可能的比率,使用所预测的等位基因比率和来自一种或多种目标细胞的DNA或RNA与样品中全部DNA或RNA的可能的比率计算的检验统计值的所预期的分布。在一些实施例中,通过比较以下来计算数据拟合:(i)所计算的等位基因比率与所预测的等位基因比率,或(ii)使用所计算的等位基因比率和DNA或RNA的可能的比率计算的检验统计值与使用所预测的等位基因比率和DNA或RNA的可能的比率计算的检验统计值的所预期的分布。在一些实施例中,根据数据拟合对一种或多种假设进行分级,且选择等级最高的假设。在一些实施例中,使用技术或算法(如搜索算法)进行以下步骤中的一个或多个:计算数据拟合、对假设进行分级或选择等级最高的假设。在一些实施例中,数据拟合是针对β-二项分布的拟合或针对二项分布的拟合。在一些实施例中,技术或算法是选自由以下组成的群组:最大似然估计、最大后验估计、贝叶斯估计(Bayesian estimation)、动态估计(如动态贝叶斯估计)和最大期望估计。在一些实施例中,所述方法包括对所获得的基因数据和所预期的基因数据应用技术或算法。
在一些实施例中,所述方法包括创建可能的比率的划分,其在来自一种或多种目标细胞的DNA或RNA与样品中全部DNA或RNA的比率的下限到上限的范围内。在一些实施例中,列举指定第一同源染色体区段的过度呈现程度的一种或多种假设的集合。在一些实施例中,所述方法包括针对划分中的DNA或RNA的每种可能的比率和每种假设,估计(i)鉴于DNA或RNA的可能的比率和由所述假设指定的过度呈现程度,在至少一种细胞中是杂合的基因座的所预测的等位基因比率,或(ii)使用所预测的等位基因比率和DNA或RNA的可能的比率计算的检验统计值的所预期的分布。在一些实施例中,所述方法包括针对划分中的DNA或RNA的每种可能的比率和每种假设,通过比较以下来计算表示假设正确的似然性:(i)所计算的等位基因比率与所预测的等位基因比率,或(ii)使用所计算的等位基因比率和DNA或RNA的可能的比率计算的检验统计值与使用所预测的等位基因比率和DNA或RNA的可能比率计算的检验统计值的所预期的分布。在一些实施例中,对于每种假设,通过组合划分中的每种可能的比率的假设的概率来确定组合概率;且选择具有最大组合概率的假设。在一些实施例中,基于可能的比率是正确比率的似然性,通过将具体可能的比率的假设的概率加权来确定每种假设的组合概率。
在一些实施例中,使用选自由以下组成的群组的技术来估计来自一种或多种目标细胞的DNA或RNA与样品中全部DNA或RNA的比率:最大似然估计、最大后验估计、贝叶斯估计、动态估计(如动态贝叶斯估计)和最大期望估计。在一些实施例中,假设两种或更多种(或所有)相关CNV的来自一种或多种目标细胞的DNA或RNA与样品中全部DNA或RNA的比率是相同的。在一些实施例中,计算每种相关CNV的来自一种或多种目标细胞的DNA或RNA与样品中全部DNA或RNA的比率。
使用不完美定相数据的例示性方法
应理解,在许多实施例中,使用不完美定相数据。举例来说,对于第一和/或第二同源染色体区段上的一个或多个基因座,可能不是在100%确定性的情况下已知存在哪些等位基因。在一些实施例中,使用个体的可能的单倍型(如以基于群体的单倍型出现率为基础的单倍型)的先验来计算每种假设的概率。在一些实施例中,通过使用另一种方法对基因数据进行定相或通过使用来自其它个体(如先验个体)的定相数据以优化用于个体的基于信息的定相的群体数据来调节可能的单倍型的先验。
在一些实施例中,定相基因数据包含定相基因数据的两个或更多个可能的集合的概率数据,其中定相数据的每个可能的集合包含第一同源染色体区段上的多态基因座集合中的每个基因座处的等位基因的可能的一致性和第二同源染色体区段上的多态基因座集合中的每个基因座处的等位基因的可能的一致性。在一些实施例中,针对定相基因数据的每个可能的集合,确定至少一种假设的概率。在一些实施例中,通过组合定相基因数据的每个可能的集合的假设的概率来确定假设的组合概率;且选择具有最大组合概率的假设。
本文中所公开的任何方法或任何已知的可以用于产生不完美定相数据的方法(如使用基于群体的单倍型出现率以推断最有可能的相)都可以用于所要求的方法中。在一些实施例中,通过概率性地组合较小区段的单倍型来获得定相数据。举例来说,可以基于来自第一区域的一个单倍型与来自相同染色体的另一区域的另一单倍型的可能的组合来确定可能的单倍型。可以使用例如基于群体的单倍型出现率和/或不同区域之间的已知的重组率来确定来自不同区域的具体单倍型是相同染色体上的相同、较大单倍型域(haplotypeblock)的一部分的概率。
在一些实施例中,单一假设拒绝检验用于二体性的零假设。在一些实施例中,计算二体性假设的概率,且如果概率低于既定阈值(如小于1/1,000),那么拒绝二体性的假设。如果拒绝零假设,那么这可以归因于不完美定相数据中的误差或归因于存在CNV。在一些实施例中,获得更精确的定相数据(如来自本文中所公开的任何用于获得实际定相数据而非基于生物信息学推断的定相数据的分子定相方法的定相数据)。在一些实施例中,使用更精确的定相数据重新计算二体性假设的概率,以确定是否仍应拒绝二体性假设。拒绝此假设表示存在染色体区段的复制或缺失。视需要,可以通过调节阈值来改变假阳性率。
使用定相数据来确定倍性的其它例示性实施例
在说明性实施例中,本文中提供用于确定个体的样品中的染色体区段的倍性的方法。所述方法包括以下步骤:接收等位基因出现率数据,其包含染色体区段上的多态基因座集合中的每个基因座处的样品中的每种等位基因的量;通过估计等位基因出现率数据的相来产生多态基因座集合的定相等位基因信息;使用等位基因出现率数据,产生不同倍性状态的多态基因座的等位基因出现率的单独概率;使用单独概率和定相等位基因信息产生多态基因座集合的联合概率;和基于联合概率,选择指示染色体倍性的最佳拟合模型,由此确定染色体区段的倍性。
如本文中所公开,可以通过所属领域中已知的方法产生等位基因出现率数据(在本文中也称为所测量的遗传等位基因数据)。举例来说,可以使用qPCR或微阵列产生数据。在一个说明性实施例中,使用核酸序列数据,尤其高通量核酸序列数据产生数据。
在某些说明性实例中,在用于产生单独概率之前,针对误差校正等位基因出现率数据。在特定说明性实施例中,所校正的误差包括等位基因扩增效率偏差。在其它实施例中,所校正的误差包括环境污染和基因型污染。在一些实施例中,所校正的误差包括等位基因扩增偏差、测序误差、环境污染和基因型污染。
在某些实施例中,使用多态基因座集合的不同倍性状态和等位基因失衡分数的模型的集合来产生单独概率。在这些实施例和其它实施例中,通过考虑染色体区段上的多态基因座之间的关联性来产生联合概率。
因此,在组合这些实施例中的一些实施例的一个说明性实施例中,本文中提供用于检测个体的样品中的染色体倍性的方法,其包括以下步骤:接收个体中的染色体区段上的多态基因座集合处的等位基因的核酸序列数据;使用核酸序列数据检测基因座集合处的等位基因出现率;校正所检测的等位基因出现率中的等位基因扩增效率偏差以产生多态基因座集合的经校正的等位基因出现率;通过估计核酸序列数据的相来产生多态基因座集合的定相等位基因信息;通过比较经校正的等位基因出现率与多态基因座集合的不同倍性状态和等位基因失衡分数的模型的集合来产生不同倍性状态的多态基因座的等位基因出现率的单独概率;考虑染色体区段上的多态基因座之间的关联性,通过组合单独概率来产生多态基因座集合的联合概率;和基于联合概率,选择指示染色体非整倍性的最佳拟合模型。
如本文中所公开,可以使用多态基因座集合的不同倍性状态和平均等位基因失衡分数的模型或假设的集合来产生单独概率。举例来说,在具体说明性实例中,通过模型化染色体区段的第一同系物和染色体区段的第二同系物的倍性状态来产生单独概率。模型化的倍性状态包括以下:(1)所有细胞不具有染色体区段的第一同系物或第二同系物的缺失或扩增;(2)至少一些细胞具有染色体区段的第一同系物的缺失或第二同系物的扩增;和(3)至少一些细胞具有染色体区段的第二同系物的缺失或第一同系物的扩增。
应理解,以上模型也可以称为用于约束模型的假设。因此,以上说明3种可以使用的假设。
模型化的平均等位基因失衡分数可以包括平均等位基因失衡的任何包括染色体区段的实际平均等位基因失衡的范围。举例来说,在某些说明性实施例中,模型化的平均等位基因失衡的范围可以在作为下端的0、0.1、0.2、0.25、0.3、0.4、0.5、0.6、0.75、1、2、2.5、3、4和5%与作为上端的1、2、2.5、3、4、5、10、15、20、25、30、40、50、60、70、80、90、95和99%之间。用于在所述范围下的模型化的间隔可以是任何取决于所使用的计算能力和允许用于分析的时间的间隔。举例来说,可以模型化0.01、0.05、0.02或0.1间隔。
在某些说明性实施例中,样品的染色体区段的平均等位基因失衡在0.4%与5%之间。在某些实施例中,平均等位基因失衡较低。在这些实施例中,平均等位基因失衡通常小于10%。在某些说明性实施例中,等位基因失衡在作为下端的0.25、0.3、0.4、0.5、0.6、0.75、1、2、2.5、3、4和5%与作为上端的1、2、2.5、3、4和5%之间。在其它例示性实施例中,平均等位基因失衡在作为下端的0.4、0.45、0.5、0.6、0.7、0.8、0.9或1.0%与作为上端的0.5、0.6、0.7、0.8、0.9、1.0、1.5、2.0、3.0、4.0或5.0%之间。举例来说,在说明性实例中,样品的平均等位基因失衡在0.45与2.5%之间。在另一实例中,在0.45、0.5、0.6、0.8、0.8、0.9或1.0%的敏感性下检测平均等位基因失衡。也就是说,检验方法能够在AAI低到0.45、0.5、0.6、0.8、0.8、0.9或1.0%的情况下检测到染色体非整倍性。在本发明的方法中,具有低等位基因失衡的例示性样品包括来自患有具有循环肿瘤DNA的癌症的个体的血浆样品或来自具有循环胚胎DNA的怀孕女性的血浆样品。
应理解,对于SNV,通常使用突变体等位基因出现率(基因座处的突变体等位基因的数目/所述基因座处的等位基因的总数)测量异常DNA的比例。因为肿瘤中的两种同系物的量之间的差是类似的,我们通过平均等位基因失衡(AAI)来测量CNV的异常DNA的比例,定义为|(H1-H2)|/(H1+H2),其中Hi是样品中同系物i的拷贝的平均数且Hi/(H1+H2)是同系物i的部分丰度或同系物比率。最大同系物比率是丰度较高的同系物的同系物比率。
分析法脱扣率是使用所有SNP估计的不具有读段的SNP的百分比。单一等位基因脱扣(ADO)率是仅使用杂合SNP估计的仅存在一个等位基因的SNP的百分比。可以通过以下方式来确定基因型置信度:针对每个SNP处的B至等位基因读段的读段数目拟合二项分布且使用SNP的焦点区域的倍性状态估计每个基因型的概率。
对于肿瘤组织样品,可以由等位基因出现率分布之间的转换来描述染色体非整倍性(本段中由CNV例示)。在癌症患者、怀疑患有癌症的个体、先前诊断患有癌症的个体或作为用于具有风险的个体或一般群体的癌症筛检的血浆样品中,可以通过最大似然算法来鉴别CNV,所述最大似然算法搜索已知在癌症中呈现非整倍性的区域和/或来自相同个体的肿瘤样品也具有CNV的位置中的血浆CNV。在说明性实施例中,算法使用个体的单倍型相信息针对所预期的等位基因计数来拟合所测量的和经校正的检验样品等位基因计数,例如使用联合分布模式,其中正在分析所述个体的样品中是否存在循环肿瘤DNA。这类单倍型相信息可以由来自个体的包括大部分或至少60、70、80、90、95、96、97、98、99%或所有正常细胞DNA的任何样品(如(但不限于)白细胞层样品、唾液样品或皮肤样品),由亲本基因型信息推导,或通过重新单倍型定相来推导,所述重新单倍型定相可以通过多种方法来实现(参见例如Snyder,M.等人,《单倍型解析基因组测序:实验方法和应用(Haplotype-resolved genomesequencing:experimental methods and applications)》,《遗传学自然综述(Nat RevGenet)》16,344-358(2015)),如通过稀释(Kaper,F.等人,《通过稀释、扩增和测序进行的全基因组单倍型分析(Whole-genomehaplotyping by dilution,amplification,andsequencing)》,《美国国家科学院院刊》110,5552-5557(2013))或长读段测序(Kuleshov,V.等人,《使用长读段和统计方法进行的全基因组单倍型分析(Whole-genome haplotypingusing long reads and statistical methods)》,《自然生物技术(Nat Biotech)》32,261-266(2014))进行的单倍型分析。这种算法可以模型化三个假设集合的在0.025%间隔下,在所有等位基因失衡比率下的所预期的等位基因出现率:(1)所有细胞都是正常的(不存在等位基因失衡),(2)一些/所有细胞具有同系物1缺失或同系物2扩增,或(3)一些/所有细胞具有同系物2缺失或同系物1扩增。可以使用贝叶斯分类器(Bayesian classifier),基于所有杂合SNP处的所预期的和所观察的等位基因出现率的β二项模型来确定每种假设的似然性,且接着可以计算多个SNP的联合似然性,在某些说明性实施例中,考虑SNP基因座的关联性,如本文中所例示。实际上,在说明性实施例中,由算法使用如上文所公开获得的正常细胞单倍型相信息以使用联合分布模型,针对所预期的等位基因计数拟合所测量的和经典型校正的检验样品等位基因计数。接着,可以所选最大似然假设。
考虑肿瘤中具有平均N个拷贝的染色体区域且假设c表示来源于二体区域中的正常细胞和肿瘤细胞的混合物的血浆中的DNA的分数。AAI计算为:
在某些说明性实例中,在用于产生单独概率之前,针对误差校正等位基因出现率数据。本文中公开不同类型的误差和/或偏差校正。在特定说明性实施例中,所校正的误差是等位基因扩增效率偏差。在其它实施例中,所校正的误差包括测序误差、环境污染和基因型污染。在一些实施例中,所校正的误差包括等位基因扩增偏差、测序误差、环境污染和基因型污染。
应理解,可以确定等位基因的等位基因扩增效率偏差作为包括检验中样品的实验或实验室确定的一部分,或其可以在不同时间使用包括等位基因的样品集合确定,其中正在计算所述等位基因的效率。通常与检验中样品分析在同一次操作中确定环境污染和基因污染。
在某些实施例中,确定样品中的纯合等位基因的环境污染和基因污染。应理解,对于任何来自个体的既定样品,即使一个基因座由于其在群体中具有相对高杂合性而被选择用于分析,但样品中的一些基因座将是杂合的且其它基因座将是纯合的。在一些实施例中,宜使用个体的杂合基因座确定染色体区段的倍性,而可以使用纯合基因座计算环境和基因型污染。
在某些说明性实例中,通过分析模型的所产生的定相等位基因信息与所估计的等位基因出现率之间的差的量值来进行选择。
在说明性实例中,基于多态基因座集合的所预期的和所观察的等位基因出现率的β二项模型来产生等位基因出现率的单独概率。在说明性实例中,使用贝叶斯分类器产生单独概率。
在某些说明性实施例中,通过对使用多重扩增反应产生的一系列扩增子的多个拷贝进行高通量DNA测序来产生核酸序列数据,其中所述扩增子系列中的每个扩增子跨越多态基因座集合中的至少一个多态基因座且其中所述集合中的每个聚合基因座被扩增。在某些实施例中,对于至少1/2的反应物,多重扩增反应是在限制性引物条件下进行。在一些实施例中,在多重反应的1/10、1/5、1/4、1/3、1/2或所有的反应物中使用限制性引物浓度。本文中提供用于考虑在扩增反应(如PCR)中实现限制性引物条件的因素。
在某些实施例中,本文中所提供的方法检测跨越多个染色体的多个染色体区段的倍性。因此,在这些实施例中,确定样品中染色体区段集合的染色体倍性。在这些实施例中,需要多重性更高的扩增反应。因此,在这些实施例中,多重扩增反应可以包括例如2,500到50,000个多重反应。在某些实施例中,进行以下范围内的多重反应:作为范围的下端的100、200、250、500、1000、2500、5000、10,000、20,000、25000、50000与作为范围的上端的200、250、500、1000、2500、5000、10,000、20,000、25000、50000和100,000之间。
在说明性实施例中,多态基因座集合是已知呈现高杂合性的基因座集合。然而,预期对于任何既定个体,这些基因座中的一些将是纯合的。在某些说明性实施例中,本发明的方法利用个体的纯合和杂合基因座的核酸序列信息。举例来说,个体的纯合基因座用于误差校正,而杂合基因座用于确定样品的等位基因失衡。在某些实施例中,个体的至少10%的多态基因座是杂合基因座。
如本文中所公开,偏好于分析已知在群体中是杂合的目标SNP基因座。因此,在某些实施例中,选择已知其中至少10、20、25、50、75、80、90、95、99或100%的多态基因座在群体中是杂合的多态基因座。
如本文中所公开,在某些实施例中,样品是来自怀孕女性的血浆样品。
在一些实例中,所述方法进一步包含对具有已知的平均等位基因失衡比率的对照样品进行所述方法。对照物可以具有在0.4和10%之间的指示染色体区段的非整倍性的具体等位基因状态的平均等位基因失衡比率,以模拟以低浓度存在的样品中的等位基因的平均等位基因失衡,如关于来自肿瘤的循环游离DNA所预期。
在一些实施例中,如本文中所公开,使用PlasmArt对照物作为对照物。因此,在某些方面中,存在通过包含以下的方法产生的样品:使已知呈现染色体非整倍性的核酸样品片段化成模拟在个体的血浆中循环的DNA片段的尺寸的片段。在某些方面中,使用对于染色体区段不具有非整倍性的对照物。
在说明性实施例中,可以在方法中分析来自一种或多种对照物和检验样品的数据。举例来说,对照物可以包括来自个体的未怀疑含有染色体非整倍性的不同样品或怀疑含有CNV或染色体非整倍性的样品。举例来说,当检验样品是怀疑含有循环游离肿瘤DNA的血浆样品时,也可以与血浆样品一起对来自个体的肿瘤的对照样品进行所述方法。如本文中所公开,可以通过将已知呈现染色体非整倍性的DNA样品片段化来制备对照样品。这类片段化可以产生模拟凋亡细胞的DNA组合物的DNA样品,尤其当样品是来自罹患癌症的个体时。来自对照样品的数据将提高染色体非整倍性的检测的置信度。
在用于确定倍性的方法的某些实施例中,样品是来自怀疑患有癌症的个体的血浆样品。在这些实施例中,所述方法进一步包含基于所述选择来确定个体的肿瘤细胞中是否存在拷贝数目变化。在这些实施例中,样品可以是来自个体的血浆样品。在这些实施例中,方法可以进一步包括基于所述选择来确定个体中是否存在癌症。
这些用于确定染色体区段的倍性的实施例可以进一步包括检测单核苷酸变异位置集合中的单核苷酸变异位置处的单核苷酸变异体,其中检测到染色体非整倍性或单核苷酸变异体或这两者指示样品中存在循环肿瘤核酸。
这些实施例可以进一步包括接收个体的肿瘤的染色体区段的单倍型信息,和使用单倍型信息以产生多态基因座集合的不同倍性状态和等位基因失衡分数的模型集合。
如本文中所公开,用于确定倍性的方法的某些实施例可以进一步包括在比较初始或经校正的等位基因出现率与模型集合之前,从初始或经校正的等位基因出现率数据去除离群值。举例来说,在某些实施例中,在用于模型化之前,从数据去除比染色体区段上的其它基因座的平均值高或低至少2或3倍标准差的基因座等位基因出现率。
如本文中所提及应理解,在本文中所提供的许多实施例中,包括用于确定染色体区段的倍性的实施例,优选使用不完美或完美定相数据。还应理解,本文中提供多种特征,其与用于检测倍性的先前方法相比提供改善,且可以使用这些特征的多种不同组合。
在某些实施例中,本文中提供计算机系统和计算机可读介质以进行本发明的任何方法。这些计算机系统和计算机可读介质包括用于进行确定倍性的方法的系统和计算机可读介质。因此且作为用于说明本文中所提供的任何方法都可以使用利用本文中的公开内容的系统和计算机可读介质进行的系统实施例的非限制性实例,在另一方面中,本文中提供用于检测个体的样品中的染色体倍性的系统,所述系统包含:输入处理器,其被配置成接收等位基因出现率数据,所述等位基因出现率数据包含染色体区段上的多态基因座集合中的每个基因座处的样品中的每种等位基因的量;建模器,其被配置成:通过估计等位基因出现率数据的相来产生多态基因座集合的定相等位基因信息;和使用等位基因出现率数据产生不同倍性状态的多态基因座的等位基因出现率的单独概率;和使用单独概率和定相等位基因信息产生多态基因座集合的联合概率;以及假设管理器,其被配置成基于联合概率选择指示染色体倍性的最佳拟合模型,由此确定染色体区段的倍性。
在这一系统实施例的某些实施例中,等位基因出现率数据是由核酸测序系统产生的数据。在某些实施例中,所述系统进一步包含误差校正单元,其被配置成校正等位基因出现率数据中的误差,其中经校正的等位基因出现率数据由建模器用于产生单独概率。在某些实施例中,误差校正单元校正等位基因扩增效率偏差。.在某些实施例中,建模器使用多态基因座集合的不同倍性状态和等位基因失衡分数的模型的集合来产生单独概率。在某些例示性实施例中,建模器通过考虑染色体区段上的多态基因座之间的关联性来产生联合概率。
在一个说明性实施例中,本文中提供用于检测个体的样品中的染色体倍性的系统,其包括以下:输入处理器,其被配置成接收个体中的染色体区段上的多态基因座集合处的等位基因的核酸序列数据,和使用核酸序列数据检测基因座集合处的等位基因出现率;误差校正单元,其被配置成校正所检测的等位基因出现率中的误差和产生多态基因座集合的经校正的等位基因出现率;建模器,其被配置成:通过估计核酸序列数据的相来产生多态基因座集合的定相等位基因信息;通过比较定相等位基因信息与多态基因座集合的不同倍性状态和等位基因失衡分数的模型的集合来产生不同倍性状态的多态基因座的等位基因出现率的单独概率;和考虑染色体区段上的多态基因座之间的相对距离,通过组合单独概率来产生多态基因座集合的联合概率;以及假设管理器,其被配置成基于联合概率选择指示染色体非整倍性的最佳拟合模型。
在本文中所提供的某些例示性系统实施例中,多态基因座集合包含1000到50,000个多态基因座。在本文中所提供的某些例示性系统实施例中,多态基因座集合包含100个已知的杂合性热点基因座。在本文中所提供的某些例示性系统实施例中,多态基因座集合包含100个在重组热点的0.5kb处或以内的基因座。
在本文中所提供的某些例示性系统实施例中,最佳拟合模型分析染色体区段的第一同系物和染色体区段的第二同系物的以下倍性状态:(1)所有细胞都不具有染色体区段的第一同系物或第二同系物的缺失或扩增;(2)一些或所有细胞具有染色体区段的第一同系物的缺失或第二同系物的扩增;和(3)一些或所有细胞染色体区段的第二同系物的缺失或第一同系物的扩增。
在本文中所提供的某些例示性系统实施例中,所校正的误差包含等位基因扩增效率偏差、污染和/或测序误差。在本文中所提供的某些例示性系统实施例中,污染包含环境污染和基因型污染。在本文中所提供的某些例示性系统实施例中,确定纯合等位基因的环境污染和基因污染物。
在本文中所提供的某些例示性系统实施例中,假设管理器被配置成分析模型的所产生的定相等位基因信息与所估计的等位基因出现率之间的差的量值。在本文中所提供的某些例示性系统实施例中,建模器基于多态基因座集合处的所预期的和所观察的等位基因出现率的β二项模型来产生等位基因出现率的单独概率。在本文中所提供的某些例示性系统实施例中,建模器使用贝叶斯分类器产生单独概率。
在本文中所提供的某些例示性系统实施例中,通过对使用多重扩增反应产生的一系列扩增子的多个拷贝进行高通量DNA测序来产生核酸序列数据,其中所述扩增子系列中的每个扩增子跨越多态基因座集合中的至少一个多态基因座且其中集合中的每个聚合基因座被扩增。在本文中所提供的某些例示性系统实施例中,其中对于至少1/2的反应物,多重扩增反应是在限制性引物条件下进行。在本文中所提供的某些例示性系统实施例中,其中样品的平均等位基因失衡在0.4%与5%之间。
在本文中所提供的某些例示性系统实施例中,样品是来自怀疑患有癌症的个体的血浆样品,且假设管理器进一步被配置成基于最佳拟合模型来确定个体的肿瘤细胞中是否存在拷贝数目变化。
在本文中所提供的某些例示性系统实施例中,样品是来自个体的血浆样品且假设管理器进一步被配置成基于最佳拟合模型来确定个体中是否存在癌症。在这些实施例中,假设管理器可以进一步被配置成检测单核苷酸变异位置集合中的单核苷酸变异位置处的单核苷酸变异体,其中检测到染色体非整倍性或单核苷酸变异体或这两者指示样品中存在循环肿瘤核酸。
在本文中所提供的某些例示性系统实施例中,输入处理器进一步被配置成接收个体的肿瘤的染色体区段的单倍型信息,且建模器被配置成使用单倍型信息以产生多态基因座集合的不同倍性状态和等位基因失衡分数的模型的集合。
在本文中所提供的某些例示性系统实施例中,建模器产生在0%到25%范围内的等位基因失衡分数的模型。
应理解,本文中所提供的任何方法都可以由储存在非暂时性计算机可读介质上的计算机可读编码来执行。因此,在一个实施例中,本文中提供用于检测个体的样品中的染色体倍性的非暂时性计算机可读介质,其包含计算机可读代码,所述计算机可读代码在由处理装置执行时引起处理装置:接收等位基因出现率数据,其包含染色体区段上的多态基因座集合中的每个基因座处的样品中的每种等位基因的量;通过估计等位基因出现率数据的相来产生多态基因座集合的定相等位基因信息;使用等位基因出现率数据产生不同倍性状态的多态基因座的等位基因出现率的单独概率;使用单独概率和定相等位基因信息产生多态基因座集合的联合概率;和基于联合概率选择指示染色体倍性的最佳拟合模型,由此确定染色体区段的倍性。
在某些计算机可读介质实施例中,等位基因出现率数据是由核酸序列数据产生。某些计算机可读介质实施例进一步包含校正等位基因出现率数据中的误差和使用经校正的等位基因出现率数据进行产生单独概率步骤。在某些计算机可读介质实施例中,所校正的误差是等位基因扩增效率偏差。在某些计算机可读介质实施例中,使用多态基因座集合的不同倍性状态和等位基因失衡分数的模型的集合来产生单独概率。在某些计算机可读介质实施例中,通过考虑染色体区段上的多态基因座之间的关联性来产生联合概率。
在一个具体实施例中,本文中提供用于检测个体的样品中的染色体倍性的非暂时性计算机可读介质,其包含计算机可读代码,所述计算机可读代码在由处理装置执行时引起处理装置:接收个体中的染色体区段上的多态基因座集合处的等位基因的核酸序列数据;使用核酸序列数据检测基因座集合处的等位基因出现率;校正所检测的等位基因出现率中的等位基因扩增效率偏差以产生多态基因座集合的经校正的等位基因出现率;通过估计核酸序列数据的相来产生多态基因座集合的定相等位基因信息;通过比较经校正的等位基因出现率与多态基因座集合的不同倍性状态和等位基因失衡分数的模型的集合来产生不同倍性状态的多态基因座的等位基因出现率的单独概率;考虑染色体区段上的多态基因座之间的关联性,通过组合单独概率来产生多态基因座集合的联合概率;和基于联合概率,选择指示染色体非整倍体的最佳拟合模型。
在某些说明性计算机可读介质实施例中,通过分析模型的所产生的定相等位基因信息与所估计的等位基因出现率之间的差的量值来进行选择。
在某些说明性计算机可读介质实施例中,基于多态基因座集合的所预期的和所观察的等位基因出现率的β二项模型来产生等位基因出现率的单独概率。
应理解,本文中所提供的任何方法实施例都可以通过执行储存在非暂时性计算机可读介质上的代码来进行。
检测癌症的例示性实施例
在某些方面中,本发明提供用于检测癌症的方法。应理解,样品可以是来自怀疑患有癌症的个体的肿瘤样品或液体样品,如血浆。所述方法在以样品中的全部DNA的分数形式检测基因突变(如单核苷酸变化,如SNV,或拷贝数目变化,如具有少量的这些基因变化的样品中的CNV)方面尤其有效。因此,在检测样品中来自癌症的DNA或RNA的敏感性方面是优越的。所述方法可以组合本文中关于检测CNV和SNV所提供的改善中的任一种或全部以实现这一优越的敏感性。
因此,在某些实施例中,本文中提供用于确定个体的样品中是否存在循环肿瘤核酸的方法,和包含计算机可读代码的非暂时性计算机可读介质,所述计算机可读代码在由处理装置执行时引起处理装置进行所述方法。所述方法包括以下步骤:分析样品以确定个体中的染色体区段上的多态基因座集合处的倍性;和基于倍性确定来确定多态基因座处的平均等位基因失衡水平,其中平均等位基因失衡等于或大于0.4%、0.45%、0.5%、0.6%、0.7%、0.75%、0.8%、0.9%或1%指示样品中存在循环肿瘤核酸,如ctDNA。
在某些说明性实例中,平均等位基因失衡大于0.4、0.45或0.5%指示存在ctDNA。在某些实施例中,用于确定是否存在循环肿瘤核酸的方法进一步包含检测单核苷酸变异位置集合中的单核苷酸变异位点处的单核苷酸变异体,其中检测到等位基因失衡等于或大于0.5%或检测到单核苷酸变异体或这两者指示样品中存在循环肿瘤核酸。应理解,所提供的任何用于检测染色体倍性或CNV的方法都可以用于确定等位基因失衡水平,通常表示为平均等位基因失衡。应理解,在本发明的这一方面中,本文中所提供的任何用于检测SNV的方法都可以用于检测单核苷酸。
在某些实施例中,用于确定是否存在循环肿瘤核酸的方法进一步包含对具有已知平均等位基因失衡比率的对照样品进行所述方法。举例来说,对照物可以是来自个体的肿瘤的样品。在一些实施例中,对照物具有关于所分析的样品所预期的平均等位基因失衡。举例来说,AAI在0.5%与5%之间或平均等位基因失衡比率是0.5%。
在某些实施例中,用于确定是否存在循环肿瘤核酸的方法中的分析步骤包括分析已知呈现癌症中的非整倍性的染色体区段集合。在某些实施例中,用于确定是否存在循环肿瘤核酸的方法中的分析步骤包括分析1,000到50,000个或100到1000个多态基因座的倍性。在某些实施例中,用于确定是否存在循环肿瘤核酸的方法中的分析步骤包括分析100到1000个单核苷酸变异体位点。举例来说,在这些实施例中,分析步骤可以包括进行多重PCR以扩增跨越1000到50,000个聚合基因座和100到1000个单核苷酸变异体位点的扩增子。这一多重反应可以设置成单一反应或不同子集多重反应的集合。本文中所提供的多重反应方法,如本文中所公开的大规模多重PCR,提供用于进行扩增反应以帮助获得改善的复用且因此获得改善的敏感性水平的例示性方法。
在某些实施例中,对于至少10%、20%、25%、50%、75%、90%、95%、98%、99%或100%的反应物,多重PCR反应是在限制性引物条件下进行。可以使用本文中所提供的改善的用于进行大规模多重反应的条件。
在某些方面中,以上用于确定个体的样品中是否存在循环肿瘤核酸的方法和其所有实施例都可以用系统来进行。本公开提供关于用于进行所述方法的特定功能和结构特征的教示内容。作为非限制性实例,所述系统包括以下:
输入处理器,其被配置成分析来自样品的数据以确定个体中的染色体区段上的多态基因座集合处的倍性;和
建模器,其被配置成基于倍性确定来确定多态基因座处的等位基因失衡水平,其中等位基因失衡等于或大于0.5%指示存在循环。
检测单核苷酸变异体的例示性实施例
在某些方面中,本文中提供用于检测样品中的单核苷酸变异体的方法。本文中所提供的改善的方法可以实现样品中的0.015%、0.017%、0.02%、0.05%、0.1%、0.2%、0.3%、0.4%或0.5%SNV的检测极限。检测SNV的所有实施例都可以用系统来进行。本公开提供关于用于进行所述方法的特定功能和结构特征的教示内容。此外,本文中提供包含非暂时性计算机可读介质的实施例,所述非暂时性计算机可读介质包含计算机可读代码,所述计算机可读代码在由处理装置执行时引起处理装置进行本文中所提供的用于检测SNV的方法。
因此,在一个实施例中,本文中提供用于确定来自个体的样品中的基因组位置集合处是否存在单核苷酸变异体的方法,所述方法包含:对于每个基因组位置,使用训练数据集产生跨越基因组位置的扩增子的效率和每个循环的误差率的估计值;接收样品中每个基因组位置的所观察的核苷酸一致性信息;通过比较每个基因组位置处的所观察的核苷酸一致性信息与不同变异体百分比的模型来确定由每个基因组位置处的一个或多个真实突变引起的单核苷酸变异体百分比的概率集合,所述模型独立地使用每个基因组位置的所估计的扩增效率和每个循环的误差率;和由每个基因组位置的概率集合确定最有可能的真实变异体百分比和置信度。
在用于确定是否存在单核苷酸变异体的方法的说明性实施例中,产生跨越基因组位置的扩增子集合的效率和每个循环的误差率的估计值。举例来说,可以包括2、3、4、5、10、15、20、25、50、100个或更多的跨越基因组位置的扩增子。
在用于确定是否存在单核苷酸变异体的方法的说明性实施例中,所观察的核苷酸一致性信息包含每个基因组位置的所观察的全部读段的数目和每个基因组位置的所观察的变异体等位基因读段的数目。
在用于确定是否存在单核苷酸变异体的方法的说明性实施例中,样品是血浆样品且样品的循环肿瘤DNA中存在单核苷酸变异体。
在另一实施例中,本文中提供用于估计来自个体的样品中的单核苷酸变异体的百分比的方法。所述方法包括以下步骤:在基因组位置集合处,使用训练数据集产生跨越这些基因组位置的一个或多个扩增子的效率和每个循环的误差率的估计值;接收样品中的每个基因组位置的所观察的核苷酸一致性信息;使用扩增子的扩增效率和每个循环的误差率,产生包含初始百分比的真实突变分子的搜索空间的分子总数、背景误差分子和真实突变分子的所估计的平均值和方差;和通过使用所估计的平均值和方差针对样品中的所观察的核苷酸一致性信息拟合分布,通过确定最有可能的真实单核苷酸变异体百分比来确定样品中由真实突变引起的单核苷酸变异体的百分比。
在这一用于估计样品中的单核苷酸变异体的百分比的方法的说明性实例中,样品是血浆样品且样品的循环肿瘤DNA中存在单核苷酸变异体。
本发明的这一实施例的训练数据集通常包括来自一名或优选一组健康个体的样品。在某些说明性实施例中,与一个或多个检验中样品在同一天或甚至在同一次操作中分析训练数据集。举例来说,来自2、3、4、5、10、15、20、25、30、36、48、96、100、192、200、250、500、1000名或更多的健康个体的组的样品可以用于产生训练数据集。当可以获得较大数目(例如96名或更多)的健康个体的数据时,即使在对检验中样品进行所述方法之前进行操作,扩增效率估计值的置信度也会提高。PCR误差率可以使用不是仅针对SNV碱基位置,而是针对SNV周围的整个扩增区域所产生的核酸序列信息,因为误差率是以每个扩增子计的。举例来说,使用来自50名个体的样品和对SNV周围的20个碱基对扩增子进行测序,可以使用来自1000个碱基读段的误差出现率数据确定误差出现率。
通常,通过估计扩增区段的扩增效率的平均值和标准差且接着将其针对分布模型(如二项分布或β二项分布)进行拟合来估计扩增效率。确定具有已知的循环数目的PCR反应的误差率且接着估计每个循环的误差率。
在某些说明性实施例中,估计检验数据集的起始分子进一步包括如果所观察的读段数目与所估计的读段数目显著不同,那么使用步骤(b)中所估计的起始数目的分子更新检验数据集的效率的估计值。接着,可以针对新的效率和/或起始分子更新估计值。
用于估计分子总数、背景误差分子和真实突变分子的搜索空间可以包括其中SNV位置处的碱基的作为下端的0.1%、0.2%、0.25%、0.5%、1%、2.5%、5%、10%、15%、20%或25%到作为上端的1%、2%、2.5%、5%、10%、12.5%、15%、20%、25%、50%、75%、90%或95%的拷贝是SNV碱基的搜索空间。当所述方法是检测循环肿瘤DNA时,较低范围(作为下端的0.1%、0.2%、0.25%、0.5%或1%到作为上端的1%、2%、2.5%、5%、10%、12.5%或15%)可以用于血浆样品的说明性实例中。将较高范围用于肿瘤样品。
针对全部分子中的全部误差分子(背景误差和真实突变)的数目拟合分布,以计算搜索空间中的每个可能的真实突变的似然性或概率。这一分布可以是二项分布或β二项分布。
通过确定最有可能的真实突变百分比和使用来自拟合分布的数据计算置信度来确定最有可能的真实突变。作为说明性实例且不意图限制本文中所提供的方法的临床解释,如果平均突变率较高,那么作出SNV的阳性确定所需的置信度百分比较低。举例来说,如果使用最有可能的假设的样品中的SNV的平均突变率是5%且置信度百分比是99%,那么将作出阳性SNV识别。在这一说明性实例的另一方面,如果使用最有可能的假设的样品中的SNV的平均突变率是1%且置信度百分比是50%,那么在某些情形下,将不作出阳性SNV识别。应理解,数据的临床解释将是敏感性、特异性、发病率、流行率和替代性产品可用性的函数。
在一个说明性实施例中,样品是循环DNA样品,如循环肿瘤DNA样品。
在另一实施例中,本文中提供用于检测来自个体的检验样品中的一种或多种单核苷酸变异体的方法。根据这一实施例的方法包括以下步骤:
对于单核苷酸变异位置集合中的每个单核苷酸变异体位置,基于测序操作中产生的结果,确定来自多个正常个体中的每一个的多个对照样品的中值变异体等位基因出现率,以鉴别正常样品中具有低于阈值的中值变异体等位基因出现率的所选择的单核苷酸变异体位置和在从每个单核苷酸变异体位置去除离群样品之后,确定每个单核苷酸变异体位置的背景误差;基于在测序操作中产生的检验样品的数据,确定检验样品的所选择的单核苷酸变异体位置的所观察的读段深度加权平均值和方差;和使用计算机,鉴别一个或多个单核苷酸变异体位置,所述一个或多个单核苷酸变异体位置与所述位置的背景误差相比具有统计显著读段深度加权平均值,由此检测一种或多种单核苷酸变异体。
在这一用于检测一种或多种SNV的方法的某些实施例中,样品是血浆样品,对照样品是血浆样品,且所检测的检测到的一种或多种单核苷酸变异体存在于样品的循环肿瘤DNA中。在这一用于检测一种或多种SNV的方法的某些实施例中,所述多个对照样品包含至少25个样品。在某些说明性实施例中,所述多个对照样品是作为下端的至少5、10、15、20、25、50、75、100、200或250个样品到作为上端的10、15、20、25、50、75、100、200、250、500和1000个样品。
在这一用于检测一种或多种SNV的方法的某些实施例中,从高通量测序操作中产生的数据去除离群值以计算所观察的读段深度加权平均值且确定所观察的方差。在这一用于检测一种或多种SNV的方法的某些实施例中,检验样品的每个单核苷酸变异体位置的读段深度是至少100个读段。
在这一用于检测一种或多种SNV的方法的某些实施例中,测序操作包含在限制性引物反应条件下进行的多重扩增反应。使用本文中所提供的改善的用于进行多重扩增反应的方法进行说明性实例中的这些实施例。
不受理论约束,本发明的实施例的方法利用使用正常血浆样品的背景误差模型以解决操作特异性假象,所述正常血浆样品是与检验中样品在同一测序操作中测序。去除具有高于阈值的正常中值变异体等位基因出现率(例如>0.1%、0.2%、0.25%、0.5%、0.75%和1.0%)的噪声位置。
从模型迭代地去除异常样品以解决噪声和污染。对于每个基因座的每个碱基取代,计算误差的读段深度加权平均值和标准差。在某些说明性实施例中,对具有至少具有阈值数目的读段(例如至少2、3、4、5、6、7、8、9、10、15、20、25、50、100、250、500或1000个变异体读段)的单核苷酸变异体位置和(在某些实施例中)针对背景误差模型的大于2.5、5、7.5或10的a1Z评分的样品(如肿瘤或游离血浆样品)作为候选突变进行计数。
在某些实施例中,对于单核苷酸变异体位置集合中的每个单核苷酸变异体位置,在测序操作中达到作为范围的下端的大于100、250、500、1,000、2000、2500、5000、10,000、20,000、25,0000、50,000或100,000个到作为上端的2000、2500、5,000、7,500、10,000、25,000、50,000、100,000、250,000或500,000个读段的读段深度。通常,测序操作是高通量测序操作。在说明性实施例中,由读段深度对检验中样品的所产生的平均值或中值进行加权。因此,具有在1000个读段中检测到的1个变异体等位基因的样品中的变异体等位基因确定为真的似然性的权重高于具有在10,000个读段中检测到的1个变异体等位基因的样品。因为变异体等位基因(即,突变)的确定未在100%置信度下进行,所鉴别的单核苷酸变异体可视为候选变异体或候选突变。
用于定相数据的分析的例示性检验统计值
下文描述用于定相数据的分析的例示性检验统计值,所述定相数据是来自已知或怀疑是混合样品的样品,所述混合样品含有来源于两种或更多种在遗传学上不一致的细胞的DNA或RNA。假设f表示相关DNA或RNA的分数,例如具有相关CNV的DNA或RNA的分数,或来自相关细胞(如癌细胞)的DNA或RNA的分数。在癌症检验的一些实施例中,f表示来自癌细胞与正常细胞的混合物中的癌细胞的DNA或RNA的分数,或f表示癌细胞与正常细胞的混合物中的癌细胞的分数。应注意,这是指来自相关细胞的DNA的分数,假设每个相关细胞提供DNA的两个拷贝。这与缺失或复制的区段处的来自相关细胞的DNA分数不同。
将每个SNP的可能的等位基因值表示为A和B。使用AA、AB、BA和BB表示所有可能的有序等位基因对。在一些实施例中,分析具有有序等位基因AB或BA的SNP。假设N
N
定义等位基因比率R
假设T表示目标SNP的数目。
在不失一般性的情况下,一些实施例关注单一染色体区段。为了更清楚起见,在本说明书中,短语“与第二同源染色体区段相比的第一同源染色体区段”意指染色体区段的第一同系物和染色体区段的第二同系物。在一些这类实施例中,所有目标SNP都包含于相关区段染色体中。在其它实施例中,分析多个染色体区段的可能的拷贝数目变化。
MAP估计
这一方法利用通过有序等位基因进行定相的知识以检测目标区段的缺失或复制。对于每个SNP i,定义
接着定义
下文描述X
二体性假设
在目标片段未缺失或复制的假设下,
其中
如果采用恒定读段深度N,那么这提供具有以下参数的二项分布S:
缺失假设
在缺失第一同系物的假设下(即,AB SNP变成B,且BA SNP变成A),那么R
因此,
如果采用恒定读段深度N,那么这提供具有以下参数的二项分布S:
在缺失第二同系物的假设下(即,AB SNP变成A,且BA SNP变成B),那么R
因此,
如果采用恒定读段深度N,那么这提供具有以下参数的二项分布S:
复制假设
在复制第一同系物的假设下(即,AB SNP变成AAB,且BA SNP变成BBA),那么R
如果采用恒定读段深度N,那么这提供具有以下参数的二项分布S:
在复制第二同系物的假设下(即,AB SNP变成ABB,且BA SNP变成BAA),那么R
如果采用恒定读段深度N,那么这提供具有以下参数的二项分布S:
分类
如以上章节中说明,X
这使得能够在每种假设下计算检验统计值S的概率。可以计算提供所测量的数据的每种假设的概率。在一些实施例中,选择具有最大概率的假设。视需要,可以通过在恒定读段深度N下取每个N
可以使用算法(例如搜索算法),如最大似然估计、最大后验估计或贝叶斯估计,通过选择提供所测量的数据的最有可能的f的值(如产生最佳数据拟合的f的值)来估计f的值。在一些实施例中,分析多个染色体区段且基于每个区段的数据估计f的值。如果所有目标细胞都具有这些复制或缺失,那么基于这些不同区段的数据的所估计的f的值是类似的。在一些实施例中,以实验方式测量f,如通过基于癌症与非癌性DNA或RNA之间的甲基化差异(低甲基化或超甲基化)来确定来自癌细胞的DNA或RNA的分数。
单一假设拒绝
二体性假设的S的分布不取决于f。因此,可以在不计算f的情况下,在二体性假设下计算所测量的数据的概率。单一假设拒绝检验可以用于二体性的零假设。在一些实施例中,计算在二体性假设下的S的概率,且如果概率低于既定阈值(如小于1/1,000),那么拒绝二体性的假设。这表示存在染色体区段的复制或缺失。视需要,可以通过调节阈值来改变假阳性率。
用于定相数据的分析的例示性方法
下文描述用于数据的分析的例示性方法,所述数据是来自已知或怀疑是混合样品的样品,所述混合样品含有来源于两种或更多种在遗传学上不一致的细胞的DNA或RNA。在一些实施例中,使用定相数据。在一些实施例中,所述方法涉及针对每个所计算的等位基因比率,确定所计算的等位基因比率是否高于或低于所预期的等位基因比率和具体基因座的差的量值。在一些实施例中,确定具体假设的基因座处的等位基因比率的似然性分布且所计算的等位基因比率越接近似然性分布的中心,假设正确的可能性越高。在一些实施例中,所述方法涉及确定假设对于每个基因座是正确的似然性。在一些实施例中,所述方法涉及确定假设对于每个基因座是正确的似然性和组合每个基因座的所述假设的概率,以及选择具有最大组合概率的假设。在一些实施例中,所述方法涉及确定假设对于每个基因座和来自一种或多种目标细胞的DNA或RNA与样品中全部DNA或RNA的每种可能的比率是正确的似然性。在一些实施例中,通过组合每个基因座和每种可能的比率的所述假设的概率来确定每种假设的组合概率,且选择具有最大组合概率的假设。
在一个实施例中,考虑以下假设:H
r(AB,H
偏差、污染和测序误差校正:
SNP处的观察结果D
假设c
可以通过校正环境污染、基因分型污染和测序误差来获得既定所预期的等位基因比率r的所观察的等位基因比率q(r、c
因为污染物基因型是未知的,因此可以使用群体出现率获得P(r(c
每个SNP处的似然性:
以下等式提供在既定等位基因比率r下,观察n
方程式(2):
假设D
搜索算法:
在一些实施例中,忽略等位基因比率似乎是离群值的SNP(如通过忽略或去除等位基因比率比平均值高或低至少2或3倍标准差的SNP)。应注意,所鉴别的这一方法的优点是在存在较高嵌合百分比的情况下,等位基因比率的可变性可以是较高的,因此这确保SNP将不会由于嵌合而被修整。
假设F={f
针对每种假设对每个f应用算法。使用搜索方法,如果存在其中缺失或复制假设的置信度高于无缺失和无复制假设的置信度的f的范围F*,那么可以得出存在嵌合的结论。在一些实施例中,确定F*中P(D
在一些实施例中,使用β二项分布代替二项分布。在一些实施例中,使用参考染色体或染色体区段确定β二项的样品特异性参数。
使用模拟的理论性能:
视需要,可以通过对具有既定读段深度(DOR)的SNP随机指定参考读段的数目来评估算法的理论性能。在正常情况下,将p=0.5用于二项概率参数,且对于缺失或复制,相应地修改p。每次模拟的例示性输入参数如下:(1)SNP的数目S,(2)每个SNP的恒定DOR D,(3)p和(4)实验数目。
第一模拟实验:
本实验关注S∈{500、1000}、D∈{500、1000}和p∈{0%、1%、2%、3%、4%、5%}。我们在每种设置下进行1,000个模拟实验(因此,24,000个实验具有相位,和24,000个不具有相位)。我们由二项分布模拟读段数目(视需要,可以使用其它分布)。在具有或不具有相信息的情况下确定假阳性率(在p=0%的情况下)和假阴性率(在p>0%的情况下)。应注意,相信息极其有帮助,尤其对于S=1000,D=1000。但对于S=500,D=500,算法在具有或不具有所检验的条件以外的相的情况下具有最高假阳性率。
相信息对于低嵌合百分比(≤3%)尤其适用。在不具有相信息的情况下,关于p=1%观察到高假阴性水平,因为缺失置信度是通过对H
第二模拟实验:
本实验关注S∈{100、200、300、400、500}、D∈{1000、2000、3000、4000、5000}和p∈{0%、1%、1.5%、2%、2.5%、3%}且在每种设置下进行10000个随机实验。在具有或不具有相信息的情况下确定假阳性率(在p=0%的情况下)和假阴性率(在p>0%的情况下)。使用单倍型信息,对于D≥3000和N≥200,假阴性率低于10%,而在D=5000和N≥400情况下达到相同性能。假阴性率之间的差在小嵌合百分比的情况下尤其显著。举例来说,当p=1%时,在不具有单倍型数据的情况下从未达到小于20%假阴性率,然而对于N≥300和D≥3000,假阴性率接近于0%。对于p=3%,在具有单倍型数据的情况下观察到0%假阴性率,而在不具有单倍型数据的情况下,需要N≥300和D≥3000才能达到相同性能。
用于在不具有定相数据的情况下检测缺失和复制的例示性方法
在一些实施例中,使用非定相基因数据确定在个体的基因组中(如在一种或多种细胞的基因组中或在cfDNA或cfRNA中),与第二同源染色体区段相比,是否存在第一同源染色体区段的拷贝数目的过度呈现。在一些实施例中,使用定相基因数据,但忽略定相。在一些实施例中,DNA或RNA的样品是来自个体的cfDNA或cfRNA的混合样品,其包括来自两种或更多种在遗传学上不同的细胞的cfDNA或cfRNA。在一些实施例中,所述方法利用每个基因座的所计算的等位基因比率与所预期的等位基因比率之间的差的量值。
在一些实施例中,方法涉及通过测量每个基因座处的每种等位基因的数量,获得DNA或RNA样品中的染色体或染色体区段上的多态基因座集合处的基因数据,所述DNA或RNA样品是来自个体的一种或多种细胞。在一些实施例中,计算至少一种衍生样品的细胞中的杂合基因座的等位基因比率。在一些实施例中,具体基因座的所计算的等位基因比率是所述基因座的一种等位基因的所测量的数量除以所有等位基因的全部所测量的数量。在一些实施例中,具体基因座的所计算的等位基因比率是所述基因座的一种等位基因(如第一同源染色体区段上的等位基因)的所测量的数量除以一种或多种其它等位基因(如第二同源染色体区段上的等位基因)的所测量的数量。所计算的等位基因比率和所预期的等位基因比率可以使用本文中所描述的任何方法或任何标准方法(如本文中所描述的所计算的等位基因比率或所预期的等位基因比率的任何数学转换)来计算。
在一些实施例中,基于每个基因座的所计算的等位基因比率与所预期的等位基因比率之间的差的量值计算检验统计值。在一些实施例中,使用下式计算检验统计值Δ:
其中δ
其中μ
其中
举例来说,当所预期的等位基因比率是0.5时,δ
μ
在一些实施例中,列举指定一种或多种细胞的基因组中的染色体或染色体区段的拷贝数目的一种或多种假设的集合。在一些实施例中,选择最有可能基于检验统计值的假设,由此确定一种或多种细胞的基因组中的染色体或染色体区段的拷贝数目。在一些实施例中,如果检验统计值属于一种假设的检验统计值的分布的概率高于上限阈值,那么选择所述假设;如果检验统计值属于一种或多种假设的检验统计值的分布的概率低于下限阈值,那么拒绝所述一种或多种假设;或如果检验统计值属于一种假设的检验统计值的分布的概率在下限阈值与上限阈值之间或如果未在足够高的置信度下确定概率,那么既不选择也不拒绝所述假设。在一些实施例中,由经验分布确定上限阈值和/或下限阈值,如来自训练数据的分布(如具有已知的拷贝数目的样品,如二倍体样品或已知具有具体缺失或复制的样品)。这类经验分布可以用于选择用于单一假设排斥检验的阈值。应注意,检验统计值Δ与S无关且因此这两者都可以视需要而独立地使用。
用于使用等位基因分布或模式来检测缺失和复制的例示性方法
本章节包括用于确定与第二同源染色体区段相比,是否存在第一同源染色体区段的拷贝数目的过度呈现的方法。在一些实施例中,所述方法涉及列举(i)指定个体的一种或多种细胞(如癌细胞)的基因组中的染色体或染色体区段的拷贝数目的多个假设,或(ii)指定在个体的一种或多种细胞的基因组中,与第二同源染色体区段相比,第一同源染色体区段的拷贝数目的过度呈现程度的多个假设。在一些实施例中,所述方法涉及从个体获得染色体或染色体区段上的多个多态基因座(如SNP基因座)处的基因数据。在一些实施例中,创建用于每种假设的个体的所预期的基因型的概率分布。在一些实施例中,计算个体的所获得的基因数据与个体的所预期的基因型的概率分布之间的数据拟合。在一些实施例中,根据数据拟合对一种或多种假设进行分级,且选择等级最高的假设。在一些实施例中,使用技术或算法(如搜索算法)进行以下步骤中的一个或多个:计算数据拟合、对假设进行分级或选择等级最高的假设。在一些实施例中,数据拟合是针对β-二项分布的拟合或针对二项分布的拟合。在一些实施例中,技术或算法是选自由以下组成的群组:最大似然估计、最大后验估计、贝叶斯估计、动态估计(如动态贝叶斯估计)和最大期望估计。在一些实施例中,所述方法包括对所获得的基因数据和所预期的基因数据应用技术或算法。
在一些实施例中,所述方法涉及列举(i)指定个体的一种或多种细胞(如癌细胞)的基因组中的染色体或染色体区段的拷贝数目的多个假设,或(ii)指定在个体的一种或多种细胞的基因组中,与第二同源染色体区段相比,第一同源染色体区段的拷贝数目的过度呈现程度的多个假设。在一些实施例中,所述方法涉及从个体获得染色体或染色体区段上的多个多态基因座(如SNP基因座)处的基因数据。在一些实施例中,基因数据包括多个多态基因座的等位基因计数。在一些实施例中,针对每种假设,创建染色体或染色体区段上的多个多态基因座处的所预期的等位基因计数的联合分布模型。在一些实施例中,使用联合分布模型和在样品上测量的等位基因计数确定一种或多种假设的相对概率,且选择具有最大概率的假设。
在一些实施例中,使用等位基因的分布或模式(如所计算的等位基因比率的模式)确定存在或不存在CNV,如缺失或复制。视需要,可以基于这一模式确定CNV的亲本来源。
例示性计数方法/定量方法
在一些实施例中,使用一种或多种计数方法(也称为定量方法)检测一种或多种CNS,如染色体区段或整个染色体的缺失或复制。在一些实施例中,使用一种或多种计数方法确定第一同源染色体区段的拷贝数目的过度呈现是否是由第一同源染色体区段的复制或第二同源染色体区段的缺失引起。在一些实施例中,使用一种或多种计数方法确定所复制的染色体区段或染色体的额外拷贝数目(如是否存在1、2、3、4个或更多的额外拷贝)。在一些实施例中,使用一种或多种计数方法来区分具有许多复制和较小肿瘤分数的样品与具有较少复制和较大肿瘤分数的样品。举例来说,可以使用一种或多种计数方法来区分具有四个额外染色体拷贝且肿瘤分数是10%的样品与具有两个额外染色体拷贝且肿瘤分数是20%的样品。例示性方法公开于例如美国公开案第2007/0184467号;第2013/0172211号;和第2012/0003637号;美国专利案第8,467,976号;第7,888,017号;第8,008,018号;第8,296,076号;和第8,195,415号;2014年6月5日提交的美国序列号62/008,235和2014年8月4日提交的美国序列号62/032,785中,其各自以全文引用的方式并入本文中。
在一些实施例中,计数方法包括对映射到一个或多个既定染色体或染色体区段的基于DNA序列的读段的数目进行计数。一些这类方法涉及产生映射到特定染色体或染色体区段的DNA序列读段的数目的参考值(截止值),其中多个读段超过所述值指示特定遗传异常。
在一些实施例中,比较一个或多个基因座的所有等位基因的总测量数量(如多态或非多态基因座的总量)与参考量。在一些实施例中,参考量是(i)阈值,或(ii)具体拷贝数目假设的所预期的量。在一些实施例中,参考量(对于不存在CNV)是已知或预期不具有缺失或复制的一个或多个染色体或染色体区段的一个或多个基因座的所有等位基因的总测量数量。在一些实施例中,参考量(对于存在CNV)是已知或预期缺失或复制的一个或多个染色体或染色体区段的一个或多个基因座的所有等位基因的总测量数量。在一些实施例中,参考量是一个或多个参考染色体或染色体区段的一个或多个基因座的所有等位基因的总测量数量。在一些实施例中,参考量是两个或更多个不同染色体、染色体区段或不同样品的所确定的值的平均值或中值。在一些实施例中,使用随机(例如大规模平行鸟枪法测序)或靶向测序来确定一种或多种多态或非多态基因座的量。
在利用参考量的一些实施例中,方法包括(a)测量相关染色体或染色体区段上的遗传物质的量;(b)比较来自步骤(a)的量与参考量;和(c)基于所述比较来鉴别存在或不存在缺失或复制。
在利用参考染色体或染色体区段的一些实施例中,方法包括对来自样品的DNA或RNA进行测序以获得与目标基因座对准的多个序列标签。在一些实施例中,序列标签具有足够的长度以分配到特定目标基因座(例如,长度是15-100个核苷酸);目标基因座是来自多个不同的染色体或染色体区段,所述多个不同的染色体或染色体区段包括至少一个怀疑在样品中具有非正态分布的第一染色体或染色体区段和至少一个假定在样品中具有正态分布的第二染色体或染色体区段。在一些实施例中,将多个序列标签分配到其相应的目标基因座。在一些实施例中,确定与第一染色体或染色体区段的目标基因座对准的序列标签的数目和与第二染色体或染色体区段的目标基因座对准的序列标签的数目。在一些实施例中,比较这些数目以确定存在或不存在第一染色体或染色体区段的非正态分布(如缺失或复制)。
在一些实施例中,将f的值(如肿瘤分数)用于CNV确定,如用于比较两个染色体或染色体区段的量之间的所观察的差异与在既定f值下的具体类型的CNV的所预期的差异(参见例如美国公开案第2012/0190020号;美国公开案第2012/0190021号;美国公开案第2012/0190557号;美国公开案第2012/0191358号,其各自以全文引用的方式并入本文中)。举例来说,与二体参考染色体区段进行比较的在肿瘤中复制的染色体区段的量的差异随肿瘤分数增加而增加。在一些实施例中,所述方法包括比较相关染色体或染色体区段针对参考染色体或染色体区段(如预期或已知具有二体性的染色体或染色体区段)的相对出现率与f的值,以确定CNV的似然性。举例来说,可以比较第一染色体或染色体区段与参考染色体或染色体区段之间的量的差与在各种可能的CNV(如相关染色体区段的一个或两个额外拷贝)的既定f值下的预期值。
以下预示性实例说明使用计数方法/定量方法区分第一同源染色体区段的复制与第二同源染色体区段的缺失。如果将宿主的正常二体基因组视为基线,那么正常细胞与癌细胞的混合物的分析产生基线与混合物中的癌症DNA之间的平均差。举例来说,设想其中样品中的10%的DNA是来源于在分析法所靶向的染色体区域中具有缺失的细胞的情况。在一些实施例中,定量方法证实对应于所述区域的读段数量预期是正常样品的预期值的95%。这是因为遗失了具有目标区域的缺失的每个肿瘤细胞中的两个目标染色体区域中的一个且因此映射到所述区域的DNA的总量是90%(对于正常细胞)加1/2×10%(对于肿瘤细胞)=95%。或者,在一些实施例中,对偶基因方法证实杂合基因座处的等位基因的平均比率是19:20。现在设想其中样品中的10%的DNA是来源于具有分析法所靶向的染色体区域的五倍局部扩增的细胞的情况。在一些实施例中,定量方法证实对应于所述区域的读段数量预期是正常样品的预期值的125%。这是因为在目标区域中,具有五倍局部扩增的每个肿瘤细胞中的两个目标染色体区域中的一个被额外复制五次,且因此映射到所述区域的DNA的总量是90%(对于正常细胞)加(2+5)×10%/2(对于肿瘤细胞)=125%。或者,在一些实施例中,对偶基因方法证实杂合基因座处的等位基因的平均比率是25:20。应注意,当单独使用对偶基因方法时,具有10%cfDNA的样品中的染色体区域的五倍局部扩增可能显得与具有40%cfDNA的样品中的相同区域的缺失相同;在这两种情况下,在缺失的情况下呈现不足的单倍型似乎是在局部复制的情况下不具有CNV的单倍型,且在缺失的情况下不具有CNV的单倍型似乎是在局部复制的情况下过度呈现的单倍型。组合由这种对偶基因方法产生的似然性与由定量方法产生的似然性可以区分两种可能性。
使用参考样品的例示性计数方法/定量方法
使用一种或多种参考样品的例示性定量方法描述于2014年6月5日提交的美国序列号62/008,235和2014年8月4日提交的美国序列号62/032,785中,其以全文引用的方式并入本文中。在一些实施例中,通过以下方式来鉴别最有可能在一种或多种染色体或相关染色体上不具有任何CNV的一种或多种参考样品(例如正常样品):选择具有最高肿瘤DNA分数的样品、选择z评分最接近于零的样品、选择其中数据以最高置信度或似然性符合对应于不存在CNV的假设的样品、选择已知是正常的样品、选择来自具有最低患癌似然性的个体(例如年龄较小、参加乳癌筛检的男性、不具有家族病史等)的样品、选择具有最高DNA输入量的样品、选择具有最高信噪比的样品、基于相信与患癌似然性相关的其它准则选择样品或使用某一准则组合选择样品。在选择参考集合后,可以作出这些情况是二体性的假设,且接着估计每个SNP的偏差,也就是说,每个基因座的实验特定扩增和其它处理偏差。接着,可以使用这种实验特定偏差估计值校正相关染色体(如染色体21基因座)和(视需要)其它染色体基因座、并非其中假设染色体21具有二体性的子集的一部分的样品的测量结果中的偏差。在校正这些具有未知倍性的样品中的偏差之后,接着可以使用相同或不同方法第二次分析这些样品的数据,以确定个体是否罹患第21对染色体三体症。举例来说,可以对其余具有未知倍性的样品使用定量方法,且可以使用染色体21的经校正的所测量的基因数据计算z评分。或者,作为染色体21的倍性状态的初步估计的一部分,可以计算来自怀疑患有癌症的个体的样品的肿瘤分数。可以计算在具有所述肿瘤分数的情况下,在二体性(二体性假设)的情况下所预期的经校正的读段的比例和在三体性(三体性假设)的情况下所预期的经校正的读段的比例。或者,如果未预先测量肿瘤分数,那么可以针对不同肿瘤分数产生二体性和三体性假设的集合。对于每种情况,考虑各种DNA基因座的选择和测量中的所预期的统计变化,可以计算经校正的读段的比例的预期分布。对于每个具有未知倍性的样品,可以比较所观察的经校正的读段比例与所预期的经校正的读段比例的分布,且可以计算二体性和三体性假设的似然比。可以选择与具有最高的似然性计算值的假设相关联的倍性状态作为正确的倍性状态。
在一些实施例中,可以选择具有足够低的患癌似然性的样品的子集作为对照样品集合。子集可以是固定数目,或其可以是基于仅选择低于阈值的样品的可变数目。可以将来自样品的子集的定量数据组合、求平均值或使用加权平均值组合,其中加权是基于样品是正常样品的似然性。可以使用定量数据确定当前批次的对照样品中的样品测序的扩增中的每个基因座的偏差。每个基因座的偏差还可以包括来自其它批次的样品的数据。每个基因座的偏差可以指示所述基因座与其它基因座相比,所观察到的相对扩增过度或扩增不足,作出样品的子集不含任何CNV以及任何所观察到的扩增过度或扩增不足是由扩增和/或测序或其它偏差引起的假设。每个基因座的偏差可以考虑扩增子的GC含量。出于计算每个基因座的偏差的目的,可以将基因座分组成基因座组。在针对多个基因座中的每个基因座计算每个基因座的偏差之后,可以通过调节每个基因座的定量测量结果以去除所述基因座处的偏差的影响来校正不属于样品的子集的一个或多个样品和任选地,属于样品的子集的一个或多个样品的测序数据。举例来说,如果在患者的子集中观察到SNP 1的读段深度是平均值的两倍,那么调节可以涉及将对应于SNP 1的读段数目替换为大小是所述数目的一半的数目。如果所讨论的基因座是SNP,那么调节可以涉及使对应于所述基因座处的每个等位基因的读段数目减小一半。在调节一个或多个样品中的每个基因座的测序数据之后,可以使用用于检测一个或多个染色体区域中是否存在CNV的方法来分析测序数据。
在一个实例中,样品A是来源于使用定量方法分析的正常细胞与癌性细胞的混合物的被扩增的DNA的混合物。下文说明例示性可能数据。发现在染色体22上的q臂的一个区域中,映射到所述区域的DNA仅是预期值的90%;发现在对应于HER2基因的局部区域中,映射到所述区域的DNA是预期值的150%;且发现在染色体5的p臂中,映射到所述p臂的DNA是预期值的105%。临床医生可推断样品具有染色体22上的q臂上的一个区域的缺失,和HER2基因的复制。临床医生可以推断因为22q缺失在乳癌中是常见的且因为在两条染色体上具有22q区域的缺失的细胞通常不能存活,样品中的约20%的DNA是来自在两条染色体中的一条上具有22q缺失的细胞。临床医生还可以推断,如果来自来源于肿瘤细胞的混合样品的DNA是来源于遗传肿瘤细胞的集合且所述遗传肿瘤细胞的HER2区域和22q区域是同源的,那么所述细胞含有HER2区域的五倍复制。
在一个实例中,还使用对偶基因方法分析样品A。下文说明例示性可能数据。染色体22上的q臂上的同一个区域中的两种单倍型以4:5的比率存在;对应于HER2基因的局部区域中的两种单倍型以1:2的比率存在;且染色体5的p臂中的两种单倍型以20:21的比率存在。基因组的所有其它分析区域都不具有统计显著过量的任何单倍型。临床医生可以推断,样品含有来自在22q区域、HER2区域和5p臂中具有CNV的肿瘤的DNA。基于对22q缺失在乳癌中极常见的了解和/或定量分析证实映射到基因组的22q区域的DNA的量的呈现不足,临床医生可以推断存在具有22q缺失的肿瘤。基于对HER2扩增在乳癌中极常见的了解和/或定量分析证实映射到基因组的HER2区域的DNA的量的过度呈现,临床医生可以推断存在具有HER2扩增的肿瘤。
例示性参考染色体或染色体区段
在一些实施例中,还对一种或多种参考染色体或染色体区段进行本文中所描述的任何方法且将结果与一种或多种相关染色体或染色体区段的结果进行比较。
在一些实施例中,使用参考染色体或染色体区段作为预期不存在CNV的情况的对照。在一些实施例中,参考物是来自一个或多个不同样品的相同染色体或染色体区段,已知或预期所述一个或多个不同样品在所述染色体或染色体区段中不具有缺失或复制。在一些实施例中,参考物是来自所检验的样品的预期具有二体性的不同染色体或染色体区段。在一些实施例中,参考物是来自相同的所检验的样品中的一种相关染色体的不同区段。举例来说,参考物可以是位于具有潜在的缺失或复制的区域的外部的一个或多个区段。参考相同的所检验的染色体可以避免不同染色体之间的可变性,如染色体之间的代谢、细胞凋亡、组蛋白、失活和/或扩增中的差异。分析与所检验的染色体相同的染色体上的不具有CNV的区段也可以用于确定同系物之间的代谢、细胞凋亡、组蛋白、失活和/或扩增中的差异,使得能够确定在不存在CNV的情况下的同系物之间的可变性水平,以与来自潜在CNV的结果进行比较。在一些实施例中,潜在的CNV的所计算的与所预期的等位基因比率之间的差的量值大于参考物的相应的量值,由此证实存在CNV。
在一些实施例中,使用参考染色体或染色体区段作为预期存在CNV的情况(如相关具体缺失或复制)的对照。在一些实施例中,参考物是来自一个或多个不同样品的相同染色体或染色体区段,已知或预期所述一个或多个不同样品在所述染色体或染色体区段中具有缺失或复制。在一些实施例中,参考物是来自已知或预期具有CNV的所检验的样品的不同染色体或染色体区段。在一些实施例中,潜在的CNV的所计算的与所预期的等位基因比率之间的差的量值与CNV的参考物的相应量值类似(如不显著不同),由此证实存在CNV。在一些实施例中,潜在的CNV的所计算的与所预期的等位基因比率之间的差的量值小于(如显著小于)CNV的参考物的相应量值,由此证实不存在CNV。在一些实施例中,使用其中癌细胞的基因型(或来自癌细胞的DNA或RNA,如cfDNA或cfRNA)与非癌性细胞的基因型(或来自非癌性细胞的DNA或RNA,如cfDNA或cfRNA)不同的一个或多个基因座确定肿瘤分数。肿瘤分数可以用于确定第一同源染色体区段的拷贝数目的过度表示是否是由第一同源染色体区段的复制或第二同源染色体区段的缺失引起。肿瘤分数还可以用于确定被复制的染色体区段或染色体的额外拷贝的数目(如是否存在1、2、3、4个或更多的额外拷贝),如用于区分具有四个额外染色体拷贝且肿瘤分数是10%的样品与具有两个额外染色体拷贝且肿瘤分数是20%的样品。肿瘤分数还可以用于确定可能的CNV的所观察的数据与所预期的数据的符合情况。在一些实施例中,使用CNV的过度表示程度来选择用于个体的具体疗法或治疗方案。举例来说,一些治疗剂仅对染色体区段的至少四个、六个或更多的拷贝有效。
在一些实施例中,用于确定肿瘤分数的一个或多个基因座位于参考染色体或染色体区段上,如已知或预期具有二体性的染色体或染色体区段、通常或在个体已知患有或具有增加的风险的具体类型的癌症的癌细胞中极少复制或缺失的染色体或染色体区段,或不太可能是非整倍体的染色体或染色体区段(如预期在缺失或复制的情况下会引起细胞死亡的区段)。在一些实施例中,使用本发明的任何方法确认参考染色体或染色体区段在癌细胞和非癌性细胞中都具有二体性。在一些实施例中,使用具有较高的二体性识别置信度的一个或多个染色体或染色体区段。
可以用于确定肿瘤分数的例示性基因座包括癌细胞(或DNA或RNA,如来自癌细胞的cfDNA或cfRNA)中的不存在于个体的非癌性细胞(或来自非癌性细胞的DNA或RNA)中的多态现象或突变(如SNP)。在一些实施例中,通过以下方式来确定肿瘤分数:在来自个体的样品(如血浆样品或肿瘤活检)中,鉴别其中癌细胞(或来自癌细胞的DNA或RNA)具有非癌性细胞(或来自非癌性细胞的DNA或RNA)中不存在的等位基因的多态基因座;和使用一个或多个所鉴别的多态基因座处的癌细胞特有的等位基因的量来确定样品中的肿瘤分数。在一些实施例中,非癌性细胞对于多态基因座处的第一等位基因来说是纯合的,且癌细胞(i)对于第一等位基因和第二等位基因来说是杂合的,或(ii)对于多态基因座处的第二等位基因来说是纯合的。在一些实施例中,非癌性细胞对于多态基因座处的第一等位基因和第二等位基因来说是杂合的,且癌细胞(i)具有多态基因座处的第三等位基因的一个或两个拷贝。在一些实施例中,假设或已知癌细胞仅具有非癌性细胞中不存在的等位基因的一个拷贝。举例来说,如果非癌性细胞的基因型是AA且癌细胞是AB且样品中的所述基因座处的5%的信号是来自B等位基因且95%是来自A等位基因,那么样品的肿瘤分数是10%。在一些实施例中,假设或已知癌细胞具有非癌性细胞中不存在的等位基因的两个拷贝。举例来说,如果非癌性细胞的基因型是AA且癌细胞是BB且样品中的所述基因座处的5%的信号是来自B等位基因且95%是来自A等位基因,那么样品的肿瘤分数是5%。在一些实施例中,分析其中癌细胞具有非癌性细胞中不存在的等位基因的多个基因座以确定癌细胞中哪些基因座是杂合的和哪些基因座是纯合的。举例来说,对于其中非癌性细胞是AA的基因座来说,如果来自B等位基因的信号在一些基因座处是约5%且在一些基因座处是约10%,那么假设癌细胞在具有约5%B等位基因的基因座处是杂合的,且在具有约10%B等位基因的基因座处是纯合的(表明肿瘤分数是约10%)。
可以用于确定肿瘤分数的例示性基因座包括其中癌细胞和非癌性细胞共同具有一个等位基因的基因座(如其中癌细胞是AB且非癌性细胞是BB或癌细胞是BB且非癌性细胞是AB的基因座)。比较混合样品(含有来自癌细胞和非癌性细胞的DNA或RNA)中A信号的量、B信号的量或A与B信号的比率与以下的对应值:(i)含有仅来自癌细胞的DNA或RNA的样品,或(ii)含有仅来自非癌性细胞的DNA或RNA的样品。使用值的差来确定混合样品的肿瘤分数。
在一些实施例中,可以用于确定肿瘤分数的基因座是基于以下的基因型来选择:(i)含有仅来自癌细胞的DNA或RNA的样品,和/或(ii)含有仅来自非癌性细胞的DNA或RNA的样品。在一些实施例中,基因座是基于混合样品的分析来选择,如满足以下条件的基因座:每种等位基因的绝对量或相对量与在癌细胞和非癌性细胞在具体基因座处都具有相同基因型的情况下的预期值不同。举例来说,如果癌细胞和非癌性细胞具有相同基因型,那么预期基因座在所有细胞是AA的情况下将产生0%B信号、在所有细胞是AB的情况下将产生50%B信号或在所有细胞是BB的情况下将产生100%B信号。B信号的其它值指示所述基因座处的癌细胞和非癌性细胞的基因型不同且因此所述基因座可以用于确定肿瘤分数。
在一些实施例中,比较基于一个或多个基因座处的等位基因所计算的肿瘤分数与使用一种或多种本文中所公开的计数方法所计算的肿瘤分数。
用于检测表型或分析多个突变的例示性方法
在一些实施例中,方法包括分析样品中与疾病或病症(例如癌症)或增加的疾病或病症风险相关联的突变的集合。在可以用于改善方法的信噪比和将肿瘤分类成不同临床子组的类别(如M或C癌症类别)内的事件之间存在强相关性。举例来说,共同考虑的一个或多个染色体或染色体区段上的少数突变(如少数CNV)的边界结果可以是极强的信号。在一些实施例中,确定存在或不存在多种相关多态现象或突变(如2、3、4、5、8、10、12、15种或更多种)可以提高存在或不存在疾病或病症(如癌症)或增加的疾病或病症(如癌症)的风险的确定的敏感性和/或特异性。在一些实施例中,使用跨越多个染色体的事件之间的相关性以与分别观察每个信号相比,更有效地观察一个信号。方法本身的设计可以优化以对肿瘤进行最佳分类。对于对一种具体突变/CNV的敏感性可能至关重要的复发来说,这可以惊人地适用于早期检测和筛检。在一些实施例中,事件未必总是相关,但具有相关的可能性。在一些实施例中,使用具有噪声协方差矩阵的矩阵估计公式,所述噪声协方差矩阵具有非对角项。
在一些实施例中,本发明提供一种检测个体中的表型(如癌症表型)的方法,其中所述表型是由存在突变集合中的至少一种突变来定义。在一些实施例中,所述方法包括获得来自个体的一种或多种细胞的DNA或RNA样品的DNA或RNA测量结果,其中怀疑所述细胞中的一种或多种具有表型;和对于突变集合中的每种突变,分析DNA或RNA测量结果以确定至少一种细胞具有所述突变的似然性。在一些实施例中,所述方法包括在以下情况下确定个体具有表型:(i)对于至少一种突变,至少一种细胞含有所述突变的似然性大于阈值,或(ii)对于至少一种突变,至少一种细胞具有所述突变的似然性小于阈值,和对于多种突变,至少一种细胞具有至少一种突变的组合似然性大于阈值。在一些实施例中,一种或多种细胞具有突变集合中的突变的子集或所有突变。在一些实施例中,突变的子集与癌症或增加的癌症风险相关联。在一些实施例中,突变集合包括M类癌症突变中的突变的子集或所有突变(Ciriello,《自然遗传学(Nat Genet.)》45(10):1127-1133,2013,doi:10.1038/ng.2762,其以全文引用的方式并入本文中)。在一些实施例中,突变集合包括C类癌症突变中的突变的子集或所有突变(Ciriello,见上文)。在一些实施例中,样品包括游离DNA或RNA。在一些实施例中,DNA或RNA测量结果包括一个或多个相关染色体或染色体区段上的多态基因座集合处的测量结果(如每个基因座处的每种等位基因的数量)。
例示性方法组合
为了提高结果的准确性,进行两种或更多种用于检测存在或不存在CNV的方法(如本发明的任何方法或任何已知的方法)。在一些实施例中,进行一种或多种用于分析指示存在或不存在疾病或病症或增加的疾病或病症风险的因子的方法(如本文中所描述的任何方法或任何已知的方法)。
在一些实施例中,使用标准数学技术计算两种或更多种方法之间的协方差和/或相关性。标准数学技术还可以用于基于两种或更多种检验来确定具体假设的组合概率。例示性技术包括元分析、用于独立检验的费舍尔组合概率检验(Fisher's combinedprobability test for independent tests)、用于组合具有已知协方差的相依性p值的布朗方法(Brown's method for combining dependent p-values with known covariance)和用于组合具有未知协方差的相依性p值的考斯特方法(Kost's method for combiningdependent p-values with unknown covariance)。在通过第一方法,以与第二方法确定似然性的方式正交或不相关的方式确定似然性的情况下,组合似然性是简单的且可以通过相乘和标准化或使用如以下的公式来进行:
R
R
用于分析多个因子或变量的例示性方法公开于2011年9月20日颁布的美国专利案第8,024,128号;2006年7月31日提交的美国公开案第2007/0027636号;和2006年12月6日提交的美国公开案第2007/0178501号中,其各自以全文引用的方式并入本文中。
在各种实施例中,具体假设或诊断的组合概率大于80、85、90、92、94、96、98、99或99.9%,或大于某一其他阈值。
检测极限
如由实例部分中提供的实验证明,本文中所提供的方法能够在检测极限或敏感性是0.45%AAI的情况下检测样品中的平均等位基因失衡,其是本发明的说明性方法的非整倍性的检测极限。类似地,在某些实施例中,本文中所提供的方法能够检测到样品中的平均等位基因失衡是0.45、0.5、0.6、0.8、0.8、0.9或1.0%。也就是说,检验方法能够在AAI低到0.45、0.5、0.6、0.8、0.8、0.9或1.0%的情况下检测到样品中的染色体非整倍性。如由实例部分中提供的实验证明,本文中所提供的方法能够在检测极限或敏感性是0.2%的情况下针对至少一些SNV来检测样品中是否存在SNV,在一个说明性实施例中,其是至少一些SNV的检测极限。类似地,在某些实施例中,所述方法能够检测到SNV的出现率或SNV AAI是0.2、0.3、0.4、0.5、0.6、0.8、0.8、0.9或1.0%。也就是说,检验方法能够在检测极限低到SNV的染色体基因座处的全部等位基因计数的0.2、0.3、0.4、0.5、0.6、0.8、0.8、0.9或1.0%的情况下检测到样品中的SNV。
在一些实施例中,本发明的方法的突变(如SNV或CNV)的检测极限小于或等于10、5、2、1、0.5、0.1、0.05、0.01或0.005%。在一些实施例中,本发明的方法的突变(如SNV或CNV)的检测极限在15到0.005%之间,如在10到0.005%、10到0.01%、10到0.1%、5到0.005%、5到0.01%、5到0.1%、1到0.005%、1到0.01%、1到0.1%、0.5到0.005%、0.5到0.01%、0.5到0.1%或0.1到0.01之间且包括端值。
在一些实施例中,检测极限使得检测到(或能够检测到)样品(如cfDNA或cfRNA样品)中存在于小于或等于10、5、2、1、0.5、0.1、0.05、0.01或0.005%的具有所述基因座的DNA或RNA分子中的突变(如SNV或CNV)。举例来说,即使小于或等于10、5、2、1、0.5、0.1、0.05、0.01或0.005%的具有所述基因座的DNA或RNA分子在所述基因座中具有突变,仍可以检测到所述突变(而不是例如基因座的野生型或非突变型版本或所述基因座处的不同突变)。在一些实施例中,检测极限使得检测到(或能够检测到)样品(如cfDNA或cfRNA样品)中存在于小于或等于10、5、2、1、0.5、0.1、0.05、0.01或0.005%的DNA或RNA分子中的突变(如SNV或CNV)。在其中CNV是缺失的一些实施例中,即使缺失仅存在于样品中的小于或等于10、5、2、1、0.5、0.1、0.05、0.01或0.005%的DNA或RNA分子中,仍可以检测到所述缺失,所述DNA或RNA分子具有可能含有或可能不含有所述缺失的相关区域。在其中CNV是缺失的一些实施例中,即使缺失仅存在于样品中的小于或等于10、5、2、1、0.5、0.1、0.05、0.01或0.005%的DNA或RNA分子中,仍可以检测到所述缺失。在其中CNV是复制的一些实施例中,即使所存在的额外复制的DNA或RNA小于或等于样品中的DNA或RNA分子的10、5、2、1、0.5、0.1、0.05、0.01或0.005%,仍可以检测到复制,所述DNA或RNA分子具有样品中可能被复制或可能不被复制的相关区域。在其中CNV是复制的一些实施例中,即使所存在的额外复制的DNA或RNA小于或等于样品中的DNA或RNA分子的10、5、2、1、0.5、0.1、0.05、0.01或0.005%,仍可以检测到复制。
例示性样品
在本发明的任何方面的一些实施例中,样品包括来自怀疑具有缺失或复制的细胞(如怀疑具有癌性的细胞)的细胞性和/或细胞外遗传物质。在一些实施例中,样品包含任何怀疑含有具有缺失或复制的细胞、DNA或RNA的组织或体液,如肿瘤或其它包括癌细胞、DNA或RNA的样品。可以对任何包含DNA或RNA的样品进行用作这些方法的一部分的基因测量,所述样品例如(但不限于)组织、血液、血清、血浆、尿液、毛发、泪液、唾液、皮肤、指甲、粪便、胆汁、淋巴、子宫颈粘液、精液、肿瘤、或其它包含核酸的细胞或物质。样品可以包括任何细胞类型或可以使用来自任何细胞类型的DNA或RNA(如来自任何怀疑具有癌性的器官或组织的细胞,或神经元)。在一些实施例中,样品包括细胞核和/或粒线体DNA。在一些实施例中,样品是来自本文中所公开的任何目标个体。在一些实施例中,目标个体是癌症患者。
例示性样品包括含有cfDNA或cfRNA的样品。在一些实施例中,cfDNA在无需溶解细胞的步骤的情况下即可用于分析。游离DNA可以从多种组织获得,如呈液体形式的组织,例如血液、血浆、淋巴、腹水或脑脊髓液。在一些情况下,cfDNA包含来源于胚胎细胞的DNA。在一些情况下,从血浆分离cfDNA,所述血浆是从已被离心以去除细胞物质的全血分离。cfDNA可以是来源于目标细胞(如癌细胞)和非目标细胞(如非癌细胞)的DNA的混合物。
在一些实施例中,样品含有或怀疑含有DNA(或RNA)的混合物,如来源于癌细胞的DNA(或RNA)与来源于非癌性(即,正常)细胞的DNA(或RNA)的混合物。在一些实施例中,样品中至少0.5、1、3、5、7、10、15、20、30、40、50、60、70、80、90、92、94、95、96、98、99或100%的细胞是癌细胞。在一些实施例中,样品中至少0.5、1、3、5、7、10、15、20、30、40、50、60、70、80、90、92、94、95、96、98、99或100%的DNA(如cfDNA)或RNA(如cfRNA)是来自癌细胞。在各种实施例中,样品中的细胞的癌性细胞百分比在0.5到99%之间,如在1到95%、5到95%、10到90%、5到70%、10到70%、20到90%或20到70%之间且包括端值。在一些实施例中,样品富含癌细胞或来自癌细胞的DNA或RNA。在其中样品富含癌细胞的一些实施例中,富含癌细胞的样品中至少0.5、1、2、3、4、5、6、7、10、15、20、30、40、50、60、70、80、90、92、94、95、96、98、99或100%的细胞是癌细胞。在其中样品富含来自癌细胞的DNA或RNA的一些实施例中,富含来自癌细胞的DNA或RNA的样品中至少0.5、1、2、3、4、5、6、7、10、15、20、30、40、50、60、70、80、90、92、94、95、96、98、99或100%的DNA或RNA是来自癌细胞。在一些实施例中,使用细胞分选(如荧光活化细胞分选(FACS))来富集癌细胞(Barteneva等人,《生物化学与生物物理学报(Biochim Biophys Acta.)》1836(1):105-22,2013年8月.doi:10.1016/j.bbcan.2013.02.004.2013年2月24日电子版,和Ibrahim等人,《生化工程/生物技术进展(Adv Biochem Eng Biotechnol.)》106:19-39,2007,其各自以全文引用的方式并入本文中)。
在一些实施例中,样品富含胚胎细胞。在其中样品富含胚胎细胞的一些实施例中,富含胚胎细胞的样品中至少0.5、1、2、3、4、5、6、7%或更多的细胞是胚胎细胞。在一些实施例中,样品中的细胞的胚胎细胞百分比在0.5到100%之间,如在1到99%、5到95%、10到95%、10到95%、20到90%或30到70%之间且包括端值。在一些实施例中,样品富含胚胎DNA。在其中样品富含胚胎DNA的一些实施例中,富含胚胎DNA的样品中至少0.5、1、2、3、4、5、6、7%或更多的DNA是胚胎DNA。在一些实施例中,样品中的DNA的胚胎DNA百分比在0.5到100%之间,如在1到99%、5到95%、10到95%、10到95%、20到90%或30到70%之间且包括端值。
在一些实施例中,样品包括单细胞或包括来自单细胞的DNA和/或RNA。在一些实施例中,平行分析多个独立细胞(例如至少5、10、20、30、40或50个来自相同个体或来自不同个体的细胞)。在一些实施例中,组合来自相同个体的多个样品的细胞,其与分开地分析样品相比减少工作量。组合多个样品还可以允许针对癌症同时检验多个组织(其可以用于提供癌症筛检或用于更彻底的癌症筛检或确定癌症是否转移到其它组织)。
在一些实施例中,样品含有单细胞或少量细胞,如2、3、5、6、7、8、9或10个细胞。在一些实施例中,样品具有1到100、100到500或500到1,000个细胞且包括端值。在一些实施例中,样品含有1到10皮克、10到100皮克、100皮克到1纳克、1到10纳克、10到100纳克或100纳克到1微克RNA和/或DNA且包括端值。
在一些实施例中,将样品包埋于石蜡膜中。在一些实施例中,样品与防腐剂(如甲醛)一起保藏且任选地包覆在石蜡中,其可以引起DNA的交联,使得较少的DNA可以用于PCR。在一些实施例中,样品是甲醛固定的石蜡包埋(FFPE)样品。在一些实施例中,样品是新鲜样品(如由1或2天的分析获得的样品)。在一些实施例中,样品在分析之前被冷冻。在一些实施例中,样品是历史样品。
这些样品可以用于本发明的任何方法中。
例示性样品制备方法
在一些实施例中,方法包括分离或纯化DNA和/或RNA。所属领域中已知多种用于实现这类目的的标准程序。在一些实施例中,可以对样品进行离心以分离各层。在一些实施例中,可以使用过滤来分离DNA或RNA。在一些实施例中,DNA或RNA的制备可以涉及扩增、分离、通过色谱纯化、液液分离、隔离、优先富集、优先扩增、靶向扩增或所属领域中已知或本文中所描述的多种其它技术中的任一种。在分离DNA的一些实施例中,使用RNA酶使RNA降解。在分离RNA的一些实施例中,使用DNA酶(如来自Invitrogen,Carlsbad,CA,USA的DNA酶I)使DNA降解。在一些实施例中,使用RNeasy小型试剂盒(Qiagen)根据制造商方案分离RNA。在一些实施例中,使用mirVana PARIS试剂盒(Ambion,Austin,TX,USA)根据制造商方案(Gu等人,《神经化学杂志(J.Neurochem.)》122:641-649,2012,其以全文引用的方式并入本文中)分离小型RNA分子。可以任选地使用Nanovue(GE Healthcare,Piscataway,NJ,USA)确定RNA的浓度和纯度,且可以任选地使用2100Bioanalyzer(Agilent Technologies,SantaClara,CA,USA)测量RNA完整性(Gu等人,《神经化学杂志》122:641-649,2012,其以全文引用的方式并入本文中)。在一些实施例中,使用TRIZOL或RNAlater(Ambion)使RNA在储存期间稳定。
在一些实施例中,添加通用标记衔接子以制备库。在接合之前,可以对样品DNA进行末端平端化,且接着向3'端添加单一腺苷碱基。在接合之前,可以使用限制酶或某种其它裂解方法使DNA裂解。在接合期间,样品片段的3'腺苷和衔接子的互补性3'酪氨酸突出端可以增强接合效率。在一些实施例中,使用在AGILENT SURESELECT试剂盒中发现的接合试剂盒进行衔接子接合。在一些实施例中,使用通用引物扩增库。在一个实施例中,通过尺寸分离或通过使用如AGENCOURT AMPURE珠粒等产物或其它类似方法来将被扩增的库分级分离。在一些实施例中,使用PCR扩增来扩增目标基因座。在一些实施例中,对被扩增的DNA进行测序(如使用ILLUMINA IIGAX或HiSeq测序器进行测序)。在一些实施例中,从被扩增的DNA的每个末端对被扩增的DNA进行测序以减少测序误差。如果当从被扩增的DNA的一端进行测序时,具体碱基中存在序列误差,那么当从被扩增的DNA的另一侧进行测序时,互补碱基中不太可能存在序列误差(与从被扩增的DNA的同一个末端进行多次测序相比)。
在一些实施例中,使用全基因组应用(WGA)以扩增核酸样品。存在多种可以用于WGA的方法:接合介导的PCR(LM-PCR)、简并寡核苷酸引物PCR(DOP-PCR)以及多重置换扩增(MDA)。在LM-PCR中,称为衔接子的短DNA序列被接合到DNA的平末端。这些衔接子含有通用扩增序列,其用于通过PCR来扩增DNA。在DOP-PCR中,在第一轮粘接和PCR中使用随机引物,所述随机引物也含有通用扩增序列。接着,使用第二轮PCR以用通用引物序列进一步扩增序列。MDA使用phi-29聚合酶,其是一种复制DNA并且已被用于单细胞分析的高度进行性和非特异性酶。在一些实施例中,不进行WGA。
在一些实施例中,使用选择性扩增或富集来扩增或富集目标基因座。在一些实施例中,扩增和/或选择性富集技术可以涉及PCR(如接合介导的PCR)、通过杂交进行的片段捕获、分子倒置探针或其它环化中探针。在一些实施例中,使用实时定量PCR(RT-qPCR)、数字PCR或乳液PCR、单一等位基因碱基延伸反应,接着进行质谱分析(Hung等人,《临床病理学杂志(J Clin Pathol)》62:308-313,2009,其以全文引用的方式并入本文中)。在一些实施例中,用杂交捕获探针通过杂交进行的捕获用于优先富集DNA。在一些实施例中,用于扩增或选择性富集的方法可以涉及使用探针,其中在与目标序列正确杂交之后,核苷酸探针的3'端或5'端通过少量核苷酸与多态等位基因的多态位点分离。这种分离会减少一个等位基因的优先扩增,称为等位基因偏差。这是优于涉及使用探针的方法(其中正确杂交的探针的3'端或5'端与等位基因的多态位点直接相邻或非常靠近)的一种改进。在一个实施例中,排除其中杂交区可以或确定含有多态位点的探针。杂交位点处的多态位点可以引起一些等位基因的不相等杂交或抑制整体杂交,引起某些等位基因的优先扩增。这些实施例优于涉及靶向扩增和/或选择性富集的其它方法的改进之处在于,其更好地保持了样品在每个多态基因座处的初始等位基因出现率,无论样品是来自单一个体还是个体混合物的纯基因组样品。
在一些实施例中,使用PCR(称为微型PCR)产生极短的扩增子(2012年11月21日提交的美国申请案第13/683,604号、美国公开案第2013/0123120号、2011年11月18日提交的美国申请案第13/300,235号、2011年11月18日提交的美国公开案第2012/0270212号和2014年5月16日提交的美国序列号61/994,791,其各自以全文引用的方式并入本文中)。cfDNA(如以坏死方式或以细胞凋亡方式释放的癌症cfDNA)是高度片段化的。对于胚胎cfDNA,片段尺寸大致以高斯(Gaussian)方式分布,其中平均值是160bp,标准偏差是15bp,最小尺寸是约100bp且最大尺寸是约220bp。一个具体目标基因座的多态位点可以占据来源于所述基因座的各种片段中的从起点到末端的任何位置。因为cfDNA片段较短,所以两个引物位点存在的似然性,包含正向和反向引物位点的具有长度L的片段的似然性是扩增子长度与片段长度的比率。在理想条件下,其中扩增子是45、50、55、60、65或70bp的分析法将分别从72%、69%、66%、63%、59%或56%的可用模板片段分子成功地扩增。在最优选与来自怀疑患有癌症的个体的样品的cfDNA相关的某些实施例中,使用引物扩增cfDNA,所述引物产生85、80、75或70bp且在某些优选实施例中,75bp的最大扩增子长度且具有50与65℃之间且在某些优选实施例中,54-60.5℃之间的熔融温度。扩增子长度是正向和反向引发位点的5'端之间的距离。比所属领域的技术人员通常所使用的更短的扩增子长度可以通过仅需要短序列读段便产生所需多态基因座的更有效的测量结果。在一个实施例中,扩增子的实质部分小于100bp、小于90bp、小于80bp、小于70bp、小于65bp、小于60bp、小于55bp、小于50bp或小于45bp。
在一些实施例中,使用直接多重PCR、连续PCR、巢式PCR、双重巢式PCR、一又二分之一边巢式PCR、完全巢式PCR、单边完全巢式PCR、单边巢式PCR、半巢式PCR、半巢式PCR、三重半巢式PCR、半巢式PCR、单边半巢式PCR、反向半巢式PCR或单边PCR进行扩增,其描述于2012年11月21日提交的美国申请案第13/683,604号、美国公开案第2013/0123120号、2011年11月18日提交的美国申请案第13/300,235号、美国公开案第2012/0270212号和2014年5月16日提交的美国序列号61/994,791中,其以全文引用的方式并入本文中。视需要,这些方法中的任何方法都可以用于微型PCR。
视需要,可以从时间观点出发来限制PCR扩增的延伸步骤以减少从长度超过200个核苷酸、300个核苷酸、400个核苷酸、500个核苷酸或1,000个核苷酸的片段进行的扩增。这可以引起片段化或较短DNA(如经历细胞凋亡或坏死的胚胎DNA或来自癌细胞的DNA)的富集和检验性能的改善。
在一些实施例中,使用多重PCR。在一些实施例中,用于扩增核酸样品中的目标基因座的方法涉及(i)使核酸样品与引物库接触,所述引物同时与至少100;200;500;750;1,000;2,000;5,000;7,500;10,000;20,000;25,000;30,000;40,000;50,000;75,000;或100,000个不同目标基因座杂交以产生反应混合物;和(ii)使反应混合物经历引物延伸反应条件(如PCR条件)以产生包括目标扩增子的扩增产物。在一些实施例中,至少50、60、70、80、90、95、96、97、98、99或99.5%的目标基因座被扩增。在各种实施例中,小于60、50、40、30、20、10、5、4、3、2、1、0.5、0.25、0.1或0.05%的扩增产物是引物二聚体。在一些实施例中,引物在溶液中(如溶解于液相中而非在固相中)。在一些实施例中,引物在溶液中且未固定在固体负载物上。在一些实施例中,引物不是微阵列的一部分。在一些实施例中,引物不包括分子倒置探针(MIP)。
在一些实施例中,使两个或更多个(如3或4个)目标扩增子(如来自本文中所公开的微型PCR方法的扩增子)接合在一起且接着对接合产物进行测序。将多个扩增子组合成单一接合产物可以提高后续测序步骤的效率。在一些实施例中,目标扩增子在接合之前的长度小于150、100、90、75或50个碱基对。选择性富集和/或扩增可以涉及用不同的标签、分子条形码、用于扩增的标签和/或用于测序的标签来标记每个单独分子。在一些实施例中,通过测序(如通过高通量测序)或通过与阵列(如SNP阵列、ILLUMINA INFINIUM阵列或AFFYMETRIX基因芯片)杂交来分析扩增产物。在一些实施例中,使用纳米孔测序,如由Genia研发的纳米孔测序技术(参见例如万维网网址geniachip.com/technology,其以全文引用的方式并入本文中)。在一些实施例中,使用双重测序(施密特等人,《通过下一代测序来检测超罕见突变(Detection of ultra-rare mutations by next-generationsequencing)》,《美国国家科学院院刊》109(36):14508-14513,2012,其以全文引用的方式并入本文中)。这种方法通过对DNA双螺旋的两条链中的每一条独立地进行标记和测序来显著减少误差。由于两条链是互补的,因此在两条链中的相同位置处发现真实突变。相比之下,PCR或测序误差仅在一条链中引起突变且因此可以作为技术误差而忽略。在一些实施例中,所述方法需要用随机但互补的双链核苷酸序列(称为双螺旋标签)来标记双螺旋DNA的两条链。通过首先将单链随机化核苷酸序列引入一个衔接子链中且接着用DNA聚合酶使相对的链延伸,得到互补、双链标签来将双链标签序列并入标准测序衔接子中。在被标记的衔接子与被剪切的DNA接合之后,单独标记的链从衔接子尾部上的不对称引物位点进行PCR扩增且经历双端测序。在一些实施例中,将样品(如DNA或RNA样品)分成多个部分,如不同的孔(例如WaferGen SmartChip的孔)。将样品分成不同的部分(如至少5、10、20、50、75、100、150、200或300个部分)可以提高分析的敏感性,因为与整个样品相比,一些孔中的具有突变的分子的百分比更高。在一些实施例中,每个部分具有小于500、400、200、100、50、20、10、5、2或1个DNA或RNA分子。在一些实施例中,单独地对每个部分中的分子进行测序。在一些实施例中,向相同部分中的所有分子中添加(如通过用含有条形码的引物进行扩增或通过条形码的接合)相同的条形码(如随机或非人类序列),且向不同部分中的分子中添加不同的条形码。可以将带条形码的分子集中起来且共同测序。在一些实施例中,分子在集中和测序之前扩增,如通过使用巢式PCR。在一些实施例中,使用一个正向和两个反向引物,或两个正向和一个反向引物。
在一些实施例中,检测到(或能够检测到)样品(如cfDNA或cfRNA样品)中存在于小于10、5、2、1、0.5、0.1、0.05、0.01或0.005%的DNA或RNA分子中的突变(如SNV或CNV)。在一些实施例中,检测到(或能够检测到)样品(如来自例如血液样品的cfDNA或cfRNA样品)中存在于小于1,000、500、100、50、20、10、5、4、3或2个原始DNA或RNA分子(在扩增之前)中的突变(如SNV或CNV)。在一些实施例中,检测到(或能够检测到)样品(如来自例如血液样品的cfDNA或cfRNA样品)中仅存在于1个原始DNA或RNA分子(在扩增之前)中的突变(如SNV或CNV)。
举例来说,如果突变(如单核苷酸变异体(SNV))的检测极限是0.1%,那么可以通过将检验部分分成多个部分(如100个孔)来检测到以0.01%存在的突变。大部分孔不具有突变的拷贝。对于极少数的具有突变的孔,突变具有显著更高的读段百分比。在一个实例中,存在来自目标基因座的DNA的20,000个初始拷贝,且这些拷贝中的两个包括相关SNV。如果将样品分成100孔,那么98个孔具有SNV,且2个孔以0.5%的概率具有SNV。可以将每个孔中的DNA加注条形码、扩增、与来自其它孔的DNA集中在一起且测序。不具有SNV的孔可以用于测量背景扩增/测序误差率,以确定来自离群孔的信号是否高于背景噪声水平。
在一些实施例中,使用阵列检测扩增产物,如具有针对一种或多种相关染色体(例如染色体13、18、21、X、Y或其任何组合)的探针的阵列,尤其微阵列。举例来说,应理解,可以使用可商购的SNP检测微阵列,例如Illumina(San Diego,CA)GoldenGate、DASL、Infinium或CytoSNP-12基因分型分析法,或来自Affymetrix的SNP检测微阵列产品,如OncoScan微阵列。
在涉及测序的一些实施例中,读段深度是映射到既定基因座的测序读段的数目。可以针对读段总数将读段深度标准化。在样品的读段深度的一些实施例中,读段深度是目标基因座的平均读段深度。在基因座的读段深度的一些实施例中,读段深度是由映射到所述基因座的测序器测量的读段数目。通常,基因座的读段深度越大,基因座处的等位基因的比率越倾向于接近原始DNA样品中的等位基因的比率。读段深度可以多种不同方式表示,包括(但不限于)百分比或比例。因此,举例来说,在例如产生1百万个克隆的序列的高度平行DNA测序器(如Illumina HISEQ)中,一个基因座的3,000次测序产生所述基因座处的3,000个读段的读段深度。所述基因座处的读段的比例是3,000除以1百万个全部读段,或全部读段的0.3%。
在一些实施例中,获得等位基因数据,其中等位基因数据包括指示多态基因座的特异性等位基因的拷贝数目的定量测量结果。在一些实施例中,等位基因数据包括指示在多态基因座处观察的每个等位基因的拷贝数目的定量测量结果。通常,获得相关多态基因座的所有可能的等位基因的定量测量结果。举例来说,先前段落中讨论的任何用于确定SNP或SNV基因座的等位基因的方法(例如微阵列、qPCR、DNA测序,如高通量DNA测序)都可以用于产生多态基因座的特异性等位基因的拷贝数目的定量测量结果。这种定量测量结果在本文中称为对偶基因出现率数据或所测量的遗传等位基因数据。使用等位基因数据的方法有时称为定量对偶基因方法;这与仅使用来自非多态基因座或来自多态基因座,但不考虑对偶基因一致性的定量数据的定量方法不同。当使用高通量测序来测量等位基因数据时,等位基因数据通常包括映射到相关基因座的每个等位基因的读段数目。
在一些实施例中,获得非等位基因数据,其中非等位基因数据包括指示特异性基因座的拷贝数目的定量测量结果。基因座可以是多态或非多态的。在一些实施例中,当基因座是非多态的时,非等位基因数据不含关于可能存在于所述基因座处的单独等位基因的相对数量或绝对数量的信息。仅使用非等位基因数据(也就是说,来自非多态等位基因的定量数据,或来自多态基因座,但不考虑每个片段的对偶基因一致性的定量数据)的方法称为定量方法。通常,获得相关多态基因座的所有可能的等位基因的定量测量结果,其中总共一个值与所述基因座处的所有等位基因的所测量的数量相关联。可以通过将所述基因座处的每个等位基因的定量对偶基因求和来获得多态基因座的非等位基因数据。当使用高通量测序来测量等位基因数据时,非等位基因数据通常包括映射到相关基因座的读段的数目。测序测量结果可以指示存在于所述基因座处的每种等位基因的相对和/或绝对数目,且非等位基因数据包括映射到基因座的读段的总数而与对偶基因一致性无关。在一些实施例中,相同的测序测量结果集合可以用于产生等位基因数据和非等位基因数据。在一些实施例中,使用等位基因数据作为用于确定相关染色体处的拷贝数目的方法的一部分,且可以使用所产生的非等位基因数据作为不同的用于确定相关染色体处的拷贝数目的方法的一部分。在一些实施例中,两种方法以统计方式正交,且组合以实现相关染色体处的拷贝数目的更精确的确定。
在一些实施例中,获得基因数据包括(i)由实验室技术获取DNA序列信息,例如通过使用自动高通量DNA测序器,或(ii)获取先前由实验室技术获得的信息,其中所述信息是以电子方式传送,例如由计算机通过因特网传送或通过由测序装置进行电子转移来传送。
其它例示性样品制备、扩增和定量方法描述于2012年11月21日提交的美国申请案第13/683,604号(美国公开案第2013/0123120号和2014年5月16日提交的美国序列号61/994,791,其以全文引用的方式并入本文中)中。这些方法可以用于分析本文中所公开的任何样品。
用于游离DNA的例示性定量方法
视需要,可以使用标准方法测量cfDNA或cfRNA的量或浓度。在一些实施例中,确定游离粒线体DNA(cf mDNA)的量或浓度。在一些实施例中,确定来源于细胞核DNA的游离DNA(cf nDNA)的量或浓度。在一些实施例中,同时确定cf mDNA和cf nDNA的量或浓度。
在一些实施例中,使用qPCR测量cf nDNA和/或cfm DNA(Kohler等人,《作为乳房肿瘤的潜在生物标记物的血浆循环游离细胞核和粒线体DNA的含量(Levels of plasmacirculating cell free nuclear and mitochondrial DNA as potential biomarkersfor breast tumors)》,《分子癌症(Mol Cancer)》8:105,2009,8:doi:10.1186/1476-4598-8-105,其以全文引用的方式并入本文中)。举例来说,可以使用多重qPCR测量来自cf nDNA的一种或多种基因座(如甘油醛-3-磷酸脱氢酶,GAPDH)和来自cf mDNA的一种或多种基因座(ATP酶8,MTATP 8)。在一些实施例中,使用荧光标记的PCR测量cf nDNA和/或cf mDNA(Schwarzenbach等人,《乳癌和良性乳房疾病患者中游离肿瘤DNA和RNA的评估(Evaluationof cell-free tumour DNA and RNA in patients with breast cancer and benignbreast disease)》,《分子生物系统(Mol Biosys)》7:2848-2854,2011,其以全文引用的方式并入本文中)。视需要,可以使用标准方法(如夏皮罗-威尔克检验(Shapiro-Wilk-Test))确定数据的正态分布。视需要,可以使用标准方法(如曼-惠特尼U检验(Mann-Whitney-U-Test))比较cf nDNA和mDNA含量。在一些实施例中,使用标准方法(如曼-惠特尼U检验或克鲁斯卡尔-沃利斯检验(Kruskal-Wallis-Test))比较cf nDNA和/或mDNA含量与其它确认的预后因子。
例示性RNA扩增、定量和分析方法
任何以下例示性方法都可以用于扩增和任选地定量RNA,如cfRNA、细胞RNA、细胞质RNA、编码细胞质RNA、非编码细胞质RNA、mRNA、miRNA、线粒体RNA、rRNA或tRNA。在一些实施例中,miRNA是可在万维网网址mirbase.org获得的miRBase数据库中列出的任何miRNA分子,其以全文引用的方式并入本文中。例示性miRNA分子包括miR-509;miR-21和miR-146a。
在一些实施例中,使用逆转录酶多重接合依赖性探针扩增(RT-MLPA)来扩增RNA。在一些实施例中,每个杂交探针集合由两个跨越SNP的短合成寡核苷酸和一个长寡核苷酸组成(Li等人,《妇产科医学档案(Arch Gynecol Obstet.)》,《用新的SNP标记物集合通过RT-MLPA进行第21对染色体三体症的非侵袭性产前诊断的发展(Development ofnoninvasive prenatal diagnosis of trisomy 21 by RT-MLPA with a new set of SNPmarkers)》,2013年7月5日,DOI 10.1007/s00404-013-2926-5;Schouten等人,《通过多重接合依赖性探针扩增进行的40个核酸序列的相对定量(Relative quantification of40nucleic acid sequences by multiplex ligation-dependent probeamplification)》,《核酸研究(Nucleic Acids Res)》30:e57,2002;Deng等人(2011)《通过逆转录酶多重接合依赖性探针扩增进行的第21对染色体三体症的非侵袭性产前诊断(Non-invasive prenatal diagnosis of trisomy 21by reverse transcriptase multiplexligation-dependent probe amplification)》,《临床化学与实验医学(Clin,Chem.LabMed.)》49:641-646,2011,其各自以全文引用的方式并入本文中)。
在一些实施例中,用逆转录酶PCR扩增RNA。在一些实施例中,如先前所描述,用实时逆转录酶PCR扩增RNA,如使用SYBR GREEN I的单步骤实时逆转录酶PCR(Li等人,《妇产科医学档案》,《用新的SNP标记物集合通过RT-MLPA进行第21对染色体三体症的非侵袭性产前诊断的发展》,2013年7月5日,DOI10.1007/s00404-013-2926-5;Lo等人,《血浆胎盘RNA等位基因比率实现非侵袭性产前染色体非整倍性检测(Plasma placental RNA allelic ratiopermits noninvasive prenatal chromosomal aneuploidy detection)》,《自然医学(NatMed)》13:218-223,2007;Tsui等人,《基于全身微阵列的母体血浆中胎盘mRNA的鉴别:针对非侵袭性产前基因表达谱分析(Systematic micro-array based identification ofplacental mRNA in maternal plasma:towards non-invasive prenatal geneexpression profiling.)》,《遗传医学杂志(J Med Genet)》41:461-467,2004;Gu等人,《神经化学杂志(J.Neurochem.)》122:641-649,2012,其各自以全文引用的方式并入本文中)。
在一些实施例中,使用微阵列检测RNA。举例来说,可根据制造商方案使用来自Agilent Technologies的人类miRNA微阵列。简单来说,将被分离的RNA脱磷酸化且与pCp-Cy3接合。基于14.0版Sanger miRBase,将被标记的RNA纯化且与含有针对人类成熟miRNA的探针的miRNA阵列杂交。清洗阵列且使用微阵列扫描仪(G2565BA,Agilent Technologies)扫描。通过Agilent提取软件v9.5.3评估每个杂交信号的强度。标记、杂交和扫描可以根据Agilent miRNA微阵列系统中的方案进行(Gu等人,《神经化学杂志(J.Neurochem.)》122:641-649,2012,其以全文引用的方式并入本文中)。
在一些实施例中,使用TaqMan分析法检测RNA。例示性分析法是TaqMan ArrayHuman MicroRNA Panel v1.0(早期访问)(Applied Biosystems),其含有157种TaqManMicroRNA分析法,包括各别逆转录引物、PCR引物和TaqMan探针(Chim等人,《母体血浆中胎盘微RNA的检测和表征(Detection and characterization of placental microRNAs inmaternal plasma)》,《临床化学(Clin Chem.)》54(3):482-90,2008,其以全文引用的方式并入本文中)。
视需要,可以使用标准方法确定一种或多种mRNA的mRNA剪接模式(Fackenthal1和Godley,《疾病模型和机制(Disease Models&Mechanisms)》1:37-42,2008,doi:10.1242/dmm.000331,其以全文引用的方式并入本文中)。举例来说,可以使用高密度微阵列和/或高通量DNA测序来检测mRNA剪接变异体。
在一些实施例中,使用完全转录组鸟枪法测序或阵列测量转录组。
例示性扩增方法
还研发了改进的PCR扩增方法,其最小化或防止由同一个反应体积中的邻近或相邻目标基因座的扩增引起的干扰(如同时扩增所有目标基因座的样品多重PCR反应的一部分)。这些方法可以用于同时扩增邻近或相邻目标基因座,其与必须将邻近的目标基因座分离成不同的反应体积使得其可以单独地扩增以避免干扰相比更快且成本更低。
在一些实施例中,使用具有低5'→3'核酸外切酶和/或低链置换活性的聚合酶(例如DNA聚合酶、RNA聚合酶或逆转录酶)进行目标基因座的扩增。在一些实施例中,少量的5'→3'核酸外切酶可以减少或防止邻近引物(例如未延伸的引物或在引物延伸期间添加有一个或多个核苷酸的引物)的降解。在一些实施例中,少量的链置换活性可以减少或防止邻近引物(例如未延伸的引物或在引物延伸期间添加有一个或多个核苷酸的引物)的置换。在一些实施例中,扩增彼此相邻(例如目标基因座之间不存在碱基)或邻近(例如基因座相距50、40、30、20、15、10、9、8、7、6、5、4、3、2或1个碱基以内)的目标基因座。在一些实施例中,一个基因座的3'端与下一个下游基因座的5'端相距50、40、30、20、15、10、9、8、7、6、5、4、3、2或1个碱基以内。
在一些实施例中,扩增至少100、200、500、750、1,000、2,000、5,000、7,500、10,000、20,000、25,000、30,000、40,000、50,000、75,000或100,000个不同的目标基因座,如通过在一个反应体积中同时扩增。在一些实施例中,至少50、60、70、80、90、95、96、97、98、99或99.5%的扩增产物是目标扩增子。在各种实施例中,作为目标扩增子的扩增产物的量在50到99.5%之间,如在60到99%、70到98%、80到98%、90到99.5%或95到99.5%之间且包括端值。在一些实施例中,扩增(例如与扩增之前的量相比扩增至少5、10、20、30、50或100倍)至少50、60、70、80、90、95、96、97、98、99或99.5%的目标基因座,如通过在一个反应体积中同时扩增。在各种实施例中,扩增(例如与扩增之前的量相比扩增至少5、10、20、30、50或100倍)的目标基因座的量在50到99.5%之间,如在60到99%、70到98%、80到99%、90到99.5%、95到99.9%或98到99.99%之间且包括端值。在一些实施例中,产生较少的非目标扩增子,如由来自第一引物对的正向引物和来自第二引物对的反向引物形成的较少扩增子。如果例如来自第一引物对的反向引物和/或来自第二引物对的正向引物发生降解和/或置换,那么这类不合需要的非目标扩增子可以使用先前扩增方法产生。
在一些实施例中,这些方法允许使用更长的延伸时间,因为鉴于聚合酶的低5'→3'核酸外切酶和/或低链置换活性,结合于被延伸的引物的聚合酶不太可能使邻近引物(如下一个下游引物)发生降解和/或置换。在各种实施例中,使用反应条件(如延伸时间和温度)使得聚合酶的延伸率允许添加到被延伸的引物中的核苷酸的数目等于或大于同一条链上的引物结合位点的3'端与下一个下游引物结合位点的5'端之间的核苷酸的数目的80、90、95、100、110、120、130、140、150、175或200%。
在一些实施例中,使用DNA作为模板,使用DNA聚合酶产生DNA扩增子。在一些实施例中,使用DNA作为模板,使用RNA聚合酶产生RNA扩增子。在一些实施例中,使用RNA作为模板,使用逆转录酶产生cDNA扩增子。
在一些实施例中,在相同条件下,聚合酶中的少量的5'→3'核酸外切酶小于相同量的
在一些实施例中,聚合酶是PUSHION DNA聚合酶,如PHUSION High Fidelity DNA聚合酶(M0530S,New England BioLabs,Inc)或PHUSION Hot Start Flex DNA聚合酶(M0535S,New England BioLabs,Inc.;Frey和Suppman,《生物化学(BioChemica.)》2:34-35,1995;Chester和Marshak,《分析生物化学(Analytical Biochemistry.)》209:284-290,1993,其各自以全文引用的方式并入本文中)。PHUSION DNA聚合酶是与持续合成能力增强域融合的火球菌(Pyrococcus)样酶。PHUSION DNA聚合酶具有5'→3'聚合酶活性和3'→5'核酸外切酶活性,且产生平端产物。PHUSION DNA聚合酶不具有5'→3'核酸外切酶活性和链置换活性。
在一些实施例中,聚合酶是
在一些实施例中,聚合酶是T4DNA聚合酶(M0203S,New England BioLabs,Inc.;Tabor和Struh.(1989).《依赖DNA的DNA聚合酶(DNA-Dependent DNA Polymerases)》,Ausebel等人(编),《现代分子生物学实验技术(Current Protocols in MolecularBiology.)》3.5.10-3.5.12.New York:John Wiley&Sons,Inc.,1989;Sambrook等人,《分子克隆:实验指南(Molecular Cloning:A Laboratory Manual)》(第2版),5.44-5.47.ColdSpring Harbor:Cold Spring Harbor Laboratory Press,1989其以全文引用的方式并入本文中)。T4DNA聚合酶以5'→3'方向催化DNA的合成且需要存在模板和引物。这种酶具有3'→5'核酸外切酶活性,其活性显著高于在DNA聚合酶I中发现的活性。T4DNA聚合酶不具有5'→3'核酸外切酶活性和链置换活性。
在一些实施例中,聚合酶是硫化叶菌(Sulfolobus)DNA聚合酶IV(M0327S,NewEngland BioLabs,Inc.;Boudsocq等人,(2001).《核酸研究》,29:4607-4616,2001;McDonald等人,(2006).《核酸研究》,34:1102-1111,2006,其各自以全文引用的方式并入本文中)。硫化叶菌DNA聚合酶IV是热稳定Y家族病变旁路DNA聚合酶,其跨越多种DNA模板病变有效合成DNA(McDonald,J.P.等人(2006).《核酸研究》,34,1102-1111,其以全文引用的方式并入本文中)。硫化叶菌DNA聚合酶IV不具有5'→3'核酸外切酶活性和链置换活性。
在一些实施例中,如果引物与具有SNP的区域结合,那么引物可以按不同效率来结合和扩增不同等位基因或可以仅结合和扩增一种等位基因。对于杂合性个体,一种等位基因可能不由引物扩增。在一些实施例中,设计用于每种等位基因的引物。举例来说,如果存在两个等位基因(例如双等位基因SNP),那么两个引物可以用于结合目标基因座的相同位置(例如用于结合“A”等位基因的正向引物和用于结合“B”等位基因的正向引物)。标准方法(如dbSNP数据库)可以用于确定已知的SNP(如具有高杂合率的SNP热点)的位置。
在一些实施例中,扩增子在尺寸方面是类似的。在一些实施例中,目标扩增子的长度范围是小于100、75、50、25、15、10或5个核苷酸。在一些实施例中(如片段化DNA或RNA中目标基因座的扩增),目标扩增子的长度在50与100个核苷酸之间,如在60与80个核苷酸或60与75个核苷酸之间且包括端值。在一些实施例中(如整个外显子或基因中的多个目标基因座的扩增),目标扩增子的长度在100与500个核苷酸之间,如在150与450核苷酸、200与400个核苷酸、200与300个核苷酸或300与400个核苷酸之间且包括端值。
在一些实施例中,使用引物对同时扩增多个目标基因座,所述引物对包括用于所述反应体积中的待扩增的每个目标基因座的正向和反向引物。在一些实施例中,每个目标基因座用单一引物进行一轮PCR,且接着每个目标基因座用一个引物对进行第二轮PCR。举例来说,可以每个目标基因座用单一引物进行第一轮PCR,使得所有引物结合相同的链(如对每个目标基因座使用正向引物)。这使得PCR以线性方式扩增且减少或消除扩增子之间的由序列或长度差而引起的扩增偏差。在一些实施例中,接着对每个目标基因座使用正向和反向引物来扩增扩增子。
例示性引物设计方法
视需要,可以使用具有降低的形成引物二聚体的似然性的引物进行多重PCR。具体地说,高度多重PCR通常会产生极高比例的由非生产性副反应(如引物二聚体形成)产生的产物DNA。在一个实施例中,可以从引物库去除最有可能引起非生产性副反应的具体引物,得到将产生更大比例的映射到基因组的扩增DNA的引物库。去除有问题的引物(也就是说,特别有可能形成二聚体的引物)的步骤已经出乎意料地实现了极其高的PCR复用水平,以便通过测序进行后续分析。
存在多种用于从库中选择使非映射引物二聚体或其它引物故障产物的量降到最低的引物的方式。经验数据表明,少量‘坏’引物造成了大量非映射引物二聚体副反应。去除这些‘坏’引物可以增加映射到目标基因座的序列读段的百分比。鉴别‘坏’引物的一种方式是查看通过目标扩增而被扩增的DNA的测序数据;可以去除所发现的具有最大出现率的引物二聚体,得到明显不太可能产生不映射到基因组的副产物DNA的引物库。还存在公开可用的可以计算各种引物组合的结合能的程序,并且去除结合能最高的引物组合也将得到明显不太可能产生不映射到基因组的副产物DNA的引物库。
在用于选择引物的一些实施例中,通过将一或多个引物或引物对设计为候选目标基因座来创建初始候选引物库。可以基于公开可用的关于目标基因座的所需参数的信息来选择一组候选目标基因座(例如SNP),所述信息是例如在目标群体内SNP的出现率或SNP的杂合率。在一个实施例中,可以使用Primer3程序(万维网网址primer3.sourceforge.net;libprimer3版本2.2.3,其以全文引用的方式并入本文中)设计PCR引物。视需要,引物可以被设计成在具体粘接温度范围内粘接、具有具体范围的GC含量、具有具体尺寸范围、产生在具体尺寸范围内的目标扩增子和/或具有其它参数特征。以每种候选目标基因座多个引物或引物对为起始物质增加了引物或引物对针对大部分或所有目标基因座将保留在库中的似然性。在一个实施例中,选择准则可能需要每个目标基因座至少一个引物对保留在库中。以这种方式,大部分或所有目标基因座将在使用最终引物库时被扩增。这正是以下应用所需要的:如筛检基因组中的大量位置处的缺失或复制,或筛检与疾病或增加的疾病风险相关联的大量序列(如多态现象或其它突变)。如果来自库的一个引物对将产生与由另一个引物对产生的目标扩增子重叠的目标扩增子,那么可以从库中去除所述引物对中的一个以防止干扰。
在一些实施例中,计算(例如在计算机上计算)来自候选引物库的两种引物的大部分或所有可能组合的“不理想评分”(越高的评分表示越小的合意性)。在不同实施例中,计算库中至少80、90、95、98、99或99.5%的可能的候选引物组合的不理想评分。每个不理想评分至少部分地基于在两种候选引物之间形成二聚体的似然性。视需要,不理想评分还可以基于一个或多个选自由以下组成的群组的其它参数:目标基因座的杂合率、与目标基因座处的序列(例如,多态现象)相关联的疾病流行率、与目标基因座处的序列(例如,多态现象)相关联的疾病外显率、候选引物对目标基因座的特异性、候选引物的尺寸、目标扩增子的熔融温度、目标扩增子的GC含量、目标扩增子的扩增效率、目标扩增子的尺寸和与重组热点的中心的距离。在一些实施例中,候选引物对目标基因座的特异性包括候选引物由于结合和扩增除其被设计成应该扩增的目标基因座以外的基因座而发生错物引发的似然性。在一些实施例中,从库中去除一种或多种或所有发生错物引发的候选引物。在一些实施例中,为了增加所选择的候选引物的数目,不从库中去除可能发生错误引发的候选引物。如果考虑多个因素,那么不理想评分可以基于各种参数的加权平均值来计算。参数可以基于其对于将使用引物的具体应用的重要性而分配不同的权重。在一些实施例中,从库中去除不理想评分最高的引物。如果所去除的引物是与一个目标基因座杂交的引物对的成员,那么可以从库中去除所述引物对的另一个成员。可以视需要重复去除引物的过程。在一些实施例中,进行所述选择方法直到库中剩余的候选引物组合的不理想评分全部等于或低于最小阈值。在一些实施例中,进行所述选择方法直到库中剩余的候选引物的数量减少到所需数量为止。
在各种实施例中,在计算不理想评分之后,从库中去除作为两种候选引物的最大数量组合中的不理想评分高于第一最小阈值的部分的候选引物。这个步骤忽略了等于或低于第一最小阈值的相互作用,因为这些相互作用不太重要。如果所去除的引物是与一个目标基因座杂交的引物对的成员,那么可以从库中去除所述引物对的另一个成员。可以视需要重复去除引物的过程。在一些实施例中,进行所述选择方法直到库中剩余的候选引物组合的不理想评分全部等于或低于第一最小阈值。如果库中剩余的候选引物的数量高于所需数量,那么可以通过将第一最小阈值降低到更低的第二最小阈值并且重复去除引物的过程来减少引物数量。如果库中剩余的候选引物的数量低于所需数量,那么可以通过将第一最小阈值增加到更高的第二最小阈值并且使用原始候选引物库重复去除引物的过程来继续进行所述方法,从而实现库中剩余更多的候选引物。在一些实施例中,进行所述选择方法直到库中剩余的候选引物组合的不理想评分全部等于或低于第二最小阈值,或直到库中剩余的候选引物的数量减少到所需数量。
视需要,可以将产生与由另一个引物对产生的目标扩增子重叠的目标扩增子的引物对分到分开的扩增反应中。对于需要分析所有候选目标基因座(而不是由于重叠目标扩增子而从分析中省略候选目标基因座)的应用,可能需要多个PCR扩增反应。
这些选择方法使必须从库中去除的候选引物的数量降到最低,实现了引物二聚体的所需减少。通过从库中去除更少数量的候选引物,可以使用所得引物库扩增更多(或所有)的目标基因座。
复用大量引物向可以被包括的分析法施加了大量限制。无意地相互作用的分析法会产生假性扩增产物。微型PCR的尺寸限制可以引起进一步限制。在一个实施例中,有可能以极大量的潜在SNP目标(在约500到大于1百万之间)为起始物质并且试图设计扩增每个SNP的引物。当可以设计引物时,有可能试图通过使用针对DNA双螺旋体形成的公开热力学参数评估在所有可能的引物对之间形成假性引物双螺旋体的似然性来鉴别可能形成假性产物的引物对。引物相互作用可以通过与相互相用相关的评分功能进行分级并且消除相互相用评分最差的引物直到满足所需引物数量。在其中SNP可能具有杂合性最适用的情况下,也有可能对分析法清单进行分级并且选择杂合相容性最高的分析法。实验已经验证,相互相用评分高的引物最有可能形成引物二聚体。在高复用下,不可能消除所有假性相互作用,但必需去除计算机模拟中相互相用评分最高的引物或引物对,因为其会主导整个反应,极大地限制预定目标的扩增。我们已经进行这个程序以创建具有多达并且在一些情况下,超过10,000个引物的多重引物组。由于这个程序,改进是显著的,与来自没有去除最差引物的反应的10%相比,实现对目标产物进行超过80%、超过90%、超过95%、超过98%且甚至超过99%的扩增,如通过所有PCR产物的测序所确定。当与如先前所述的部分半巢式方法组合时,超过90%且甚至超过95%的扩增子可以映射到目标序列。
应注意,存在用于确定哪些PCR探针可能形成二聚体的其它方法。在一个实施例中,分析已经使用非优化的引物集合扩增的DNA池可能足以确定有问题的引物。举例来说,可以使用测序进行分析,并且确定以最大数量存在的二聚体最有可能形成二聚体且可以将其去除。在一个实施例中,引物设计方法可以与本文中所描述的微型PCR方法组合使用。
在引物上使用标签可以减少引物二聚体产物的扩增和测序。在一些实施例中,引物含有与标签形成环结构的内部区域。在具体实施例中,引物包括对目标基因座具有特异性的5'区域、对目标基因座不具有特异性且形成环结构的内部区域以及对目标基因座具有特异性的3'区域。在一些实施例中,环区域可以处于两个结合区之间,其中两个结合区被设计成结合于模板DNA的邻近或相邻区域。在各种实施例中,3'区域的长度是至少7个核苷酸。在一些实施例中,3'区域的长度在7与20个核苷酸之间,例如在7到15个核苷酸或7到10个核苷酸之间且包括端值。在各种实施例中,引物包括对目标基因座不具有特异性的5'区域(如标签或通用引物结合位点),接着是对目标基因座具有特异性的区域、对目标基因座不具有特异性且形成环结构的内部区域以及对目标基因座具有特异性的3'区域。标签-引物可以用于将必需的目标特异性序列缩短到少于20、少于15、少于12且甚至少于10个碱基对。这可以是在标准引物设计的情况下,当使引物结合位点内的目标序列片段化或其可以被设计到引物设计中时偶然发现的。这种方法的优点包括:其增加了可以被设计用于某一最大扩增子长度的分析法的数量,并且其缩短了引物序列的“非信息性”测序。其也可以与内部标记组合使用。
在一个实施例中,多重靶向PCR扩增中的非生产性产物的相对量可以通过升高粘接温度来减少。在含有与目标特异性引物相同的标签的扩增库的情况下,粘接温度可以相比于基因组DNA有所提高,因为标签将有助于引物结合。在一些实施例中,使用降低的引物浓度,任选地与更长的粘接时间一起。在一些实施例中,粘接时间可以超过3分钟、超过5分钟、超过8分钟、超过10分钟、超过15分钟、超过20分钟、超过30分钟、超过60分钟、超过120分钟、超过240分钟、超过480分钟且甚至超过960分钟。在某些说明性实施例中,使用更长的粘接时间和降低的引物浓度。在各种实施例中,使用超过正常延伸的时间,例如超过3、5、8、10或15分钟。在一些实施例中,引物浓度低到50nM、20nM、10nM、5nM、1nM以及低于1nM。这意外地产生了高度多重反应的稳定性能,例如1,000重反应、2,000重反应、5,000重反应、10,000重反应、20,000重反应、50,000重反应且甚至100,000重反应。在一个实施例中,扩增使用一个、两个、三个、四个或五个用长粘接时间操作的循环,接着是用更常用的粘接时间和被标记的引物进行的PCR循环。
为了选择目标位置,可以从一池候选引物对设计开始并且创建引物对之间的潜在不利相互作用的热力学模型,且接着使用所述模型消除与池中的其它设计不相容的设计。
在一个实施例中,本发明提供用于降低目标基因座(如可能含有与疾病或病症或增加的疾病或病症(如癌症)风险相关联的多态现象或突变的基因座)的数目和/或增加所检测的疾病负荷(例如增加所检测的多态现象或突变的数目)的方法。在一些实施例中,所述方法包括由患有疾病或病症(如癌症)的个体中的每个基因座中的多态现象或突变(如单核苷酸变异、插入或缺失,或本文中所描述的任何其它变异)的出现率或复发对基因座进行分级(如从最高到最低分级)。在一些实施例中,PCR引物被设计成针对一些或全部基因座。在选择引物库的PCR引物期间,与具有较低出现率或复发的基因座(分级较低的基因座)相比,针对具有较高出现率或复发的基因座(分级较高的基因座)的引物是有利的。在一些实施例中,包括这一参数作为本文中所描述的不理想评分的计算中的一个参数。视需要,与库中的其它设计不相容的引物(如针对高分级基因座的引物)可以包括在不同的PCR库/池中。在一些实施例中,在单独的PCR反应中使用多个库/池(如2、3、4、5个或更多个)以实现由所有库/池表示的所有(或大部分)基因座的扩增。在一些实施例中,持续进行这一方法直到一个或多个库/池中包括足够的引物,使得全部引物能够实现捕获疾病或病症的所需疾病负荷(例如通过检测至少80、85、90、95或99%的疾病负荷)。
例示性引物库
在一个方面中,本发明提供引物库,如使用本发明的任何方法从候选引物库选择的引物。在一些实施例中,所述库包括在一个反应体积中同时杂交(或能够同时杂交)或同时扩增(或能够同时扩增)至少100、200、500、750、1,000、2,000、5,000、7,500、10,000、20,000、25,000、30,000、40,000、50,000、75,000或100,000个不同的目标基因座的引物。在各种实施例中,所述库包括在一个反应体积中同时扩增(或能够同时扩增)100到500、500到1,000、1,000到2,000、2,000到5,000、5,000到7,500、7,500到10,000、10,000到20,000、20,000到25,000、25,000到30,000、30,000到40,000、40,000到50,000、50,000到75,000或75,000到100,000个不同目标基因座的引物且包括端值。在各种实施例中,所述库包括在一个反应体积中同时扩增(或能够同时扩增)1,000到100,000个不同的目标基因座,如1,000到50,000、1,000到30,000、1,000到20,000、1,000到10,000、2,000到30,000、2,000到20,000、2,000到10,000、5,000到30,000、5,000到20,000或5,000到10,000个不同的目标基因座且包括端值。在一些实施例中,所述库包括在一个反应体积中同时扩增(或能够同时扩增)目标基因座以使得小于60、40、30、20、10、5、4、3、2、1、0.5、0.25、0.1或0.5%的扩增产物是引物二聚体的引物。在各个实施例中,作为引物二聚体的扩增产物的量在0.5到60%之间,例如在0.1到40%、0.1到20%、0.25到20%、0.25到10%、0.5到20%、0.5到10%、1到20%或1到10%之间且包括端值。在一些实施例中,引物在一个反应体积中同时扩增(或能够同时扩增)目标基因座,使得至少50、60、70、80、90、95、96、97、98、99或99.5%的扩增产物是目标扩增子。在各种实施例中,作为目标扩增子的扩增产物的量在50到99.5%之间,如在60到99%、70到98%、80到98%、90到99.5%或95到99.5%之间且包括端值。在一些实施例中,引物在一个反应体积中同时扩增(或能够同时扩增)目标基因座,使得至少50、60、70、80、90、95、96、97、98、99或99.5%的目标基因座被扩增(例如与扩增之前的量相比,扩增至少5、10、20、30、50或100倍)。在各种实施例中,扩增(例如与扩增之前的量相比扩增至少5、10、20、30、50或100倍)的目标基因座的量在50到99.5%之间,如在60到99%、70到98%、80到99%、90到99.5%、95到99.9%或98到99.99%之间且包括端值。在一些实施例中,引物库包括至少100、200、500、750、1,000、2,000、5,000、7,500、10,000、20,000、25,000、30,000、40,000、50,000、75,000或100,000个引物对,其中每对引物包括正向检验引物和反向检验引物,其中每对检验引物与目标基因座杂交。在一些实施例中,引物库包括至少100、200、500、750、1,000、2,000、5,000、7,500、10,000、20,000、25,000、30,000、40,000、50,000、75,000或100,000个各自与不同目标基因座杂交的单独引物对,其中单独引物不是引物对的一部分。
在各种实施例中,每种引物的浓度小于100、75、50、25、20、10、5、2或1nM,或小于500、100、10或1μM。在各种实施例中,每种引物的浓度在1μM到100nM之间,如在1μM到1nM、1到75nM、2到50nM或5到50nM之间且包括端值。在各种实施例中,引物的GC含量在30到80%之间,如在40到70%或50到60%之间且包括端值。在一些实施例中,引物的GC含量的范围是小于30、20、10或5%。在一些实施例中,引物的GC含量的范围在5到30%,如5到20%或5到10%之间且包括端值。在一些实施例中,检验引物的熔融温度(Tm)在40到80℃,如50到70℃、55到65℃或57到60.5℃之间且包括端值。在一些实施例中,使用Primer3程序(libprimer3版本2.2.3),使用内置式圣塔露琪亚(SantaLucia)参数(万维网网址primer3.sourceforge.net)计算T
在一些实施例中,除天然存在的磷酸二酯键以外,一些(如至少80、90或95%)或所有衔接子或引物在相邻核苷酸之间包括一个或多个键。这类键的实例包括磷酰胺、硫代磷酸酯和二硫代磷酸酯键。在一些实施例中,一些(如至少80、90或95%)或所有衔接子或引物在最后一个3'核苷酸与倒数第二个3'核苷酸之间包括硫代磷酸酯(如单硫代磷酸酯)。在一些实施例中,一些(如至少80、90或95%)或所有衔接子或引物在3'端处的最后2、3、4或5个核苷酸之间包括硫代磷酸酯(如单硫代磷酸酯)。在一些实施例中,一些(如至少80、90或95%)或所有衔接子或引物在3'端处的最后10个核苷酸中的至少1、2、3、4或5个核苷酸之间包括硫代磷酸酯(如单硫代磷酸酯)。在一些实施例中,这类引物不太可能裂解或降解。在一些实施例中,引物不含酶裂解位点(如蛋白酶裂解位点)。
其它例示性多重PCR方法和库描述于2012年11月21日提交的美国申请案第13/683,604号(美国公开案第2013/0123120号)和2014年5月16日提交的美国序列号61/994,791中,其各自以全文引用的方式并入本文中。这些方法和库可以用于分析本文中所公开的任何样品和用于本发明的任何方法中。
用于检测重组的例示性引物库
在一些实施例中,引物库中的引物被设计成确定一个或多个已知的重组热点处是否发生重组(如同源人类染色体之间的交叉)。知道染色体之间发生何种交叉便可以确定个体的更精确的定相基因数据。重组热点是染色体中的重组事件倾向于集中的局部区域。通常,其由“冷点”侧接,所述冷点是低于平均重组出现率的区域。重组热点倾向于共有类似形态且长度是约1到2kb。热点分布与GC含量和重复元素分布正相关。部分变性的13聚体模体CCNCCNTNNCCNC在一些热点活性中起作用。已证实称为PRDM9的锌指蛋白质与这一模体结合且引发其位置处的重组。报道重组热点的中心之间的平均距离是约80kb。在一些实施例中,重组热点的中心之间的距离范围在约3kb到约100kb之间。公共数据库包括大量已知的人类重组热点,如HUMHOT和国际单倍型图计划(International HapMap Project)数据库(参见例如,Nishant等人,《HUMHOT:人类减数分裂重组热点数据库(HUMHOT:a database ofhuman meiotic recombination hot spots)》,《核酸研究》,34:D25-D28,2006,《数据库期刊(Database issue)》;Mackiewicz等人,《用真实数据进行的计算机模拟的人类基因组-A比较中的重组热点的分布(Distribution of Recombination Hotspots in the HumanGenome-A Comparison of Computer Simulations with Real Data)》,《公共科学图书馆综合卷(PLoS ONE)》8(6):e65272,doi:10.1371/journal.pone.0065272;和万维网网址hapmap.ncbi.nlm.nih.gov/downloads/index.html.en,其各自以全文引用的方式并入本文中)。
在一些实施例中,引物库中的引物在重组热点(已知的人类重组热点)处或附近聚集。在一些实施例中,使用相应的扩增子确定重组热点内或附近的序列,以确定所述具体热点处是否发生重组(如扩增子的序列是否是在发生重组的情况下所预期的序列或在未发生重组的情况下所预期的序列)。在一些实施例中,引物被设计成扩增部分或全部重组热点(和任选地,侧接重组热点的序列)。在一些实施例中,使用长读段测序(如使用由Illumina研发的Moleculo Technology以测序多达约10kb的测序)或成对端测序,以对部分或全部重组热点进行测序。是否发生重组事件的知识可以用于确定哪些单倍型域侧接热点。视需要,可以使用对单倍型域内的区域具有特异性的引物证实存在具体单倍型域。在一些实施例中,假设已知的重组热点之间不存在交叉。在一些实施例中,引物库中的引物在染色体的末端处或附近聚集。举例来说,这类引物可以用于确定染色体的末端处是否存在具体的臂或部分。在一些实施例中,引物库中的引物在重组热点处或附近和染色体的末端处或附近聚集。
在一些实施例中,引物库包括对重组热点(如已知的人类重组热点)具有特异性和/或对重组热点附近的区域(如与重组热点的5'或3'端相距10、8、5、3、2、1或0.5kb以内)具有特异性的一个或多个引物(如至少5、10、50、100、200、500、750、1,000、2,000、5,000、7,500、10,000、20,000、25,000、30,000、40,000或50,000个不同引物或不同引物对)。在一些实施例中,至少1、5、10、20、40、60、80、100或150个不同引物(或引物对)对相同的重组热点具有特异性,或对相同的重组热点或重组热点附近的区域具有特异性。在一些实施例中,至少1、5、10、20、40、60、80、100或150个不同引物(或引物对)对重组热点之间的区域(如不太可能经历重组的区域)具有特异性;这些引物可以用于确认是否存在单倍型域(如将取决于是否发生重组来预期的单倍型域)。在一些实施例中,引物库中的至少10、20、30、40、50、60、70、80或90%的引物对重组热点具有特异性和/或对重组热点附近的区域(如与重组热点的5'或3'端相距10、8、5、3、2、1或0.5kb以内)具有特异性。在一些实施例中,使用引物库确定大于或等于5、10、50、100、200、500、750、1,000、2,000、5,000、7,500、10,000、20,000、25,000、30,000、40,000或50,000个不同重组热点(如已知的人类重组热点)处是否发生重组。在一些实施例中,引物针对重组热点或邻近区域所靶向的区域沿基因组的所述部分大致均匀分布。在一些实施例中,至少1、5、10、20、40、60、80、100或150个不同引物(或引物对)对染色体的末端处或附近的区域(如与染色体的末端相距20、10、5、1、0.5、0.1、0.01或0.001mb以内的区域)具有特异性。在一些实施例中,引物库中的至少10、20、30、40、50、60、70、80或90%的引物对染色体的末端处或附近的区域(如与染色体的末端相距20、10、5、1、0.5、0.1、0.01或0.001mb以内的区域)具有特异性。在一些实施例中,至少1、5、10、20、40、60、80、100或150个不同引物(或引物对)对染色体中的潜在微缺失内的区域具有特异性。在一些实施例中,引物库中的至少10、20、30、40、50、60、70、80或90%的引物对染色体中的潜在微缺失内的区域具有特异性。在一些实施例中,引物库中的至少10、20、30、40、50、60、70、80或90%的引物对重组热点、重组热点附近的区域、染色体的末端处或附近的区域或染色体中的潜在微缺失内的区域具有特异性。
例示性多重PCR方法
在一个方面中,本发明提供用于扩增核酸样品中的目标基因座的方法,所述方法涉及(i)使核酸样品与同时与至少1,000、2,000、5,000、7,500、10,000、15,000、19,000、20,000、25,000、27,000、28,000、30,000、40,000、50,000、75,000或100,000个不同的目标基因座杂交的引物库接触以产生反应混合物;和(ii)使反应混合物经历引物延伸反应条件(如PCR条件)以产生包括目标扩增子的扩增产物。在一些实施例中,所述方法还包括确定存在或不存在至少一种目标扩增子(例如至少50、60、70、80、90、95、96、97、98、99或99.5%的目标扩增子)。在一些实施例中,所述方法还包括确定至少一种目标扩增子(例如至少50、60、70、80、90、95、96、97、98、99或99.5%的目标扩增子)的序列。在一些实施例中,至少50、60、70、80、90、95、96、97、98、99或99.5%的目标基因座被扩增。在一些实施例中,至少25、50、75、100、300、500、750、1,000、2,000、5,000、7,500、10,000、15,000、19,000、20,000、25,000、27,000、28,000、30,000、40,000、50,000、75,000或100,000个不同的目标基因座被扩增至少5、10、20、40、50、60、80、100、120、150、200、300或400倍。在一些实施例中,至少50、60、70、80、90、95、96、97、98、99、99.5或100%的目标基因座被扩增至少5、10、20、40、50、60、80、100、120、150、200、300或400倍。在各种实施例中,小于60、50、40、30、20、10、5、4、3、2、1、0.5、0.25、0.1或0.05%的扩增产物是引物二聚体。在一些实施例中,所述方法涉及多重PCR和测序(如高通量测序)。
在各种实施例中,使用长粘接时间和/或低引物浓度。在各种实施例中,粘接步骤的长度大于3、5、8、10、15、20、30、45、60、75、90、120、150或180分钟。在各种实施例中,粘接步骤(每个PCR循环)的长度在5与180分钟,如5到60、10到60、5到30或10到30分钟之间且包括端值。在各种实施例中,粘接步骤的长度大于5分钟(如大于10或15分钟),且每种引物的浓度小于20nM。在各种实施例中,粘接步骤的长度大于5分钟(如大于10或15分钟),且每种引物的浓度在1到20nM或1到10nM之间且包括端值。在各种实施例中,粘接步骤的长度大于20分钟(如大于30、45、60或90分钟),且每种引物的浓度小于1nM。
在高度复用的情况下,溶液可能因为溶液中的大量引物而变得粘稠。如果溶液太粘稠,那么可以将引物浓度降低到仍足以使引物结合模板DNA的量。在各种实施例中,使用小于60,000个不同的引物且每种引物的浓度小于20nM,如小于10nM或在1与10nM之间且包括端值。在各种实施例中,使用超过60,000个不同的引物(如60,000到120,000个不同的引物)且每种引物的浓度小于10nM,如小于5nM或在1与10nM之间且包括端值。
发现粘接温度可以任选地高于一些或全部引物的熔融温度(与使用低于引物的熔融温度的粘接温度的其它方法不同)。熔融温度(Tm)是满足以下条件的温度:寡核苷酸(如引物)和其完美互补物的二分之一(50%)的DNA双螺旋解离且变成单链DNA。粘接温度(T
在各种实施例中,粘接温度比至少25、50、75、100、300、500、750、1,000、2,000、5,000、7,500、10,000、15,000、19,000、20,000、25,000、27,000、28,000、30,000、40,000、50,000、75,000、100,000种或所有非一致引物的熔融温度(如凭经验测量或计算的T
在各种实施例中,粘接温度比至少25、50、75、100、300、500、750、1,000、2,000、5,000、7,500、10,000、15,000、19,000、20,000、25,000、27,000、28,000、30,000、40,000、50,000、75,000、100,000种或所有非一致引物的熔融温度(如凭经验测量或计算的T
在一些实施例中,粘接温度比引物的最高熔融温度(如凭经验测量或计算的T
在一些实施例中,粘接温度比引物的最高熔融温度(如凭经验测量或计算的T
在一些实施例中,粘接温度比至少25、50、75、100、300、500、750、1,000、2,000、5,000、7,500、10,000、15,000、19,000、20,000、25,000、27,000、28,000、30,000、40,000、50,000、75,000、100,000种或所有非一致引物的平均熔融温度(如凭经验测量或计算的T
在一些实施例中,粘接温度比至少25、50、75、100、300、500、750、1,000、2,000、5,000、7,500、10,000、15,000、19,000、20,000、25,000、27,000、28,000、30,000、40,000、50,000、75,000、100,000种或所有非一致引物的平均熔融温度(如凭经验测量或计算的T
在一些实施例中,粘接温度在50到70℃之间,如在55到60、60到65或65到70℃之间且包括端值。在一些实施例中,粘接温度在50到70℃之间,如在55到60、60到65或65到70℃之间且包括端值,且(i)粘接步骤(每个PCR循环)的长度大于3、5、8、10、15、20、30、45、60、75、90、120、150或180分钟,或(ii)粘接步骤(每个PCR循环)的长度在5与180分钟,如5到60、10到60、5到30或10到30分钟之间且包括端值。
在一些实施例中,以下条件中的一个或多个用于T
在一些实施例中,用ultrospec 2100 pr UV/可见光分光光度计(Amershambiosciences)以温度的函数形式测量在260nm下的吸光度(参见例如Takiya等人,《用于PNA/DNA双螺旋的热稳定性(Tm)预测的经验方法(An empirical approach forthermal stability(Tm)prediction of PNA/DNA duplexes)》,《核酸研讨会丛刊(奥克斯福德)(Nucleic Acids Symp Ser(Oxf))》;(48):131-2,2004,其以全文引用的方式并入本文中)。在一些实施例中,通过使温度以2℃/分钟的步长从95℃降低到20℃来测量在260nm下的吸光度。在一些实施例中,混合引物与其完美互补物(如每个成对的寡聚物2μM)且接着通过以下方式进行粘接:将样品加热到95℃,在所述温度下保持5分钟,接着在30分钟期间冷却到室温且使样品在95℃下保持至少60分钟。在一些实施例中,使用SWIFT Tm软件通过分析数据来确定熔融温度。在本发明的任何方法的一些实施例中,所述方法包括在引物用于目标基因座的PCR扩增之前或之后,凭经验测量或计算(如用计算机计算)库中的至少50、80、90、92、94、96、98、99或100%的引物的熔融温度。
在一些实施例中,库包含微阵列。在一些实施例中,库不包含微阵列。
在一些实施例中,大部分或所有引物延伸以形成扩增产物。在PCR反应中耗尽所有引物可以增加不同目标基因座的扩增的均匀性,因为相同或类似数目的引物分子转化成每个目标基因座的目标扩增子。在一些实施例中,至少80、90、92、94、96、98、99或100%的引物分子延伸以形成扩增产物。在一些实施例中,对于至少80、90、92、94、96、98、99或100%的目标基因座,至少80、90、92、94、96、98、99或100%的针对所述目标基因座的引物分子延伸以形成扩增产物。在一些实施例中,进行多个循环直到耗尽这一百分比的引物。在一些实施例中,进行多个循环直到耗尽所有或基本上所有引物。视需要,可以通过降低初始引物浓度和/或增加所进行的PCR循环的数目来消耗更高百分比的引物。
在一些实施例中,可以使用微升反应体积进行PCR方法,其与微流体应用中使用的纳升或皮升反应体积相比更难以实现特异性PCR扩增(归因于模板核酸的较低的局部浓度)。在一些实施例中,反应体积在1与60μl之间,如在5与50μl、10与50μl、10与20μl、20与30μl、30与40μl或40到50μl之间且包括端值。
在一个实施例中,本文中所公开的方法使用高效高度多重靶向PCR来扩增DNA,接着使用高通量测序来确定每个目标基因座处的等位基因出现率。在一个反应体积中以大部分所得序列读段映射到目标基因座的方式复用超过约50或100个PCR引物的能力是新颖并且非显而易见的。实现以高效方式进行高度多重靶向PCR的一种技术涉及设计不太可能彼此杂交的引物。通过以下方式来选择通常称为引物的PCR探针:创建至少300、至少500、至少750、至少1,000、至少2,000、至少5,000、至少7,500、至少10,000、至少20,000、至少25,000、至少30,000、至少40,000、至少50,000、至少75,000或至少100,000个潜在引物对之间的潜在不良相互作用或引物与样品DNA之间的不希望的相互作用的热力学模型,且接着使用所述模型消除与池中的其它设计不相容的设计。另一种实现以高效方式进行高度多重靶向PCR的技术是使用靶向PCR的部分或完全巢式方法。使用这些方法中的一种或组合允许复用单一池中至少300、至少800、至少1,200、至少4,000或至少10,000个引物,其中所得被扩增的DNA包含大部分的在测序时将映射到目标基因座的DNA分子。使用这些方法中的一种或组合允许复用单一池中的大量引物,其中所得被扩增的DNA包含超过50%、超过60%、超过67%、超过80%、超过90%、超过95%、超过96%、超过97%、超过98%、超过99%或超过99.5%的映射到目标基因座的DNA分子。
在一些实施例中,目标遗传物质的检测可以按多重方式进行。可以平行操作的基因目标序列的数量可以在一到十、十到一百、一百到一千、一千到一万、一万到十万、十万到一百万或一百万到一千万的范围内。每个池复用超过100个引物的先前尝试已经产生了显著问题和不合需要的副反应,如引物-二聚体形成。
靶向PCR
在一些实施例中,PCR可以用于靶向基因组的特定位置。在血浆样品中,使原始DNA高度片段化(通常小于500bp,平均长度小于200bp)。在PCR中,正向和反向引物粘接到相同片段以实现扩增。因此,如果片段较短,那么PCR分析法必须也扩增相对较短的区域。与MIPS相同,如果多态位置太靠近聚合酶结合位点,那么其可能引起不同等位基因的扩增偏差。当前,靶向多态区域的PCR引物(如含有SNP的引物)通常被设计成使得引物的3'端将和与多态碱基紧密相邻的碱基杂交。在本公开的实施例中,正向和反向PCR引物的3'端被设计成用于与远离目标等位基因的变异体位置(多态位点)的一个或几个位置的碱基杂交。多态位点(SNP或其它多态位点)之间的碱基和与所设计的引物的3'端杂交的碱基的数量可以是一个碱基,其可以是两个碱基,其可以是三个碱基,其可以是四个碱基,其可以是五个碱基,其可以是六个碱基,其可以是七到十个碱基,其可以是十一到十五个碱基,或其可以是十六到二十个碱基。正向和反向引物可以被设计成与不同数目的远离多态位点的碱基杂交。
可以产生大量PCR分析法,然而,不同PCR分析法之间的相互作用使得难以将其复用超过约一百个分析法。可以使用各种复合分子方法来提高复用水平,但其仍然可能限于每个反应少于100,或许200或可能500个分析法。具有大量DNA的样品可以被分到多个子反应中且接着在测序之前重组。对于DNA分子的整个样品或一些子群体受限的样品,拆分样品将引入统计噪声。在一个实施例中,少量或有限数量的DNA可以指少于10pg、在10与100pg之间、在100pg与1ng之间、在1与10ng之间或在10与100ng之间的量。应注意,虽然这种方法尤其适用于少量DNA,其中涉及分成多个池的其它方法会引起与所引入的随机噪声相关的显著问题,但这种方法在其在具有任何数量DNA的样品上操作时仍然提供使偏差降到最低的益处。在这些情形下,可以使用通用预扩增步骤来增加整体样品数量。理想地,这个预扩增步骤不应该明显地改变等位基因分布。
在一个实施例中,本公开的方法可以从有限样品(如来自体液的单细胞或DNA)产生对大量目标基因座,具体地说1,000到5,000个基因座、5,000到10,000个基因座或超过10,000个基因座具有特异性的PCR产物,用于通过测序进行基因分型或一些其它基因分型方法。当前,进行超过5到10个目标的多重PCR反应提出了一项重大挑战并且通常受到例如引物二聚体的引物副产物和其它假象的阻挠。当使用微阵列,用杂交探针检测目标序列时,可以忽略引物二聚体和其它假象,因为不检测这些物质。然而,当使用测序作为检测方法时,绝大部分测序读段将对这类假象而不是样品中所需目标序列进行测序。现有技术中所描述的用于在一个反应体积中复用超过50或100个反应,接着进行测序的方法通常将产生超过20%且通常超过50%,在许多情况下超过80%且在一些情况下超过90%的脱靶序列读段。
通常,为了进行样品的多个(n个)目标(超过50、超过100、超过500或超过1,000)的靶向测序,可以将样品分到多个扩增一个单独目标的平行反应中。这已经在PCR多孔盘中进行或可以在商业平台中进行,如FLUIDIGM ACCESS ARRAY(在微流体芯片中每个样品48个反应)或由RAIN DANCE TECHNOLOGY进行的DROPLET PCR(数百到数千个目标)。不幸的是,这些拆分和合并(split-and-pool)方法对于具有有限量的DNA的样品是有问题的,因为通常不存在足够的基因组拷贝以确保每个孔中存在基因组的每个区域的一个拷贝。当靶向多态基因座并且需要多态基因座处的等位基因的相对比例时,这是尤其严重的问题,因为通过拆分和合并所引入的随机噪声将引起存在于原始DNA样品中的等位基因的比例的测量结果非常不准确。本文中描述一种可以有效地且高效地扩增多个PCR反应的方法,所述方法适用于仅可使用有限量的DNA的情况。在一个实施例中,所述方法可以适用于分析单细胞、体液、DNA混合物(如在血浆、活检、环境和/或法医样品中发现的自由浮动DNA)。
在一个实施例中,靶向测序可以涉及以下步骤中的一个、多个或全部。a)用DNA片段的两端上的衔接子序列产生和扩增库。b)在库扩增之后分成多个反应。c)用DNA片段的两端上的衔接子序列产生和任选地扩增库。d)使用每个目标一个目标特异性“正向”引物和一个标签特异性引物进行所选目标的1000到10,000重扩增。e)使用“反向”目标特异性引物和一个(或更多个)对以第一轮中的目标特异性正向引物的一部分的形式引入的通用标签具有特异性的引物,从这一产物进行第二扩增。f)进行所选目标的1000重预扩增持续有限数目的循环。g)将产物分成多个等分试样并且在单独的反应(例如,50到500重)中扩增目标的子池,但这可以一直使用直到单重。h)合并平行子池反应的产物。i)在这些扩增期间,引物可以进行对相容的标签(部分或全长)进行测序以使得可以对产物进行测序。
高度多重PCR
本文中公开实现超过一百到数万个来自核酸样品(如从血浆获得的基因组DNA)的目标序列(例如,SNP基因座)的靶向扩增的方法。扩增样品可以相对不含引物二聚体产物并且在目标基因座处具有低等位基因偏差。如果在扩增期间或在扩增之后,产物与测序相容衔接子附接,那么这些产物的分析可以通过测序来进行。
使用所属领域中已知的方法进行高度多重PCR扩增引起所产生的引物二聚体产物超过所需扩增产物并且不适用于测序。这些可以凭经验通过消除形成这些产物的引物或通过进行引物的计算机模拟选择来减少。然而,分析法的数目越大,这个问题变得越难。
一种解决方案是将5000重反应拆分成若干个重数更低的扩增,例如一百个50重或五十个100重反应,或使用微流体或甚至将样品拆分成单独的PCR反应。然而,如果样品DNA是有限的,例如在怀孕血浆的非侵袭性产前诊断中,那么应该避免在多个反应之间分割样品,因为这将产生瓶颈效应。
本文中描述用于首先总体地扩增样品的血浆DNA且接着将样品分成多个多重目标富集反应的方法,每个反应具有更适中的数目的目标序列。在一个实施例中,本公开的方法可以用于优先富集多个基因座处的DNA混合物,所述方法包含以下步骤中的一或多个:从DNA混合物产生和扩增库,其中库中的分子具有接合在DNA片段的两端上的衔接子序列;将扩增的库分成多个反应,使用每个目标一个目标特异性“正向”引物和一个或多个衔接子特异性通用“反向”引物进行所选目标的第一轮多重扩增。在一个实施例中,本公开的方法进一步包括使用“反向”目标特异性引物和一个或多个对以第一轮中的目标特异性正向引物的一部分的形式引入的通用标签具有特异性的引物,执行第二扩增。在一个实施例中,所述方法可以涉及全巢式、半巢式(hemi-nested)、半巢式(semi-nested)、单边全巢式、单边半巢式(one sided hemi-nested)或单边半巢式(one sided semi-nested)PCR方法。在一个实施例中,本公开的方法用于优先富集多个基因座处的DNA混合物,所述方法包含进行所选目标的多重预扩增持续有限数目的循环,将产物分成多个等分试样并且在单独的反应中扩增目标的子池,以及合并平行子池反应的产物。应注意,对于50到500个基因座、对于500到5,000个基因座、对于5,000到50,000个基因座或甚至对于50,000到500,000个基因座,这种方法可以用于以将产生低水平等位基因偏差的方式进行目标扩增。在一个实施例中,引物具有部分或全长测序相容标签。
工作流程可能需要(1)提取DNA,如血浆DNA,(2)制备在片段的两端上具有通用衔接子的片段库,(3)使用对衔接子具有特异性的通用引物扩增库,(4)将扩增样品“库”分成多个等分试样,(5)对等分试样进行多重(例如约100重、1,000或10,000重,其中使用每个目标一个目标特异性引物和标签特异性引物)扩增,(6)合并一个样品的等分试样,(7)将样品加注条形码,(8)混合样品并且调节浓度,(9)对样品进行测序。工作流程可以包含多个含有所列步骤中的一个的子步骤(例如步骤(2)制备库步骤可能需要三个酶促步骤(末端平端化、dA加尾和衔接子接合)和三个纯化步骤)。工作流程的步骤可以组合、分割或按不同顺序(例如加注条形码和合并样品)执行。
重要的是应注意,可以按偏向于更高效地扩增短片段的方式进行库扩增。以这种方式,有可能优先扩增更短的序列,例如单核小体DNA片段,如在孕妇的循环中发现的(胎盘来源的)游离胚胎DNA。应注意,PCR分析法可以具有标签,例如测序标签(通常是15到25个碱基的截短形式)。在复用之后,合并样品的PCR复用结果且接着通过标签特异性PCR(也可以通过接合进行)完成(包括加注条形码)标签。此外,可以在与复用相同的反应中添加完整测序标签。在第一循环中,可以用目标特异性引物扩增目标,接着由标签特异性引物接管以完成SQ-衔接子序列。PCR引物可以不具有标签。测序标签可以通过接合来附接到扩增产物。
在一个实施例中,对于如胚胎非整倍性的检测等各种应用,可以使用高度多重PCR,接着通过克隆测序来评估扩增物质。尽管传统的多重PCR同时评估多达五十个基因座,但是本文中所描述的方法可以用于实现同时评估超过50个基因座、同时评估超过100个基因座、同时评估超过500个基因座、同时评估超过1,000个基因座、同时评估超过5,000个基因座、同时评估超过10,000个基因座、同时评估超过50,000个基因座以及同时评估超过100,000个基因座。实验已证实,可以在单一反应中以足够好的效率和特异性同时评估多达(包括)和超过10,000个不同的基因座,从而作出具有高准确性的非侵袭性产前非整倍性诊断和/或拷贝数目识别。可以在单一反应中将分析法与整个样品组合,所述样品是如从血浆分离的cfDNA样品、其一部分或cfDNA样品的其它经过处理的衍生物。样品(例如cfDNA或衍生物)还可以被分成多个平行的多重反应。最佳的样品拆分和多重数是通过权衡各种性能规格来确定。由于材料数量有限,所以将样品分成多个部分会引入采样噪声、操作时间,并且增加误差可能性。相反,更高的复用会产生更大量的假性扩增和更大的扩增不平等,这两者都会降低检验性能。
在本文中所描述的方法的应用中的两个关键相关考虑因素是原始样品(例如,血浆)的有限量和所述材料中用于获得等位基因出现率或其它测量结果的原始分子的数目。如果原始分子的数目下降到低于某一水平,那么随机采样噪声变得显著,并且会影响检验的准确性。通常,如果对每个目标基因座包含相等的500-1000个原始分子的样品进行测量,那么可以获得质量足以作出非侵袭性产前非整倍性诊断的数据。存在多种用于增加不同测量的数目的方式,例如增加样品体积。应用于样品的每个操作也潜在地引起材料损失。必需表征由各种操作所引起的损失且加以避免,或视需要改善某些操作的结果以避免可能降低检验性能的损失。
在一个实施例中,有可能在后续步骤中通过扩增所有或一部分原始样品(例如,cfDNA样品)来减少潜在损失。多种方法可以用于扩增样品中的所有遗传物质,增加可以用于下游程序的量。在一个实施例中,在一个不同衔接子、两个不同衔接子或多个不同衔接子的接合之后,通过PCR来扩增接合介导的PCR(LM-PCR)DNA片段。在一个实施例中,使用多重置换扩增(MDA)phi-29聚合酶来等温扩增所有DNA。在DOP-PCR和变体中,使用随机引发来扩增原始物质DNA。每种方法具有某些特征,如在基因组的所有呈现区域内扩增的均匀性、原始DNA的捕获和扩增的效率,以及随片段长度而变的扩增性能。
在一个实施例中,LM-PCR可以与具有3'酪氨酸的单一异源双链衔接子一起使用。异源双链衔接子能够使用可以在第一轮PCR期间被转化为原始DNA片段的5'和3'端上的两个不同序列的单一衔接子分子。在一个实施例中,有可能通过尺寸分离或产物(如AMPURE、TASS)或其它类似方法对扩增的库进行分级。在接合之前,可以对样品DNA进行末端平端化,且接着向3'端添加单一腺苷碱基。在接合之前,可以使用限制酶或某种其它裂解方法使DNA裂解。在接合期间,样品片段的3'腺苷和衔接子的互补性3'酪氨酸突出端可以增强接合效率。PCR扩增的延伸步骤从时间观点来看可能限于减少长度超过约200bp、约300bp、约400bp、约500bp或约1,000bp的片段的扩增。使用如通过可商购的试剂盒说明的条件操作多个反应;引起少于10%的样品DNA分子的成功接合。关于这一点的反应条件的一系列优化将接合提高到约70%。
微型PCR
以下微型PCR方法适用于含有短核酸、被消化的核酸或片段化核酸(如cfDNA)的样品。传统的PCR分析法设计引起不同胚胎分子大量损失,但是可以通过设计称为微型PCR分析法的极短PCR分析法来显著减少损失。使母体血清中的胚胎cfDNA高度片段化并且片段尺寸大致以高斯方式分布,其中平均值是160bp,标准偏差是15bp,最小尺寸是约100bp且最大尺寸是约220bp。片段起点和末端位置相对于目标多态现象的分布虽然不一定是随机的,但是在单独的目标中和在全体所有目标中大幅变化并且一个具体目标基因座的多态位点可以占据来源于所述基因座的各个片段中从起点到末端的任何位置。应注意,术语微型PCR同样可以指不具有额外约束或限制的普通PCR。
在PCR期间,扩增将仅从包含正向和反向引物位点的模板DNA片段发生。因为胚胎cfDNA片段较短,所以存在两个引物位点的似然性,包含正向和反向引物位点的具有长度L的胚胎片段的似然性是扩增子长度与片段长度的比率。在理想条件下,其中扩增子是45、50、55、60、65或70bp的分析法将分别从72%、69%、66%、63%、59%或56%的可用模板片段分子成功地扩增。扩增子长度是正向和反向引发位点的5'端之间的距离。比所属领域的技术人员通常所使用的更短的扩增子长度可以通过仅需要短序列读段便产生所需多态基因座的更有效的测量结果。在一个实施例中,扩增子的实质部分应小于100bp、小于90bp、小于80bp、小于70bp、小于65bp、小于60bp、小于55bp、小于50bp或小于45bp。
应注意,在现有技术中已知的方法中,通常避免如本文中所描述的短分析法,因为其不是所需的并且其通过限制引物长度、粘接特征和正向与反向引物之间的距离对引物设计施加了大量限制。
还应注意,如果任一个引物的3'端与多态位点相距约1-6个碱基以内,那么存在偏差扩增的可能性。在初始聚合酶结合位点处的这种单一碱基差异可以引起一个等位基因优先扩增,这可以改变所观察到的等位基因出现率且降低性能。所有这些限制都使鉴别将成功地扩增具体基因座的引物并且此外,设计在同一个多重反应中相容的大型引物集合变得非常具有挑战性。在一个实施例中,内部正向和反向引物的3'端被设计成与多态位点上游的DNA区域杂交,并且通过少数碱基与多态位点隔开。理想地,碱基的数目可以在6个与10个碱基之间,但是同样可以在4个与15个碱基之间、在三个与20个碱基之间、在两个与30个碱基之间或在1个与60个碱基之间,并且实现基本上相同的目的。
多重PCR可能涉及扩增所有目标的单轮PCR或其可能涉及一轮PCR,接着是一轮或多轮巢式PCR或巢式PCR的一些变体。巢式PCR由后续一轮或多轮PCR扩增组成,所述PCR扩增使用一种或多种通过至少一个碱基与前一轮中所使用的引物内部结合的新引物。巢式PCR通过在后续反应中仅扩增来自前一个反应的具有正确内部序列的扩增产物来减少假性扩增目标的数目。减少杂散扩增目标可以改善可以获得的有效测量结果的数目,尤其在测序中。巢式PCR通常需要设计完全在先前引物结合位点内部的引物,必定会增加扩增所需的最小DNA区段尺寸。对于其中DNA被高度片段化的如血浆cfDNA等样品,更大的分析法尺寸会减少可以用于获得测量结果的不同cfDNA分子的数目。在一个实施例中,为了抵消这种作用,可以使用部分巢式方法,其中第二轮引物中的一个或两个与第一结合位点重叠,内部延伸一定数量的碱基,从而获得额外特异性同时最低限度地增加总分析法尺寸。
在一个实施例中,PCR分析法的多重池被设计成潜在地扩增一条或多条染色体上的杂合SNP或其它多态或非多态基因座并且这些分析法被用于单一反应中以扩增DNA。PCR分析法的数目可以在50个与200个PCR分析法之间、在200个与1,000个PCR分析法之间、在1,000个与5,000个PCR分析法之间或在5,000个与20,000个PCR分析法之间(分别是50到200重、200到1,000重、1,000到5,000重、5,000到20,000重、超过20,000重)。在一个实施例中,约10,000个PCR分析法(10,000重)的多重池被设计成潜在地扩增X、Y、13、18和21以及1或2号染色体上的杂合SNP基因座,并且这些分析法被用于单一反应中以扩增从以下物质获得的cfDNA:血浆样品、绒毛样品、羊膜穿刺术样品、单一或少量细胞、其它体液或组织、癌症或其它遗传物质。每个基因座的SNP出现率可以通过克隆或扩增子的一些其它测序方法来确定。所有分析法的等位基因出现率分布或比率的统计分析都可以用于确定样品是否含有检验中所包括的染色体中的一种或多种的三体性。在另一实施例中,将原始cfDNA样品分成两个样品并且进行平行5,000重分析法。在另一实施例中,将原始cfDNA样品分成n个样品且进行平行(约10,000/n)重分析法,其中n在2与12之间,或在12与24之间,或在24与48之间,或在48与96之间。以与已经描述的方式类似的方式收集和分析数据。应注意,这种方法同样适用于检测易位、缺失、复制和其它染色体异常。
在一个实施例中,还可以向任何引物的3'或5'端添加与目标基因组不具有同源性的尾部。这些尾部有助于后续操作、程序或测量。在一个实施例中,尾部序列对于正向和反向目标特异性引物来说可以是相同的。在一个实施例中,可以针对正向和反向目标特异性引物使用不同尾部。在一个实施例中,可以针对不同基因座或基因座集合使用多个不同尾部。某些尾部可以在所有基因座中或在基因座子集中共用。举例来说,使用对应于任何当前测序平台所需的正向和反向序列的正向和反向尾部可以实现在扩增之后的直接测序。在一个实施例中,尾部可以用作可以用于添加其它适用序列的所有扩增目标中的共同引发位点。在一些实施例中,内部引物可以含有被设计成与目标基因座(例如多态基因座)的上游或下游杂交的区域。在一些实施例中,引物可以含有分子条形码。在一些实施例中,引物可以含有被设计成实现PCR扩增的通用引发序列。
在一个实施例中,创建10,000重PCR分析法池使得正向和反向引物具有对应于高通量测序仪器(通常称为大规模平行测序仪器,如可以从ILLUMINA获得的HISEQ、GAIIX或MYSEQ)所需要的所需正向和后向序列的尾部。此外,测序尾部所包括的5'是可以用作后续PCR中的引发位点的额外序列,用于向扩增子添加核苷酸条形码序列,实现在高通量测序仪器的单一泳道中进行多个样品的多重测序。
在一个实施例中,创建10,000重PCR分析法池使得反向引物具有对应于高通量测序仪器所需要的所需反向序列的尾部。在用第一个10,000重分析法扩增之后,可以使用另一个具有针对所有目标的部分巢式正向引物(例如6碱基巢式)和对应于第一轮中所包括的反向测序尾部的反向引物的10,000重池来进行后续PCR扩增。仅使用一个目标特异性引物和通用引物进行的这一后续轮次的部分巢式扩增限制所需的分析法尺寸,减少抽样噪声,但显著减少假性扩增子的数目。可以将测序标签添加到所附接的接合衔接子和/或作为PCR探针的一部分,使得所述标签是最终扩增子的一部分。
肿瘤分数影响检验的性能。存在多种用于富集在患者血浆中发现的DNA的肿瘤分数的方式。可以通过先前所描述的已经讨论的LM-PCR方法以及通过靶向去除长片段来增加肿瘤分数。在一个实施例中,在目标基因座的多重PCR扩增之前,可以进行额外的多重PCR反应以选择性地去除对应于后续多重PCR中所靶向的基因座的长并且很大程度上源于母体的片段。额外引物被设计成粘接与游离胚胎DNA片段中预期存在的相比,与多态现象相距更远的位点。这些引物可以在目标多态基因座的多重PCR之前用于一个循环多重PCR反应中。这些远端引物标记有可以允许选择性识别被标记的DNA碎片的分子或部分。在一个实施例中,这些DNA分子可以用生物素分子共价修饰,所述生物素分子允许在一个PCR循环之后去除新形成的包含这些引物的双链DNA。在所述第一轮期间形成的双链DNA可能是源于母体的。可以通过使用磁性抗生蛋白链菌素珠粒来实现杂交物质的去除。存在可以同样起作用的其它标记方法。在一个实施例中,可以使用尺寸选择方法来富集样品中更短的DNA链;例如小于约800bp、小于约500bp或小于约300bp的DNA链。接着可以像往常一样进行短片段的扩增。
本公开中所描述的微型PCR方法实现了来自单一样品的数百到数千或甚至数百万个基因座在单一反应中的高度多重扩增和分析。同样地,可以复用扩增DNA的检测;可以通过使用条形码PCR在一个测序通道中复用数十到数百个样品。这种多重检测已经成功地检验了多达49重,并且高得多的程度的复用是可能的。实际上,这允许数百个样品在单一测序操作中在数千个SNP处进行基因分型。对于这些样品,所述方法允许确定基因型和杂合率并且同时确定拷贝数目,两者都可以用于非整倍性检测目的。其可以用作用于突变剂量的方法的一部分。这种方法可以用于任何量的DNA或RNA,并且目标区域可以是SNP、其它多态区域、非多态区域以及其组合。
在一些实施例中,可以使用片段化DNA的接合介导的通用PCR扩增。接合介导的通用PCR扩增可以用于扩增血浆DNA,接着可以将其分成多个平行反应。其还可以用于优先扩增短片段,从而富集肿瘤分数。在一些实施例中,通过接合向片段中添加标签可以实现较短的片段的检测,使用引物的较短的目标序列特异性部分和/或在减少非特异性反应的更高温度下粘接。
本文中所描述的方法可以用于其中存在与一定量的污染DNA混合的目标DNA目标的多个目的。在一些实施例中,目标DNA和污染DNA可以来自遗传相关个体。举例来说,可以从含有胚胎(目标)DNA以及母体(污染)DNA的母体血浆检测胚胎(目标)中的基因异常;所述异常包括整个染色体异常(例如非整倍性)、部分染色体异常(例如缺失、复制、倒置、易位)、聚核苷酸多态现象(例如STR)、单核苷酸多态现象和/或其它基因异常或差异。在一些实施例中,目标和污染DNA可以来自同一个体,但是其中目标和污染DNA因一个或多个突变而不同,例如在癌症的情况下。(参见例如H.Mamon等人,《优先扩增来自血浆的细胞凋亡DNA:增强循环DNA中的次要DNA变化的检测的可能性(Preferential Amplification ofApoptotic DNA from Plasma:Potential for Enhancing Detection of Minor DNAAlterations in Circulating DNA)》,《临床化学(Clinical Chemistry)》54:9(2008))。在一些实施例中,可以在细胞培养(细胞凋亡)上清液中发现DNA。在一些实施例中,有可能在生物样品(例如,血液)中诱导细胞凋亡以用于后续库制备、扩增和/或测序。在本公开中的其它地方呈现用于实现这一目的的多种工作流程和方案。
在一些实施例中,目标DNA可以来源于单一细胞、由小于一个目标基因组拷贝组成的DNA的样品、少量DNA、来自混合来源(例如癌症患者血浆和肿瘤:健康与癌症DNA之间的混合物、移植等)的DNA、其它体液、细胞培养物、培养物上清液、法医DNA样品、古老DNA样品(例如在琥珀中捕获的昆虫)、其它DNA样品以及其组合。
在一些实施例中,可以使用短扩增子尺寸。短扩增子尺寸尤其适合于片段化DNA(参见例如A.Sikora等人,《用短PCR扩增子检测增加量的游离胚胎DNA(Detection ofincreased amounts of cell-free fetal DNA with short PCR amplicons)》,《临床化学(Clin Chem.)》,2010年1月;56(1):136-8)。
短扩增子尺寸的使用可以产生一些明显益处。短扩增子尺寸可以产生优化的扩增效率。短扩增子尺寸通常产生更短的产物,因此非特异性引发的机率更低。更短的产物可以更密集地聚集在测序流动细胞上,因为簇将更小。应注意,本文中所描述的方法可以同样适用于更长的PCR扩增子。可以视需要增加扩增子长度,例如当对更大的序列伸长部进行测序时。对单一细胞并且对基因组DNA进行以100bp到200bp长度的分析法作为巢式PCR方案中的第一步骤的146重靶向扩增实验,得到阳性结果。
在一些实施例中,本文中所描述的方法可以用于扩增和/或检测SNP、拷贝数目、核苷酸甲基化、mRNA含量、其它类型的RNA表达水平、其它遗传特征和/或表观遗传特征。本文中所描述的微型PCR方法可以与下一代测序一起使用;其可以与其它下游方法一起使用,如微阵列、由数字PCR进行的计数、实时PCR、质谱分析等。
在一些实施例中,本文中所描述的微型PCR扩增方法可以用作用于准确定量少数群体的方法的一部分。其可以用于使用尖峰校准器进行绝对定量。其可以用于通过极深测序进行突变/次要等位基因定量,并且可以按高度多重方式操作。其可以用于人类、动物、植物或其它生物中的亲戚或祖先的标准父子关系和身份检验。其可以用于法医检验。其可以用于任何类型材料的快速基因分型和拷贝数目分析(CN),所述材料是例如羊水和CVS、精子、受孕产物(POC)。其可以用于单细胞分析,如来自胚胎的活检样品的基因分型。其可以用于通过使用微型PCR的目标测序进行的快速胚胎分析(在活检不到一天、一天或两天内)。
在一些实施例中,微型PCR扩增方法可以用于肿瘤分析:肿瘤活检通常是健康细胞和肿瘤细胞的混合物。靶向PCR允许在几乎无背景序列的情况下对SNP和基因座进行深度测序。其可以用于肿瘤DNA的拷贝数目和杂合性丢失分析。所述肿瘤DNA可能存在于肿瘤患者的多个不同体液或组织中。其可以用于检测肿瘤复发和/或肿瘤筛检。其可以用于种子的质量控制检验。其可以用于繁殖或捕鱼目的。应注意,出于倍性识别的目的,这些方法中的任一种可以同样用于靶向非多态基因座。
一些描述作为本文中所公开的方法的基础的一些基本方法的文献包括:(1)WangHY,Luo M,Tereshchenko IV,Frikker DM,Cui X,Li JY,Hu G,Chu Y,Azaro MA,Lin Y,Shen L,Yang Q,Kambouris ME,Gao R,Shih W,Li H.,《基因组研究(Genome Res.)》2005年2月;15(2):276-83。分子遗传学、微生物学和免疫学部门/新泽西癌症研究所,罗伯特伍德约翰逊医学院(Robert Wood Johnson Medical School),New Brunswick,NewJersey08903,USA。(2)《以高敏感性对单核苷酸多态现象进行高通量基因分型(High-throughput genotyping of single nucleotide polymorphisms with highsensitivity)》,Li H,Wang HY,Cui X,Luo M,Hu G,Greenawalt DM,Tereshchenko IV,LiJY,Chu Y,Gao R.,《分子生物学方法(Methods Mol Biol.)》2007;396-PubMed PMID:18025699。(3)包含复用平均9个分析法以用于测序的方法描述于:《巢式补丁PCR实现了候选基因中的高度多重突变探索(Nested Patch PCR enables highly multiplexedmutation discovery in candidate genes)》,Varley KE,Mitra RD.,《基因组研究(Genome Res.)》2008年11月;18(11):1844-50。电子版2008年10月10日。应注意,本文中所公开的方法允许复用的数量级超过以上参考文献。
例示性试剂盒
在一个方面中,本发明提供一种试剂盒,如用于使用本文中所描述的任何方法扩增核酸样品中的目标基因座以用于检测染色体区段或整个染色体的缺失和/或复制的试剂盒。在一些实施例中,试剂盒可以包括本发明的任何引物库。在一个实施例中,试剂盒包含多个内部正向引物和任选的多个内部反向引物,以及任选的外部正向引物和外部反向引物,其中每个引物被设计成与紧靠着目标染色体或染色体区段以及任选其它染色体或染色体区段上的一个目标位点(例如多态位点)的上游和/或下游的DNA的区域杂交。在一些实施例中,试剂盒包括使用引物库扩增目标基因座的说明,如用于使用本文中所描述的任何方法检测一个或多个染色体区段或整个染色体的一个或多个缺失和/或复制。
在某些实施例中,本发明的试剂盒提供用于检测染色体非整倍性和CNV确定的引物对,如用于用以检测染色体非整倍性(如CNV(CoNVERGe)(以基因型方式显示拷贝数目变异事件(Copy Number Variant Events Revealed Genotypically))和/或SNV)的大规模多重反应的引物对。在这些实施例中,试剂盒可以包括至少100、200、250、300、500、1000、2000、2500、3000、5000、10,000、20,000、25,000、28,000、50,000或75,000个且最多200、250、300、500、1000、2000、2500、3000、5000、10,000、20,000、25,000、28,000、50,000、75,000或100,000个共同装运的引物对。引物对可以包含于单一容器(如单一管状物或盒子)或多个管状物或盒子中。在某些实施例中,由商业提供者预先证明引物对合格且共同出售,且在其它实施例中,客户选择定制基因目标和/或引物且商业提供者制备引物池且运送给客户(既不在一个管状物中也不在多个管状物中)。在某些例示性实施例中,试剂盒包括用于检测CNV和SNV,尤其已知与至少一种类型的癌症相关的CNV和SNV的引物。
根据本发明的一些实施例,用于循环DNA检测的试剂盒包括用于循环DNA检测的标准物和/或对照物。举例来说,在某些实施例中,标准物和/或对照物是与本文中所提供的用于进行扩增反应的引物(如用于进行CoNVERGe的引物)一起出售以及(任选的)装运和包装。在某些实施例中,对照物包括聚核苷酸,如DNA,包括呈现一种或多种染色体非整倍体(如CNV)和/或包括一种或多种SNV的被分离的基因组DNA。在某些实施例中,标准物和/或对照物被称为PlasmArt标准物且包括与已知呈现CNV(尤其在某些遗传性疾病中和在某些疾病状态(如癌症)中)的基因组的区域具有序列一致性以及反映在血浆中天然发现的cfDNA片段的尺寸分布的聚核苷酸。用于制备PlasmArt标准物的例示性方法提供于本文中的实例中。通常,将来自已知包括染色体非整倍体的来源的基因组DNA分离、片段化、纯化且进行尺寸选择。
因此,可以通过将如上文所概括制备的被分离的聚核苷酸样品以与在体内关于cfDNA所观察到的类似的浓度(如在例如所述体液中的DNA的0.01%与20%、0.1与15%或0.4与10%之间)刺入已知不呈现染色体非整倍性和/或SNV的DNA样品中来制备人工cfDNA聚核苷酸标准物和/或对照物。这些标准物/对照物可以用作分析法设计、表征、研发和/或验证的对照物,以及作为检验(如在CLIA实验室中进行的癌症检验)期间的质量控制标准物和/或作为仅供研究使用或诊断检验试剂盒中所包括的标准物。
例示性标准化/校正方法
在一些实施例中,针对偏差(如由GC含量的差异引起的偏差或由扩增效率的其它差异引起的偏差)调节或针对测序误差调节不同基因座、染色体区段或染色体的测量结果。在一些实施例中,针对等位基因之间的代谢、细胞凋亡、组蛋白、失活和/或扩增的差异来调节相同基因座的不同等位基因的测量结果。在一些实施例中,针对不同RNA等位基因之间的转录率或稳定性的差异来调节RNA中的相同基因座的不同等位基因的测量结果。
用于定相基因数据的例示性方法
在一些实施例中,使用本文中所描述的方法或任何已知的用于定相基因数据的方法来对基因数据进行定相(参见例如2009年2月9日提交的PCT公开案第WO2009/105531号和2009年8月4日提交的PCT公开案第WO2010/017214号;2012年11月21日提交的美国公开案第2013/0123120号;2010年10月7日提交的美国公开案第2011/0033862号;2010年8月19日提交的美国公开案第2011/0033862号;2011年2月3日提交的美国公开案第2011/0178719号;2008年3月17日提交的美国专利案第8,515,679号;2006年11月22日提交的美国公开案第2007/0184467号;2008年3月17日提交的美国公开案第2008/0243398号和2014年5月16日提交的美国序列号61/994,791,其各自以全文引用的方式并入本文中)。在一些实施例中,确定一个或多个已知或怀疑含有相关CNV的区域的相。在一些实施例中,还确定一个或多个侧接CNV区域的区域和/或一个或多个参考区域的相。在一个实施例中,通过测量来自个体的单倍组织(例如通过测量一个或多个精子或卵)来进行推断,对个体的基因数据进行定相。在一个实施例中,通过使用一个或多个一级亲属(如个体的父母(例如来自个体的父亲的精子)或同胞)的所测量的基因型数据进行推断,对个体的基因数据进行定相。
在一个实施例中,通过稀释来对个体的基因数据进行定相,其中在一个或多个孔中稀释DNA或RNA,如通过使用数字PCR。在一些实施例中,将DNA或RNA稀释到预期每个孔中存在不超过每个单倍型的约一个拷贝的点,且接着测量一个或多个孔中的DNA或RNA。在一些实施例中,当染色体是紧密的束时,细胞停滞在有丝分裂期,且使用微流体在单独的孔中放置单独的染色体。因为DNA或RNA被稀释,所以同一个部分(或管)中不太可能存在超过一个单倍型。因此,在管中可以有效地存在单一DNA分子,这使得能够确定单一DNA或RNA分子上的单倍型。在一些实施例中,所述方法包括将DNA或RNA样品分成多个部分使得至少一个所述部分包括来自一对染色体的一条染色体或一个染色体区段,并且对至少一个所述部分中的DNA或RNA样品进行基因分型(例如,确定两个或更多个多态基因座的存在),由此确定单倍型。在一些实施例中,基因分型涉及测序(如鸟枪法测序或单分子测序)、用于检测多态基因座的SNP阵列或多重PCR。在一些实施例中,基因分型涉及使用SNP阵列来检测多态基因座,如至少100、200、500、750、1,000、2,000、5,000、7,500、10,000、20,000、25,000、30,000、40,000、50,000、75,000或100,000个不同的多态基因座。在一些实施例中,基因分型涉及使用多重PCR。在一些实施例中,所述方法涉及使一部分样品与引物库接触以产生反应混合物,所述引物库同时与至少100、200、500、750、1,000、2,000、5,000、7,500、10,000、20,000、25,000、30,000、40,000、50,000、75,000或100,000个不同的多态基因座(如SNP)杂交;并且使反应混合物经历引物延伸反应条件以产生扩增产物,用高通量测序仪测量所述扩增产物以产生测序数据。在一些实施例中,对RNA(如mRNA)进行测序。因为mRNA仅含有外显子,对mRNA进行测序使得能够确定基因组中的较大距离(如数兆碱基)内的多态基因座(如SNP)的等位基因。在一些实施例中,通过染色体分选来确定个体的单倍型。例示性染色体分选方法包括当染色体是紧密的束时,使细胞停滞在有丝分裂期,和使用微流体在单独的孔中放置单独的染色体。另一种方法涉及使用FACS介导的单一染色体分选来收集单一染色体。可以使用标准方法(如测序或阵列)鉴别单一染色体上的等位基因,以确定个体的单倍型。
在一些实施例中,通过长读段测序来确定个体的单倍型,如通过使用由Illumina研发的Moleculo Technology。在一些实施例中,库制备步骤涉及将DNA剪切成片段,如尺寸是约10kb的片段,稀释片段且将其放置在孔中(使得约3,000个片段在单一孔中),通过长范围PCR扩增每个孔中的片段且切割成短片段且将片段加注条形码,以及将来自每个孔的带条形码的片段合并在一起以对其全部进行测序。在测序之后,计算步骤涉及基于所连接的条形码来分离来自每个孔的读段且将其分组成片段,在片段的重叠杂合SNV处将片段组装成单倍型域,以及基于定相参考图以统计方式对单倍型域进行定相和产生长单倍型重叠群。
在一些实施例中,使用来自个体的亲属的数据确定个体的单倍型。在一些实施例中,使用SNP阵列确定来自个体和个体的亲属的DNA或RNA样品中存在至少100、200、500、750、1,000、2,000、5,000、7,500、10,000、20,000、25,000、30,000、40,000、50,000、75,000或100,000个不同的多态基因座。在一些实施例中,所述方法涉及使来自个体和/或个体的亲属的DNA样品与引物库接触以产生反应混合物,所述引物库同时与至少100、200、500、750、1,000、2,000、5,000、7,500、10,000、20,000、25,000、30,000、40,000、50,000、75,000或100,000个不同的多态基因座(如SNP)杂交;和使反应混合物经历引物延伸反应条件以产生扩增产物,使用高通量测序仪测量所述扩增产物以产生测序数据。
在一个实施例中,使用计算机程序对个体的基因数据进行定相,所述计算机程序使用基于群体的单倍型出现率以推断最有可能的相,如基于HapMap的定相。举例来说,可以使用统计方法从二倍体数据直接推导单倍数据集,所述统计方法利用一般群体中已知的单倍型域(如被创建用于公共单倍型图计划(public HapMap Project)和Perlegen人类单倍型计划(Perlegen Human Haplotype Project)的单倍型域)。单倍型域基本上是在多种群体中重复出现的一系列相关等位基因。因为这些单倍型域通常是古老和普遍的,所以其可以用于由二倍体基因型预测单倍型。实现这一任务的可公开获得的算法包括不完全系统发生方法、基于共轭先验的贝叶斯方法(Bayesian approaches based on conjugatepriors)和来自群体遗传学的先验。这些算法中的一些使用隐式马尔可夫模型(hiddenMarkov model)。
在一个实施例中,使用由基因型数据估计单倍型的算法对个体的基因数据进行定相,如使用局部单倍型聚类的算法(参见例如Browning和Browning,《使用局部单倍型聚类进行的全基因组关联研究的快速和准确单倍型定相和遗失数据推断(Rapid and AccurateHaplotype Phasing and Missing-Data Inference for Whole-Genome AssociationStudies By Use of Localized Haplotype Clustering)》,《美国人类遗传学杂志(Am JHum Genet.)》,2007年11月;81(5):1084-1097,其以全文引用的方式并入本文中)。例示性程序是Beagle版本:3.3.2或版本4(可以在万维网网址hfaculty.washington.edu/browning/beagle/beagle.html获得,其以全文引用的方式并入本文中)。
在一个实施例中,使用由基因型数据估计单倍型的算法对个体的基因数据进行定相,如使用连锁不平衡随距离的衰减、基因分型标记物的顺序和间隔、遗失数据估算、重组率估计或其组合的算法(参见例如Stephens和Scheet,《单倍型推断中连锁不平衡的衰减的解释和遗失数据估算(Accounting for Decay of Linkage Disequilibrium inHaplotype Inference and Missing-Data Imputation)》,《美国人类遗传学杂志(Am.J.Hum.Genet.)》,76:449-462,2005,其以全文引用的方式并入本文中)。例示性程序是PHASE v.2.1或v2.1.1.(可以在万维网网址stephenslab.uchicago.edu/software.html获得,其以全文引用的方式并入本文中)。
在一个实施例中,使用由群体基因型数据估计单倍型的算法对个体的基因数据进行定相,如使得簇成员根据隐式马尔可夫模型沿染色体连续地改变的算法。这种方法是灵活的,实现连锁不平衡的“域样”模式和连锁不平衡随距离的逐渐降低(参见例如Scheet和Stephens,《用于大规模群体基因型数据的快速和灵活的统计模型:推断遗失基因型和单倍型相的应用(A fast and flexible statistical model for large-scale populationgenotype data:applications to inferring missing genotypes and haplotypicphase)》,《美国人类遗传学杂志》,78:629-644,2006,其以全文引用的方式并入本文中)。例示性程序是fastPHASE(可以在万维网网址stephenslab.uchicago.edu/software.html获得,其以全文引用的方式并入本文中)。
在一个实施例中,使用基因型估算方法对个体的基因数据进行定相,如使用以下参考数据集中的一个或多个的方法:HapMap数据集、在多个SNP芯片上进行基因分型的对照物的数据集和来自1,000个基因组计划的密集分型样品。例示性方法是灵活的模型化构架,其提高准确性且组合跨越多个参考图的信息(参见例如Howie,Donnelly和Marchini(2009)《用于下一代基因组广泛关联研究的灵活和准确的基因型估算方法(A flexible andaccurate genotype imputation method for the next generation of genome-wideassociation studies)》,《公共科学图书馆遗传学(PLoS Genetics)》5(6):e1000529,2009,其以全文引用的方式并入本文中)。例示性程序是IMPUTE或IMPUTE版本2(也称为IMPUTE2)(可以在万维网网址mathgen.stats.ox.ac.uk/impute/impute_v2.html获得,其以全文引用的方式并入本文中)。
在一个实施例中,使用推断单倍型的算法对个体的基因数据进行定相,如在通过重组进行聚结的遗传模型下推断单倍型的算法,如由Stephens在PHASE v2.1中研发的算法。主要算法改进依赖于使用二进制树表示每个个体的候选单倍型的集合。这些二进制树表示:(1)通过避免PHASE v2.1中的冗余操作来加速单倍型的后验概率的计算,和(2)通过在二进制树中智能探索似乎最合理的路径(即,单倍型)来解决单倍型推断问题的指数方面(参见例如Delaneau,Coulonges和Zagury,《Shape-IT:用于单倍型推断的新的快速和准确算法(Shape-IT:new rapid and accurate algorithm for haplotype inference)》,《BMC生物信息学(BMC Bioinformatics)》9:540,2008 doi:10.1186/1471-2105-9-540,其以全文引用的方式并入本文中)。例示性程序是SHAPEIT(可以在万维网网址mathgen.stats.ox.ac.uk/genetics_software/shapeit/shapeit.html获得,其以全文引用的方式并入本文中)。
在一个实施例中,使用由群体基因型数据估计单倍型的算法对个体的基因数据进行定相,如使用单倍型片段出现率获得更长的单倍型的基于经验的概率的算法。在一些实施例中,算法重构单倍型使得其具有最大局部相干性(参见例如Eronen,Geerts和Toivonen,《HaploRec:单倍型的有效和准确大规模重构(HaploRec:Efficient andaccurate large-scale reconstruction of haplotypes)》,《BMC生物信息学》7:542,2006,其以全文引用的方式并入本文中)。例示性程序是HaploRec,如HaploRec版本2.3(可以在万维网网址cs.helsinki.fi/group/genetics/haplotyping.html获得,其以全文引用的方式并入本文中)。
在一个实施例中,使用由群体基因型数据估计单倍型的算法对个体的基因数据进行定相,如使用分割-接合策略的算法和基于预期-最大化的算法(参见例如
在一个实施例中,使用由群体基因型数据估计单倍型的算法对个体的基因数据进行定相,如将基因型同时定相成单倍型和域分割的算法。在一些实施例中,使用预期-最大化算法(参见例如Kimmel和Shamir,《GERBIL:使用似然性的基因型分辨和域鉴别(GERBIL:Genotype Resolution and Block Identification Using Likelihood)》,《美国国家科学院院刊(Proceedings of the National Academy of Sciences of the United Statesof America;PNAS)》102:158-162,2005,其以全文引用的方式并入本文中)。例示性程序是GERBIL,其可以作为GEVALT版本2程序的一部分获得(可以在万维网网址acgt.cs.tau.ac.il/gevalt/获得,其以全文引用的方式并入本文中)。
在一个实施例中,使用由群体基因型数据估计单倍型的算法对个体的基因数据进行定相,如使用EM算法计算单倍型出现率的ML估计值,由此提供未指定相的基因型测量结果的算法。算法还允许遗失一些基因型测量结果(举例来说,由于PCR失败)。其还实现个体单倍型的多重估算(参见例如Clayton,D.(2002),《SNPHAP:用于估计SNP的大型单倍型的出现率的程序(SNPHAP:A Program for Estimating Frequencies of Large Haplotypes ofSNPs)》,其以全文引用的方式并入本文中)。例示性程序是SNPHAP(可以在万维网网址
在一个实施例中,使用由群体基因型数据估计单倍型的算法对个体的基因数据进行定相,如基于所收集的SNP对的基因型统计数据进行单倍型推断的算法。这一软件可以用于大量长基因组序列(例如从DNA阵列获得)的相对准确的定相。例示性程序使用基因型矩阵作为输入且输出相应的单倍型矩阵(参见例如Brinza和Zelikovsky,《2SNP:基于2-SNP单倍型的可扩展的定相(2SNP:scalable phasing based on 2-SNP haplotypes)》,《生物信息学》,22(3):371-3,2006,其以全文引用的方式并入本文中)。例示性程序是2SNP(可以在万维网网址alla.cs.gsu.edu/~software/2SNP获得,其以全文引用的方式并入本文中)。
在各种实施例中,使用关于染色体在染色体或染色体区段中的不同位置处交叉的概率的数据对个体的基因数据进行定相(如使用重组数据(如可在HapMap数据库中获得)创建任何间隔的重组风险评分),以模型化染色体或染色体区段上的多态等位基因之间的相关性。在一些实施例中,基于测序数据或SNP阵列数据,在计算机上计算多态基因座处的等位基因计数。在一些实施例中,创建(如在计算机上创建)各自关于染色体或染色体区段的不同的可能的状态的多个假设(如来自个体的一个或多个细胞的基因组中,与第二同源染色体区段相比,第一同源染色体区段的拷贝数目的过度表示;第一同源染色体区段的复制;第二同源染色体区段的缺失;或第一和第二同源染色体区段的同等表示);针对每种假设构建(如在计算机上构建)染色体上的多态基因座处的所预期的等位基因计数的模型(如联合分布模型);使用联合分布模型和等位基因计数确定(如在计算机上确定)每种假设的相对概率;和选择具有最大概率的假设。在一些实施例中,使用无需使用参考染色体的方法进行构建等位基因计数的联合分布模型和确定每种假设的相对概率的步骤。
在一些实施例中,分析来自个体的样品(例如活检(如肿瘤活检)、血液样品、血浆样品、血清样品或另一种可能主要含有或仅含有具有相关CNV的细胞、DNA或RNA的样品)以确定已知或怀疑含有相关CNV(如缺失或复制)的一个或多个区域的相。在一些实施例中,样品具有高肿瘤分数(如30、40、50、60、70、80、90、95、98、99或100%)。
在一些实施例中,样品具有单倍型不平衡或任何非整倍性。在一些实施例中,样品包括两种类型的DNA的任何混合物,其中所述两种类型具有两种单倍型的不同比率且共有至少一种单倍型。举例来说,在肿瘤情况下,正常组织是1:1,且肿瘤组织是1:0或1:2、1:3、1:4等。在一些实施例中,分析至少10、100、500、1,000、2,000、3,000、5,000、8,000或10,000个多态基因座以确定一些或全部基因座处的等位基因的相。在一些实施例中,样品是来自经过处理以变成非整倍性(如由长期细胞培养诱导的非整倍性)的细胞或组织。
在一些实施例中,样品中较大百分比或所有的DNA或RNA具有相关CNV。在一些实施例中,来自一种或多种目标细胞的含有相关CNV的DNA或RNA与样品中全部DNA或RNA的比率是至少80、85、90、95或100%。对于具有缺失的样品,针对具有缺失的细胞(或DNA或RNA)仅存在一个单倍型。这个第一单倍型可以使用标准方法确定,以确定缺失区域中的等位基因的一致性。在仅含有具有缺失的细胞(或DNA或RNA)的样品中,将仅存在来自存在于这些细胞中的第一单倍型的信号。在还含有少量的不具有缺失的细胞(或DNA或RNA)的样品(如少量非癌性细胞)中,可以忽略来自这些细胞(或DNA或RNA)中的第二单倍型的弱信号。可以通过推断来确定存在于来自个体的不具有缺失的其它细胞、DNA或RNA中的第二单倍型。举例来说,如果来自个体的不具有缺失的细胞的基因型是(AB,AB)且个体的定相数据指示第一单倍型是(A,A),那么可以推断另一单倍型是(B,B)。
对于其中存在具有缺失的细胞(或DNA或RNA)和不具有缺失的细胞(或DNA或RNA)的样品,仍然可以确定相。举例来说,可以产生其中x轴表示各个基因座沿染色体的线性位置且y轴表示作为全部(A+B)等位基因读段的一部分的A等位基因读段的数目的图。在缺失的一些实施例中,模式包括两条中央谱带,其表示杂合个体的SNP(上部谱带表示来自不具有缺失的细胞的AB和来自具有缺失的细胞的A,且下部谱带表示来自不具有缺失的细胞的AB和来自具有缺失的细胞的B)。在一些实施例中,这两条谱带的分离程度随着具有缺失的细胞、DNA或RNA的分数增加而增加。因此,A等位基因的一致性可以用于确定第一单倍型,且B等位基因的一致性可以用于确定第二单倍型。
对于具有复制的样品,针对具有复制的细胞(或DNA或RNA)存在单倍型的额外拷贝。可以使用标准方法确定被复制的区域的这一单倍型,以确定复制区域中以增加的量存在的等位基因的一致性,或可以使用标准方法确定未被复制的区域的单倍型,以确定以降低的量存在的等位基因的一致性。在确定一个单倍型之后,可以通过推断来确定另一单倍型。
对于其中存在具有复制的细胞(或DNA或RNA)和不具有复制的细胞(或DNA或RNA)的样品,仍然可以使用与上文关于缺失所描述类似的方法确定相。举例来说,可以产生其中x轴表示各个基因座沿染色体的线性位置且y轴表示作为全部(A+B)等位基因读段的一部分的A等位基因读段的数目的图。在缺失的一些实施例中,模式包括两条中央谱带,其表示杂合个体的SNP(上部谱带表示来自不具有复制的细胞的AB和来自具有复制的细胞的AAB,且下部谱带表示来自不具有复制的细胞的AB和来自具有复制的细胞的ABB)。在一些实施例中,这两条谱带的分离程度随着具有复制的细胞、DNA或RNA的分数增加而增加。因此,A等位基因的一致性可以用于确定第一单倍型,且B等位基因的一致性可以用于确定第二单倍型。在一些实施例中,确定来自已知患有癌症的个体的样品(如肿瘤活检或血浆样品)的一个或多个CNV区域的相(如所测量的区域中的至少50、60、70、80、90、95或100%的多态基因座的相),且用于分析来自同一名个体的后续样品以监测癌症的进展(如监测癌症的缓解或复发)。在一些实施例中,使用具有高肿瘤分数的样品(如来自具有高肿瘤负荷的个体的肿瘤活检或血浆样品)获得定相数据,其用于分析具有较低肿瘤分数的后续样品(如来自正在经历癌症治疗或缓解的个体的血浆样品)。
在一些实施例中,使用两种或更多种本文中所描述的方法对个体的基因数据进行定相。在一些实施例中,使用生物信息学方法(如使用基于群体的单倍型出现率以推断最有可能的相)和分子生物学方法(如本文中所公开的任何用于获得实际定相数据而非基于生物信息学推断的定相数据的分子定相方法)。在一些实施例中,使用来自其它个体(如先验个体)的定相数据优化群体数据。举例来说,可以将来自其它个体的定相数据添加到群体数据中以计算另一个体的可能的单倍型的先验。在一些实施例中,使用来自其它个体(如先验个体)的定相数据计算另一个体的可能的单倍型的先验。
在一些实施例中,可以使用概率数据。举例来说,归因于样品中DNA分子的表示的概率性质以及各种扩增和测量偏差,由两个不同的基因座或既定基因座处的不同等位基因测量的DNA分子的相对数目未必总是表示混合物或个体中的分子的相对数目。如果试图通过对来自个体的血浆的DNA进行测序来确定正常二倍体个体的常染色体上的既定基因座处的基因型,那么预期将观察到仅一种等位基因(纯合)或大致相等数目的两种等位基因(杂合)。如果在所述等位基因处,观察到十个A等位基因分子且观察到两个B等位基因分子,那么将不清楚个体在所述基因座处是否是纯合的且两个B等位基因分子是否归因于噪声或污染,或如果个体是否是杂合的且较低数目的B等位基因分子是否归因于血浆中的DNA分子的数目的随机、统计变化、扩增偏差、污染或许多其它原因。在这种情况下,可以计算个体的纯合概率和相应的个体的杂合概率,且这些概率基因型可以用于其它计算中。
应注意,对于既定等位基因比率,所观察的分子数目越大,所述比率紧密表示个体中的DNA分子的比率的似然性越大。举例来说,如果测量100个A分子和100个B分子,那么实际比率是50%的似然性显著大于测量10个A分子和10个B分子的情况。在一个实施例中,使用贝叶斯理论与详细数据模型的组合以确定在既定观察结果下,具体假设是正确的似然性。举例来说,如果考虑两种假设,一种对应于三体个体且一种对应于二体个体,那么与观察两种等位基因中的每一种的10个分子的情况相比,在观察两种等位基因中的每一种的100个分子的情况下,二体假设正确的概率将显著更高。随着数据中的噪声由于偏差、污染或一些其它噪声来源而变大,或随着既定基因座处的观察数目降低,鉴于所观察的数据,最大似然假设为真的概率降低。在实践中,有可能合计多个基因座的概率以增加可以将最大似然假设确定为正确假设的置信度。在一些实施例中,简单地合计概率而不考虑重组。在一些实施例中,计算考虑交叉现象。
在一个实施例中,使用以概率方式定相的数据确定拷贝数目变化。在一些实施例中,以概率方式定相的数据是来自数据源(如HapMap数据库)的基于群体的单倍型域出现率数据。在一些实施例中,以概率方式定相的数据是由分子方法获得的单倍型数据,例如通过稀释进行定相,其中将染色体的各个区段稀释到单一分子/反应,但其中由于随机噪声,单倍型的身份可能不是绝对已知的。在一些实施例中,以概率方式定相的数据是由分子方法获得的单倍型数据,其中可以在高度确定性下已知单倍型的身份。
设想以下假设的情况:医生想要通过测量来自个体的血浆DNA来确定个体的身体中是否具有一些在具体染色体区段处具有缺失的细胞。医生可以使用以下知识:如果用于提取血浆DNA的所有细胞都是二倍体且具有相同基因型,那么对于杂合基因座,关于两种等位基因中的每一种所观察的DNA分子的相对数目将服从以50%A等位基因和50%B等位基因为中心的一种分布。然而,如果一部分用于提取血浆DNA的细胞在具体染色体区段处具有缺失,那么对于杂合基因座,将预期关于两种等位基因中的每一种所观察的DNA分子的相对数目将服从两种分布,一种以超过50%A等位基因为中心(对于存在含有B等位基因的染色体区段的缺失的基因座)且一种以低于50%为中心(对于存在含有A等位基因的染色体区段的缺失的基因座)。含有缺失的用于提取血浆DNA的细胞的比例越大,这两种分布将越远离50%。
在这种假设的情况中,设想临床医生想要确定个体是否在个体体内的一定比例的细胞中具有染色体区域的缺失。临床医生可以从个体抽取血液到真空采血系统或其它类型的血液管中,将血液离心且分离血浆层。临床医生可以从血浆分离DNA,富集目标基因座处的DNA,可能通过靶向或其它扩增、基因座捕获技术、尺寸富集或其它富集技术。临床医生可以使用如qPCR、测序、微阵列或其它测量样品中的DNA数量的技术等分析法,通过测量SNP集合处的等位基因的数目,换句话说,产生等位基因出现率数据来分析被富集和/或扩增的DNA。我们将考虑在以下情况中的数据分析:临床医生使用靶向扩增技术扩增游离血浆DNA,且接着对被扩增的DNA进行测序以获得以下在染色体区段上发现的六个SNP处的指示癌症的例示性可能数据,其中个体在这些SNP处是杂合的:
SNP 1:460个读段A等位基因;540个读段B等位基因(46%A)
SNP 2:530个读段A等位基因;470个读段B等位基因(53%A)
SNP 3:40个读段A等位基因;60个读段B等位基因(40%A)
SNP 4:46个读段A等位基因;54个读段B等位基因(46%A)
SNP 5:520个读段A等位基因;480个读段B等位基因(52%A)
SNP 6:200个读段A等位基因;200个读段B等位基因(50%A)
由这一数据集,可能难以区分个体正常且所有细胞具有二体性的情况与个体可能患有癌症且某一部分细胞的DNA对在血浆中发现的在染色体处具有缺失或复制的游离DNA具有贡献的情况。举例来说,两种具有最大似然性的假设可以是个体在这一染色体区段处具有缺失,其中肿瘤分数是6%,和染色体的所缺失的区段在六个SNP上具有基因型(A,B,A,A,B,B)或(A,B,A,A,B,A)。在SNP集合上的个体的基因型的这种表示中,括号中的第一个字母对应于SNP 1的单倍型的基因型,第二个字母对应于SNP 2等。
如果使用一种方法确定所述染色体区段处的个体的单倍型且发现两个染色体中的一个的单倍型是(A,B,A,A,B,B),那么这将与最大似然假设一致且所计算的个体在所述区段处具有缺失且因此可能具有癌性或癌变前细胞的似然性将显著提高。另一方面,如果发现个体具有单倍型(A,A,A,A,A,A),那么个体在所述染色体区段处具有缺失的似然性将显著降低,且可能无缺失假设的似然性将较高(实际似然值将取决于其它参数,尤其如系统中所测量的噪声)。
存在多种用于确定个体的单倍型的方式,其中许多方式描述于本文中的其它地方。本文中提供部分列表且不意味是穷尽性的。一种方法是生物学方法,其中稀释单独的DNA分子直到任何既定反应体积中具有约一个来自每个染色体区域的分子,且接着使用如测序等方法测量基因型。另一种方法是基于信息学的,其中可以按概率方式使用各种单倍型和其出现率的群体数据。另一种方法是测量个体以及预期与所述个体共有单倍型域的一个或多个相关个体的二倍体数据且推断单倍型域。另一种方法是获得具有高浓度的缺失或复制区段的组织样品且基于等位基因失衡来确定单倍型,举例来说,来自具有缺失的肿瘤组织样品的基因型测量结果可以用于确定所述缺失区域的定相数据,且这一数据接着可以用于确定癌症在切除术后是否重新生长。
在实践中,通常在既定染色体区段上测量超过20个SNP、超过50个SNP、超过100个SNP、超过500个SNP、超过1,000个SNP或超过5,000个SNP。
例示性突变
与疾病或病症(如癌症)或增加的疾病或病症(如癌症)风险(如高于正常风险等级)相关联的例示性突变包括单核苷酸变异体(SNV)、多核苷酸突变、缺失(如2百万到3千万个碱基对区域的缺失)、复制或串联重复序列。在一些实施例中,突变是在DNA中,如cfDNA、游离线粒体DNA(cf mDNA)、来源于细胞核DNA的游离DNA(cf nDNA)、细胞DNA或线粒体DNA。在一些实施例中,突变是在RNA中,如cfRNA、细胞RNA、细胞质RNA、编码细胞质RNA、非编码细胞质RNA、mRNA、miRNA、线粒体RNA、rRNA或tRNA。在一些实施例中,与未患有疾病或病症(如癌症)的个体相比,突变在患有疾病或病症(如癌症)的个体中以更高的出现率存在。在一些实施例中,突变指示癌症,如致病性突变。在一些实施例中,突变是驱动子突变,其在疾病或病症中具有致病作用。在一些实施例中,突变不是致病性突变。举例来说,在一些癌症中,多个突变积聚,但其中一些不是致病性突变。不致病的突变(如与未患有疾病或病症的个体相比,在患有疾病或病症的个体中以更高的出现率存在的突变)仍适用于诊断疾病或病症。在一些实施例中,突变是一个或多个微卫星处的杂合性丢失(LOH)。
在一些实施例中,针对已知个体具有的一种或多种多态现象或突变对个体进行筛检(例如检验是否存在多态现象或突变;具有这些多态现象或突变的细胞、DNA或RNA的量的变化;或癌症缓解或复发)。在一些实施例中,针对已知个体具有风险的一种或多种多态现象或突变对个体进行筛检(如具有携带所述多态现象或突变的亲属的个体)。在一些实施例中,针对一组与疾病或病症(如癌症)相关联的多态现象或突变(例如至少5、10、50、100、200、300、500、750、1,000、1,500、2,000或5,000种多态现象或突变)对个体进行筛检。
许多与癌症相关联的编码变异体描述于Abaan等人,《NCI-60图的外显子组:癌症生物学和系统药理学的基因组资源(The Exomes of the NCI-60Panel:A GenomicResource for Cancer Biology and Systems Pharmacology)》,《癌症研究(CancerResearch)》,2013年7月15日,和万维网网址dtp.nci.nih.gov/branches/btb/characterizationNCI60.html,其各自以全文引用的方式并入本文中)。NCI-60人类癌细胞系集合由60种不同的表示肺、结肠、脑部、卵巢、乳房、前列腺和肾脏的癌症以及白血病和黑素瘤的细胞系组成。在这些细胞系中发现的基因变异由两种类型组成:在正常群体中发现的I型变异体和具有癌症特异性的II型变异体。
例示性多态现象或突变(如缺失或复制)是在以下基因中的一个或多个和其组合中:TP53、PTEN、PIK3CA、APC、EGFR、NRAS、NF2、FBXW7、ERBBs、ATAD5、KRAS、BRAF、VEGF、EGFR、HER2、ALK、p53、BRCA、BRCA1、BRCA2、SETD2、LRP1B、PBRM、SPTA1、DNMT3A、ARID1A、GRIN2A、TRRAP、STAG2、EPHA3/5/7、POLE、SYNE1、C20orf80、CSMD1、CTNNB1、ERBB2.FBXW7、KIT、MUC4、ATM、CDH1、DDX11、DDX12、DSPP、EPPK1、FAM186A、GNAS、HRNR、KRTAP4-11、MAP2K4、MLL3、NRAS、RB1、SMAD4、TTN、ABCC9、ACVR1B、ADAM29、ADAMTS19、AGAP10、AKT1、AMBN、AMPD2、ANKRD30A、ANKRD40、APOBR、AR、BIRC6、BMP2、BRAT1、BTNL8、C12orf4、C1QTNF7、C20orf186、CAPRIN2、CBWD1、CCDC30、CCDC93、CD5L、CDC27、CDC42BPA、CDH9、CDKN2A、CHD8、CHEK2、CHRNA9、CIZ1、CLSPN、CNTN6、COL14A1、CREBBP、CROCC、CTSF、CYP1A2、DCLK1、DHDDS、DHX32、DKK2、DLEC1、DNAH14、DNAH5、DNAH9、DNASE1L3、DUSP16、DYNC2H1、ECT2、EFHB、RRN3P2、TRIM49B、TUBB8P5、EPHA7、ERBB3、ERCC6、FAM21A、FAM21C、FCGBP、FGFR2、FLG2、FLT1、FOLR2、FRYL、FSCB、GAB1、GABRA4、GABRP、GH2、GOLGA6L1、GPHB5、GPR32、GPX5、GTF3C3、HECW1、HIST1H3B、HLA-A、HRAS、HS3ST1、HS6ST1、HSPD1、IDH1、JAK2、KDM5B、KIAA0528、KRT15、KRT38、KRTAP21-1、KRTAP4-5、KRTAP4-7、KRTAP5-4、KRTAP5-5、LAMA4、LATS1、LMF1、LPAR4、LPPR4、LRRFIP1、LUM、LYST、MAP2K1、MARCH1、MARCO、MB21D2、MEGF10、MMP16、MORC1、MRE11A、MTMR3、MUC12、MUC17、MUC2、MUC20、NBPF10、NBPF20、NEK1、NFE2L2、NLRP4、NOTCH2、NRK、NUP93、OBSCN、OR11H1、OR2B11、OR2M4、OR4Q3、OR5D13、OR8I2、OXSM、PIK3R1、PPP2R5C、PRAME、PRF1、PRG4、PRPF19、PTH2、PTPRC、PTPRJ、RAC1、RAD50、RBM12、RGPD3、RGS22、ROR1、RP11-671M22.1、RP13-996F3.4、RP1L1、RSBN1L、RYR3、SAMD3、SCN3A、SEC31A、SF1、SF3B1、SLC25A2、SLC44A1、SLC4A11、SMAD2、SPTA1、ST6GAL2、STK11、SZT2、TAF1L、TAX1BP1、TBP、TGFBI、TIF1、TMEM14B、TMEM74、TPTE、TRAPPC8、TRPS1、TXNDC6、USP32、UTP20、VASN、VPS72、WASH3P、WWTR1、XPO1、ZFHX4、ZMIZ1、ZNF167、ZNF436、ZNF492、ZNF598、ZRSR2、ABL1、AKT2、AKT3、ARAF、ARFRP1、ARID2、ASXL1、ATR、ATRX、AURKA、AURKB、AXL、BAP1、BARD1、BCL2、BCL2L2、BCL6、BCOR、BCORL1、BLM、BRIP1、BTK、CARD11、CBFB、CBL、CCND1、CCND2、CCND3、CCNE1、CD79A、CD79B、CDC73、CDK12、CDK4、CDK6、CDK8、CDKN1B、CDKN2B、CDKN2C、CEBPA、CHEK1、CIC、CRKL、CRLF2、CSF1R、CTCF、CTNNA1、DAXX、DDR2、DOT1L、EMSY(C11orf30)、EP300、EPHA3、EPHA5、EPHB1、ERBB4、ERG、ESR1、EZH2、FAM123B(WTX)、FAM46C、FANCA、FANCC、FANCD2、FANCE、FANCF、FANCG、FANCL、FGF10、FGF14、FGF19、FGF23、FGF3、FGF4、FGF6、FGFR1、FGFR2、FGFR3、FGFR4、FLT3、FLT4、FOXL2、GATA1、GATA2、GATA3、GID4(C17orf39)、GNA11、GNA13、GNAQ、GNAS、GPR124、GSK3B、HGF、IDH1、IDH2、IGF1R、IKBKE、IKZF1、IL7R、INHBA、IRF4、IRS2、JAK1、JAK3、JUN、KAT6A(MYST3)、KDM5A、KDM5C、KDM6A、KDR、KEAP1、KLHL6、MAP2K2、MAP2K4、MAP3K1、MCL1、MDM2、MDM4、MED12、MEF2B、MEN1、MET、MITF、MLH1、MLL、MLL2、MPL、MSH2、MSH6、MTOR、MUTYH、MYC、MYCL1、MYCN、MYD88、NF1、NFKBIA、NKX2-1、NOTCH1、NPM1、NRAS、NTRK1、NTRK2、NTRK3、PAK3、PALB2、PAX5、PBRM1、PDGFRA、PDGFRB、PDK1、PIK3CG、PIK3R2、PPP2R1A、PRDM1、PRKAR1A、PRKDC、PTCH1、PTPN11、RAD51、RAF1、RARA、RET、RICTOR、RNF43、RPTOR、RUNX1、SMARCA4、SMARCB1、SMO、SOCS1、SOX10、SOX2、SPEN、SPOP、SRC、STAT4、SUFU、TET2、TGFBR2、TNFAIP3、TNFRSF14、TOP1、TP53、TSC1、TSC2、TSHR、VHL、WISP3、WT1、ZNF217、ZNF703(Su等人,《分子诊断学杂志(J Mol Diagn)》2011,13:74-84;DOI:10.1016/j.jmoldx.2010.11.010;和Abaan等人,《NCI-60图的外显子组:癌症生物学和系统药理学的基因组资源》,《癌症研究》,2013年7月15日,其各自以全文引用的方式并入本文中)。在一些实施例中,复制是与乳癌相关联的染色体1p(“Chr1p”)复制。在一些实施例中,一种或多种多态现象或突变是在BRAF中,如V600E突变。在一些实施例中,一种或多种多态现象或突变是在K-ras中。在一些实施例中,K-ras和APC中存在一种或多种多态现象或突变的组合。在一些实施例中,K-ras和p53中存在一种或多种多态现象或突变的组合。在一些实施例中,APC和p53中存在一种或多种多态现象或突变的组合。在一些实施例中,K-ras、APC和p53中存在一种或多种多态现象或突变的组合。在一些实施例中,K-ras和EGFR中存在一种或多种多态现象或突变的组合。例示性多态现象或突变是在以下微RNA中的一个或多个中:miR-15a、miR-16-1、miR-23a、miR-23b、miR-24-1、miR-24-2、miR-27a、miR-27b、miR-29b-2、miR-29c、miR-146、miR-155、miR-221、miR-222和miR-223(Calin等人,《与慢性淋巴细胞性白血病的预后和进展相关联的微RNA标签(A microRNAsignature associated with prognosis and progression in chronic lymphocyticleukemia)》,《新英格兰医学杂志《N Engl J Med》》353:1793-801,2005,其以全文引用的方式并入本文中)。
在一些实施例中,缺失是至少0.01kb、0.1kb、1kb、10kb、100kb、1mb、2mb、3mb、5mb、10mb、15mb、20mb、30mb或40mb的缺失。在一些实施例中,缺失是1kb到40mb之间的缺失,如在1kb到100kb、100kb到1mb、1到5mb、5到10mb、10到15mb、15到20mb、20到25mb、25到30mb或30到40mb之间且包括端值。
在一些实施例中,复制是至少0.01kb、0.1kb、1kb、10kb、100kb、1mb、2mb、3mb、5mb、10mb、15mb、20mb、30mb或40mb的复制。在一些实施例中,复制是在1kb到40mb之间的复制,如在1kb到100kb、100kb到1mb、1到5mb、5到10mb、10到15mb、15到20mb、20到25mb、25到30mb或30到40mb之间且包括端值。
在一些实施例中,串联重复序列是2与60个核苷酸之间的重复序列,如2到6、7到10、10到20、20到30、30到40、40到50或50到60个核苷酸且包括端值。在一些实施例中,串联重复序列是2个核苷酸的重复序列(二核苷酸重复序列)。在一些实施例中,串联重复序列是3个核苷酸的重复序列(三核苷酸重复序列)。
在一些实施例中,多态现象或突变是预后的。例示性预后突变包括K-ras突变,如指示结肠直肠癌中的手术后疾病复发的K-ras突变(Ryan等人,《结肠直肠赘瘤形成患者的血清中循环突变体KRAS2的前瞻性研究:手术后随访中的强预后性指示(A prospectivestudy of circulating mutant KRAS2 in the serum of patients with colorectalneoplasia:strong prognostic indicator in postoperative follow up)》,《肠道学(Gut)》52:101-108,2003;和Lecomte T等人,《结肠直肠癌患者的血浆中自由循环肿瘤相关DNA的检测和其与预后的相关性(Detection of free-circulating tumor-associatedDNA in plasma of colorectal cancer patients and its association withprognosis)》,《国际癌症杂志(Int J Cancer)》100:542-548,2002,其各自以全文引用的方式并入本文中)。
在一些实施例中,多态现象或突变与对具体治疗的反应改变(如功效或副作用增加或降低)相关联。实例包括非小细胞肺癌中K-ras突变与对基于EGFR的治疗的反应降低相关联(Wang等人,《晚期非小细胞肺癌患者中基于血浆的KRAS突变分析的潜在临床显著性(Potential clinical significance of a plasma-based KRAS mutation analysis inpatients with advanced non-small cell lung cancer)》,《临床癌症研究》16:1324-1330,2010,其以全文引用的方式并入本文中)。
K-ras是在多种癌症中活化的致癌基因。例示性K-ras突变是密码子12、13和61中的突变。已经在胰腺、肺、结肠直肠、膀胱和胃部癌症中发现K-ras cfDNA突变(Fleischhacker和Schmidt,《循环核酸(CNAs)和癌症调查(Circulating nucleic acids(CNAs)and caner-a survey)》,《生物化学与生物物理学报(Biochim Biophys Acta)》1775:181-232,2007,其以全文引用的方式并入本文中)。
p53是在许多癌症中突变的肿瘤抑制因子且促进肿瘤恶化(Levine和Oren,《p53的第一个30年:不断变复杂(The first 30years of p53:growing ever more complex)》,《自然癌症综述(Nature Rev Cancer)》,9:749-758,2009,其以全文引用的方式并入本文中)。许多不同的密码子可以突变,如Ser249。已在乳房、肺、卵巢、膀胱、胃、胰腺、结肠直肠、肠和肝细胞癌症中发现p53 cfDNA突变(Fleischhacker和Schmidt,《循环核酸(CNAs)和癌症调查》,《生物化学与生物物理学报》1775:181-232,2007,其以全文引用的方式并入本文中)。
BRAF是Ras的下游致癌基因。已在神经胶质赘瘤、黑素瘤、甲状腺和肺癌中发现BRAF突变(Dias-Santagata等人,《在多形态黄星形细胞瘤中常见的BRAF V600E突变:诊断性和治疗性含义(BRAF V600E mutations are common in pleomorphicxanthoastrocytoma:diagnostic and therapeutic implications)》,《科学公共图书馆综合卷(PLOS ONE)》2011;6:e17948,2011;Shinozaki等人,《血清中的循环B-RAF DNA突变用于监测接收生物化学疗法的黑素瘤患者的效用(Utility of circulating B-RAF DNAmutation in serum for monitoring melanoma patients receivingbiochemotherapy)》,《临床癌症研究(Clin Canc Res)》13:2068-2074,2007;和Board等人,《参与AZD6244(ARRY-142886)晚期黑素瘤II期研究的患者的肿瘤和血清中BRAF突变的检测(Detection of BRAF mutations in the tumor and serum of patients enrolled inthe AZD6244(ARRY-142886)advanced melanoma phase II study)》,《英国癌症杂志(BritJ Canc)》2009;101:1724-1730,其各自以全文引用的方式并入本文中)。BRAF V600E突变在例如黑素瘤中发生且在晚期更常见。已在cfDNA中检测到V600E突变。
EGFR促进细胞增殖且在许多癌症中失调(Downward J.,《癌症疗法中的靶向RAS信号传导路径(Targeting RAS signalling pathways in cancer therapy)》,《自然癌症综述》3:11-22,2003;以及Levine和Oren,《p53的第一个30年:不断变复杂》,《自然癌症综述》9:749-758,2009,其以全文引用的方式并入本文中)。例示性EGFR突变包括外显子18-21中的突变,其已在肺癌患者中被发现。已在肺癌患者中发现EGFR cfDNA突变(Jia等人,《晚期非小细胞肺癌中吉非替尼治疗对血浆/胸腔积液中的表皮生长因子受体突变的功效的预测(Prediction of epidermal growth factor receptor mutations in the plasma/pleural effusion to efficacy of gefitinib treatment in advanced non-smallcell lung cancer)》,《临床肿瘤学癌症研究杂志(J Canc Res Clin Oncol)》2010;136:1341-1347,2010,其以全文引用的方式并入本文中)。
与乳癌相关联的例示性多态现象或突变包括微卫星处的LOH(Kohler等人,《作为乳房肿瘤的潜在生物标记的血浆循环游离细胞核和线粒体DNA的含量(Levels of plasmacirculating cell free nuclear and mitochondrial DNA as potential biomarkersfor breast tumors)》,《分子癌症》8:doi:10.1186/1476-4598-8-105,2009,其以全文引用的方式并入本文中)、p53突变(如外显子5-8中的突变)(Garcia等人,《血浆中的细胞外肿瘤DNA和乳癌患者的总存活率(Extracellular tumor DNA in plasma and overallsurvival in breast cancer patients)》,《基因、染色体和癌症(Genes Chromosomes&Cancer)》45:692-701,2006,其以全文引用的方式并入本文中)、HER2(Sorensen等人,《曲妥珠单抗治疗之后的循环HER2DNA预测乳癌中的存活率和反应(Circulating HER2 DNAafter trastuzumab treatment predicts survival and response in breastcancer)》,《抗癌剂研究(Anticancer Res)》30:2463-2468,2010,其以全文引用的方式并入本文中)、PIK3CA、MED1和GAS6多态现象或突变(Murtaza等人,《通过血浆DNA的测序进行的对后天性癌症疗法抗性的非侵袭性分析(Non-invasive analysis of acquiredresistance to cancer therapy by sequencing of plasma DNA)》,《自然(Nature)》2013;doi:10.1038/nature12065,2013,其以全文引用的方式并入本文中)。
cfDNA含量和LOH增加与整体和无疾病存活率相关联。p53突变(外显子5-8)与总存活率降低相关联。循环HER2cfDNA含量降低与HER2阳性乳房肿瘤个体中更好的对HER2靶向治疗的反应相关联。PIK3CA中的活化突变、MED1的截短和GAS6中的剪接突变引起对治疗的抗性。
与结肠直肠癌相关联的例示性多态现象或突变包括p53、APC、K-ras和胸苷酸合成酶突变以及p16基因甲基化(Wang等人,《结肠直肠癌患者的血清中作为循环生物标记物的APC、K-ras和p53突变的分子检测(Molecular detection of APC,K-ras,andp53mutations in the serum of colorectal cancer patients as circulatingbiomarkers)》,《世界外科杂志(World J Surg)》28:721-726,2004;Ryan等人,《结肠直肠赘瘤形成患者的血清中循环突变体KRAS2的前瞻性研究:手术后随访中的强预后性指示》,《肠道学》52:101-108,2003;Lecomte等人,《结肠直肠癌患者的血浆中游离循环肿瘤相关DNA的检测和其与预后的相关性》,《国际癌症杂志》100:542-548,2002;Schwarzenbach等人,《晚期结肠直肠癌患者的血液中游离循环DNA上的胸苷酸合成酶的多态现象的分子分析(Molecular analysis of the polymorphisms of thymidylate synthase on cell-freecirculating DNA in blood of patients with advanced colorectal carcinoma)》,《国际癌症杂志》127:881-888,2009,其各自以全文引用的方式并入本文中)。血清中K-ras突变的手术后检测是疾病复发的强预测因子。K-ras突变和p16基因甲基化的检测与存活率降低和疾病复发增加相关联。K-ras、APC和/或p53突变的检测与复发和/或癌转移相关联。使用cfDNA的胸苷酸合成酶(基于氟嘧啶的化学疗法的目标)中的多态现象(包括LOH、SNP、可变数目串联重复序列和缺失)可能与治疗反应相关联。
与肺癌(如非小细胞肺癌)相关联的例示性多态现象或突变包括K-ras(如密码子12中的突变)和EGFR突变。例示性预后突变包括与整体和无进展存活期延长相关联的EGFR突变(外显子19缺失或外显子21突变)以及与无进展存活期缩短相关联的K-ras突变(密码子12和13中)(Jian等人,《晚期非小细胞肺癌中吉非替尼治疗对血浆/胸腔积液中的表皮生长因子受体突变的功效的预测》,《临床肿瘤学癌症研究》136:1341-1347,2010;Wang等人,《晚期非小细胞肺癌患者中基于血浆的KRAS突变分析的潜在临床显著性》,《临床癌症研究》16:1324-1330,2010,其各自以全文引用的方式并入本文中)。指示对治疗的反应的例示性多态现象或突变包括改善对治疗的反应的EGFR突变(外显子19缺失或外显子21突变)和降低对治疗的反应的K-ras突变(密码子12和13)。已鉴别EFGR中赋予抗性的突变(Murtaza等人,《通过血浆DNA的测序进行的对后天性癌症疗法抗性的非侵袭性分析》,《自然(Nature)》doi:10.1038/nature12065,2013,其以全文引用的方式并入本文中)。
与黑素瘤(如葡萄膜黑素瘤)相关联的例示性多态现象或突变包括GNAQ、GNA11、BRAF和p53中的多态现象或突变。例示性GNAQ和GNA11突变包括R183和Q209突变。GNAQ或GNA11中的Q209突变与对骨骼的癌转移相关联。可以在转移性/晚期黑素瘤患者中检测到BRAF V600E突变。BRAF V600E是侵袭性黑素瘤的指示物。在化学疗法之后存在BRAF V600E突变与对治疗不起反应相关联。
与胰腺癌瘤相关联的例示性多态现象或突变包括K-ras和p53中的多态现象或突变(如p53Ser249)。p53Ser249还与B型肝炎感染和肝细胞癌以及卵巢癌和非霍奇金淋巴瘤(non-Hodgkin's lymphoma)相关联。
本发明的方法甚至可以检测到样品中以低出现率存在的多态现象或突变。举例来说,通过进行1千万个测序读段,可以观察到10倍的以百万分之1的出现率存在的多态现象或突变。视需要,可以视所需敏感性的水平来改变测序读段的数目。在一些实施例中,重新分析样品或使用更大数目的测序读段分析来自个体的另一样品以改善敏感性。举例来说,如果未检测到或仅检测到较少数目(如1、2、3、4或5种)的与癌症或增加的癌症风险相关联的多态现象或突变,那么重新分析样品或检验另一样品。
在一些实施例中,癌症或转移性癌症需要多种多态现象或突变。在这类情况下,筛检多种多态现象或突变可以提高准确地诊断癌症或转移性癌症的能力。在一些实施例中,当个体具有癌症或转移性癌症所需的多种多态现象或突变的子集时,可以随后重新筛检个体以观察个体是否获取其它突变。
在其中癌症或转移性癌症需要多种多态现象或突变的一些实施例中,可以比较每种多态现象或突变的出现率以观察其是否以类似出现率出现。举例来说,如果癌症需要两种突变(表示为“A”和“B”),那么一些细胞将不具有突变,一些细胞具有A,一些细胞具有B且一些细胞具有A和B。如果以类似的出现率观察到A和B,那么个体更可能具有一些具有A和B两者的细胞。如果以相异的出现率观察到A和B,那么个体更可能具有不同的细胞群体。
在其中癌症或转移性癌症需要多种多态现象或突变的一些实施例中,个体中的这类多态现象或突变的数目或一致性可以用于预测个体可能发生疾病或病症的可能性或时间。在其中多态现象或突变倾向于以某一顺序发生的一些实施例中,可以周期性地检验个体以观察个体是否获取其它多态现象或突变。
在一些实施例中,确定存在或不存在多种多态现象或突变(如2、3、4、5、8、10、12、15种或更多)可以提高存在或不存在疾病或病症(如癌症)或增加的疾病或病症(如癌症)的风险的确定的敏感性和/或特异性。
在一些实施例中,直接检测多态现象或突变。在一些实施例中,通过检测与多态现象或突变相关的一个或多个序列(例如多态基因座,如SNP)来间接地检测多态现象或突变。
例示性核酸变化
在一些实施例中,存在与疾病或病症(如癌症)或增加的疾病或病症(如癌症)风险相关联的RNA或DNA的完整性的变化(如片段化cfRNA或cfDNA的尺寸的变化或核小体组成的变化)。在一些实施例中,存在与疾病或病症(如癌症)或增加的疾病或病症(如癌症)风险相关联的甲基化模式RNA或DNA的变化(例如肿瘤抑制基因的超甲基化)。举例来说,已提出肿瘤抑制基因的启动子区域中的CpG岛的甲基化会触发局部基因沉默。在患有肝癌、肺癌和乳癌的个体中发生p16肿瘤抑制基因的异常甲基化。已在各种类型的癌症(例如鼻咽癌、结肠直肠癌、肺癌、食道癌、前列腺癌、膀胱癌、黑素瘤和急性白血病)中检测到其它频繁甲基化的肿瘤抑制基因,包括APC、Ras关联域家族蛋白质1A(RASSF1A)、谷胱甘肽S-转移酶P1(GSTP1)和DAPK。某些肿瘤抑制基因(如p16)的甲基化已被描述为癌症形成中的早期事件且因此适用于早期癌症筛检。
在一些实施例中,使用甲基化敏感性限制酶消化的基于亚硫酸氢盐转化或非亚硫酸氢盐的策略用于确定甲基化模式(Hung等人,《临床病理学杂志(J Clin Pathol)》62:308-313,2009,其以全文引用的方式并入本文中)。在亚硫酸氢盐转化中,甲基化胞嘧啶保留为胞嘧啶,而未甲基化的胞嘧啶转化成尿嘧啶。甲基化敏感性限制酶(例如BstUI)使特异性识别位点(例如5'-CG∨CG-3',对于BstUI)处的未甲基化的DNA序列裂解,而甲基化序列保持完整。在一些实施例中,检测到完整的甲基化序列。在一些实施例中,使用茎-环引物选择性地扩增限制酶消化的未甲基化片段而不共同扩增非酶消化的甲基化DNA。
mRNA剪接中的例示性变化
在一些实施例中,mRNA剪接的变化与疾病或病症(如癌症)或增加的疾病或病症(如癌症)风险相关联。在一些实施例中,mRNA剪接的变化是在以下与癌症或增加的癌症风险相关联的核酸中的一个或多个中:DNMT3B、BRCA1、KLF6、Ron或Gemin5。在一些实施例中,所检测到的mRNA剪接变异体与疾病或病症(如癌症)相关联。在一些实施例中,由健康细胞(如非癌性细胞)产生多种mRNA剪接变异体,但mRNA剪接变异体的相对量的变化与疾病或病症(如癌症)相关联。在一些实施例中,mRNA剪接的变化是由以下引起:mRNA序列的变化(如剪接位点中的突变)、剪接因子含量的变化、可用的剪接因子的量的变化(如由剪接因子与重复序列的结合引起的可用的剪接因子的量降低)、剪接调节改变或肿瘤微环境。
剪接反应是由称为剪接体的多蛋白质/RNA复合物进行(Fackenthal1和Godley,《疾病模型和机制(Disease Models&Mechanisms)》1:37-42,2008,doi:10.1242/dmm.000331,其以全文引用的方式并入本文中)。剪接体识别内含子-外显子边界且通过两种引起两个相邻外显子接合的酯基转移反应去除介入内含子。这一反应的保真性必须是优良的,因为如果接合不当地进行,那么正常蛋白质编码潜力可能受损。举例来说,在外显子跳跃保持指定翻译期间氨基酸的一致性和顺序的三重峰密码子的阅读框架的情况下,交替剪接的mRNA可以指定不具有关键氨基酸残基的蛋白质。更通常地,外显子跳跃将破坏翻译阅读框架,产生未成熟的终止密码子。这些mRNA通常通过称为无意义介导的mRNA降解的过程降解达至少90%,由此降低这类缺陷性消息将积聚以产生截短的蛋白质产物的似然性。如果错误剪接的mRNA逃离这一路径,那么将产生截短、突变或不稳定的蛋白质。
替代性剪接是一种表达来自相同基因组DNA的若干或多种不同转录物的手段且由包含具体蛋白质的可用的外显子的子集引起。通过排除一个或多个外显子,某些蛋白质域可能损失被编码的蛋白质,其可以引起蛋白质功能损失或增加。已描述若干类型的替代性剪接:外显子跳跃;替代性5'或3'剪接位点;相互排斥外显子;和显著更罕见的,内含子留存。已使用生物信息学方法比较癌症和正常细胞中的替代性剪接的量且确定与正常细胞相比,癌症呈现低替代性剪接水平。此外,与正常细胞相比,癌症中的替代性剪接事件的类型的分布不同。与正常细胞相比,癌细胞显示更少的外显子跳跃,但更多的替代性5'和3'剪接位点选择以及内含子留存。当检验外显子化现象时(使用主要由其它组织用作内含子的序列作为外显子),与癌细胞中的外显子化相关联的基因优先与mRNA处理相关联,指示癌细胞与产生异常mRNA剪接形式之间的直接相关。
DNA或RNA含量的例示性变化
在一些实施例中,存在一种或多种类型的DNA(如cfDNA cf mDNA、cf nDNA、细胞DNA或线粒体DNA)或RNA(cfRNA、细胞RNA、细胞质RNA、编码细胞质RNA、非编码细胞质RNA、mRNA、miRNA、线粒体RNA、rRNA或tRNA)的总量或浓度的变化。在一些实施例中,存在一种或多种特异性DNA(如cfDNA cf mDNA、cf nDNA、细胞DNA或线粒体DNA)或RNA(cfRNA、细胞RNA、细胞质RNA、编码细胞质RNA、非编码细胞质RNA、mRNA、miRNA、线粒体RNA、rRNA或tRNA)分子的量或浓度的变化。在一些实施例中,一种等位基因的表达水平高于相关基因座的另一种等位基因。例示性miRNA是具有20-22个核苷酸的短RNA分子,其调节基因的表达。在一些实施例中,存在转录组的变化,如一种或多种RNA分子的一致性或量的变化。
在一些实施例中,cfDNA或cfRNA的总量或浓度的增加与疾病或病症(如癌症)或增加的疾病或病症(如癌症)风险相关联。在一些实施例中,一种类型的DNA(如cfDNA cfmDNA、cf nDNA、细胞DNA或线粒体DNA)或RNA(cfRNA、细胞RNA、细胞质RNA、编码细胞质RNA、非编码细胞质RNA、mRNA、miRNA、线粒体RNA、rRNA或tRNA)的总浓度与健康(如非癌性)个体中所述类型的DNA或RNA的总浓度相比增加至少2、3、4、5、6、7、8、9、10倍或更多倍。在一些实施例中,在75到100ng/mL、100到150ng/mL、150到200ng/mL、200到300ng/mL、300到400ng/mgL、400到600ng/mL、600到800ng/mL、800到1,000ng/mL之间且包括端值的cfDNA的总浓度或超过100ng/mL,如超过200、300、400、500、600、700、800、900或1,000ng/mL的cfDNA的总浓度指示癌症、增加的癌症风险、增加的恶性而非良性肿瘤风险、癌症缓解的可能性降低或癌症的较差预后。在一些实施例中,一种类型的具有一种或多种与疾病或病症(如癌症)或增加的疾病或病症(如癌症)风险相关联的多态现象/突变(如缺失或复制)的DNA(如cfDNA cfmDNA、cf nDNA、细胞DNA或线粒体DNA)或RNA(cfRNA、细胞RNA、细胞质RNA、编码细胞质RNA、非编码细胞质RNA、mRNA、miRNA、线粒体RNA、rRNA或tRNA)的量是所述类型的DNA或RNA的总量的至少2、3、4、5、6、7、8、9、10、11、12、14、16、18、20或25%。在一些实施例中,一种类型的DNA(如cfDNA cf mDNA、cf nDNA、细胞DNA或线粒体DNA)或RNA(cfRNA、细胞RNA、细胞质RNA、编码细胞质RNA、非编码细胞质RNA、mRNA、miRNA、线粒体RNA、rRNA或tRNA)的总量中的至少2、3、4、5、6、7、8、9、10、11、12、14、16、18、20或25%具有与疾病或病症(如癌症)或增加的疾病或病症(如癌症)风险相关联的具体多态现象或突变(如缺失或复制)。
在一些实施例中,cfDNA被包裹。在一些实施例中,cfDNA未被包裹。
在一些实施例中,确定全部DNA中的肿瘤DNA的分数(如全部cfDNA中的肿瘤cfDNA的分数或全部cfDNA中具有具体突变的肿瘤cfDNA的分数)。在一些实施例中,可以确定多种突变的肿瘤DNA的分数,其中突变可以是单核苷酸变异体、拷贝数目变异体、差异甲基化或其组合。在一些实施例中,将所计算的具有最高的所计算的肿瘤分数的一种突变或突变集合的平均肿瘤分数视为样品中的实际肿瘤分数。在一些实施例中,将所计算的所有突变的平均肿瘤分数视为样品中的实际肿瘤分数。在一些实施例中,使用这一肿瘤分数对癌症进行分期(因为较高的肿瘤分数与癌症的更晚期阶段相关联)。在一些实施例中,使用肿瘤分数确定癌症的尺寸,因为较大的肿瘤可能与血浆中肿瘤DNA的分数相关。在一些实施例中,使用肿瘤分数确定具有单一或多种突变的肿瘤的比例的尺寸,因为血浆样品中所测量的肿瘤分数与具有既定突变基因型的组织的尺寸之间可能存在相关性。举例来说,具有既定突变基因型的组织的尺寸可能与可以通过关注所述具体突变来计算的肿瘤DNA的分数相关。
例示性数据库
本发明还提供含有一种或多种来自本发明的方法的结果的数据库。举例来说,数据库可以包括具有一个或多个个体的任何以下信息的记录:所鉴别的任何多态现象/突变(如CNV);多态现象/突变与疾病或病症或增加的疾病或病症风险的任何已知的关联性;多态现象/突变对被编码的mRNA或蛋白质的表达或活性水平的作用;样品中的全部DNA、RNA或细胞中的与疾病或病症相关联的DNA、RNA或细胞(如具有与疾病或病症相关联的多态现象/突变的DNA、RNA或细胞)的分数;用于鉴别多态现象/突变的样品的来源(如血液样品或来自具体组织的样品);病变细胞的数目;来自后续重复检验(如重复用于监测疾病或病症的进展或缓解的检验)的结果;其它疾病或病症检验的结果;个体被诊断患有的疾病或病症的类型;所给予的治疗;对这类治疗的反应;这类治疗的副作用;症状(如与疾病或病症相关联的症状);缓解的长度和数目;存活期的长度(如从初始检验直到死亡的时间长度或从诊断直到死亡的时间长度);死亡原因;以及其组合。
在一些实施例中,数据库包括具有一个或多个个体的任何以下信息的记录:所鉴别的任何多态现象/突变;多态现象/突变与癌症或增加的癌症风险的任何已知的关联性;多态现象/突变对被编码的mRNA或蛋白质的表达或活性水平的作用;样品中的全部DNA、RNA或细胞中的癌性DNA、RNA或细胞的分数;用于鉴别多态现象/突变的样品的来源(如血液样品或来自具体组织的样品);癌性细胞的数目;肿瘤尺寸;来自后续重复检验(如重复用于监测癌症的进展或缓解的检验)的结果;其它癌症检验的结果;个体被诊断患有的癌症的类型;所给予的治疗;对这类治疗的反应;这类治疗的副作用;症状(如与癌症相关联的症状);缓解的长度和数目;存活期的长度(如从初始检验直到死亡的时间长度或从诊断直到死亡的时间长度);死亡原因;以及其组合。在一些实施例中,对治疗的反应包括以下中的任一种:肿瘤(例如良性或癌性肿瘤)的尺寸减小或稳定;减缓或防止肿瘤尺寸增加;肿瘤细胞数目减少或稳定;延长肿瘤消失与其再现之间的无疾病存活时间;防止肿瘤的初始或后续发生;与肿瘤相关联的不良症状减少或稳定;或其组合。在一些实施例中,包括来自疾病或病症(如癌症)的一种或多种其它检验的结果,如来自组织样品的筛检检验、医学成像或微观检查的结果。
在一个这类方面中,本发明提供包括至少5、10、10
在另一方面中,本发明提供包括本发明的数据库和用户界面的计算机。在一些实施例中,用户界面能够呈现一条或多条记录中所含的一部分或所有信息。在一些实施例中,用户界面能够显示(i)已鉴别为含有多态现象或突变的一种或多种类型的癌症,其记录储存于计算机中,(ii)已在具体类型的癌症中鉴别的一种或多种多态现象或突变,其记录储存于计算机中,(iii)具体类型的癌症或具体多态现象或突变的预后信息,其记录储存于计算机中,(iv)适用于具有多态现象或突变的癌症的一种或多种化合物或其它治疗,其记录储存于计算机中,(v)一种或多种调节mRNA或蛋白质的表达或活性的化合物,其记录储存于计算机中,和(vi)一种或多种mRNA分子或蛋白质,其表达或活性由化合物调节,所述一种或多种mRNA分子或蛋白质的记录储存于计算机中。计算机的内部组件通常包括与存储器耦合的处理器。外部组分通常包括大容量存储装置,例如硬盘驱动器;用户输入装置,例如键盘和鼠标;显示器,例如监测器;和任选地,能够使计算机系统与其它计算机连接以实现数据和处理任务的共享的网络连接。可以在操作期间将程序加载到这一系统的存储器中。
在另一方面中,本发明提供一种计算机实施方法,其包括本发明的任何方法的一个或多个步骤。
例示性风险因子
在一些实施例中,还评估个体的疾病或病症(如癌症)的一种或多种风险因子。例示性风险因子包括疾病或病症的家族病史、生活方式(如吸烟和暴露于致癌物)和一种或多种激素或血清蛋白的含量(如肝癌中的α-胎蛋白(AFP)、结肠直肠癌中的癌胚抗原(CEA)或前列腺癌中的前列腺特异性抗原(PSA))。在一些实施例中,测量肿瘤的尺寸和/或数目且用于确定个体的预后或选择用于个体的治疗。
例示性筛检方法
视需要,可以确认存在或不存在疾病或病症,如癌症,或可以使用任何标准方法将疾病或病症(如癌症)分类。举例来说,可以按多种方式检测疾病或病症,如癌症,包括存在某些迹象和症状、肿瘤活检、筛检检验或医学成像(如乳房X光检查或超声波)。在检测到可能的癌症之后,可以通过组织样品的微观检查来进行诊断。在一些实施例中,被诊断的个体在多个时间点时经历使用本发明的方法进行的重复序列检验或已知的疾病或病症检验,以监测疾病或病症的进展或疾病或病症的缓解或复发。
例示性癌症
可以使用本发明的任何方法诊断、预后、稳定、治疗、预防、预测或监测治疗反应的例示性癌症包括实体肿瘤、癌瘤、肉瘤、淋巴瘤、白血病、生殖细胞肿瘤或母细胞瘤。在各种实施例中,癌症是急性淋巴母细胞性白血病、急性骨髓性白血病、肾上腺皮质癌、AIDS相关癌症、AIDS相关淋巴瘤、肛门癌、阑尾癌、星形细胞瘤(如儿童小脑或大脑星形细胞瘤)、基础细胞癌瘤、胆管癌(如肝外胆管癌)、膀胱癌、骨骼肿瘤(如骨肉瘤或恶性纤维组织细胞瘤)、脑干神经胶质瘤、脑癌(如小脑星形细胞瘤、大脑星形细胞瘤/恶性神经胶质瘤、室管膜瘤、神经管胚细胞瘤、幕上原始神经外胚层肿瘤或视路和下丘脑神经胶质瘤)、神经胶母细胞瘤、乳癌、支气管腺瘤或类癌、伯基特氏淋巴瘤、类癌肿瘤(如儿童或胃肠道类癌肿瘤)、癌瘤中枢神经系统淋巴瘤、小脑星形细胞瘤或恶性神经胶质瘤(如儿童小脑星形细胞瘤或恶性神经胶质瘤)、子宫颈癌、儿童癌症、慢性淋巴细胞性白血病、慢性骨髓性白血病、慢性骨髓增生性病症、结肠癌、皮肤T细胞淋巴瘤、促结缔组织增生性小圆细胞肿瘤、子宫内膜癌、室管膜瘤、食道癌、尤文氏肉瘤、尤文氏肿瘤家族中的肿瘤、颅外生殖细胞肿瘤(如儿童颅外生殖细胞肿瘤)、性腺外生殖细胞肿瘤、眼癌(如眼内黑素瘤或成视网膜细胞瘤眼癌)、胆囊癌、胃癌、胃肠道类癌肿瘤、胃肠道基质瘤、生殖细胞肿瘤(如颅外、性腺外或卵巢生殖细胞肿瘤)、妊娠期滋养细胞肿瘤、神经胶质瘤(如脑干、儿童大脑星形细胞瘤或儿童视路和下丘脑神经胶质瘤)、胃类癌、毛状细胞白血病、头颈癌、心脏癌症、肝细胞(肝脏)癌症、霍奇金氏淋巴瘤、下咽癌症、下丘脑和视通路神经胶质瘤(如儿童视通路神经胶质瘤)、胰岛细胞癌瘤(如内分泌或胰岛细胞癌瘤)、卡波西肉瘤、肾脏癌、喉癌、白血病(如急性成淋巴细胞性、急性骨髓、慢性淋巴细胞性、慢性骨髓性或毛状细胞白血病)、嘴唇或口腔癌症、脂肉瘤、肝癌(如非小细胞或小细胞癌症)、肺癌、淋巴瘤(如AIDS相关、伯基特氏、皮肤T细胞、霍奇金氏、非霍奇金氏或中枢神经系统淋巴瘤)、巨球蛋白血症(如瓦尔登斯特伦巨球蛋白血症、骨骼恶性纤维组织细胞瘤或骨肉瘤、神经管胚细胞瘤(如儿童神经管胚细胞瘤)、黑素瘤、梅克尔细胞癌、间皮瘤(如成年人或儿童间皮瘤)、隐性转移性鳞状颈部癌症、口腔癌症、多发性内分泌腺瘤综合症(如儿童多发性内分泌腺瘤综合症)、多发性骨髓瘤或血浆细胞赘瘤、蕈样真菌病、骨髓发育不良综合症、骨髓发育不良或骨髓增生性疾病、骨髓性白血病(如慢性骨髓性白血病)、骨髓性白血病(如成年人急性或儿童急性骨髓性白血病)、骨髓增生性病症(如慢性骨髓增生性病症)、鼻腔或副鼻窦癌症、鼻咽癌、神经母细胞瘤、口部癌症、口咽癌症、骨肉瘤或骨骼恶性纤维组织细胞瘤、卵巢癌、卵巢上皮癌症、卵巢生殖细胞肿瘤、卵巢低恶性潜在肿瘤、胰腺癌(如胰岛细胞胰腺癌)、副鼻窦或鼻腔癌症、副甲状腺癌症、阴茎癌、咽癌、嗜铬细胞瘤、松果体星形细胞瘤、松果体胚细胞瘤、成松果体细胞瘤或幕上原始神经外胚层肿瘤(如儿童成松果体细胞瘤或幕上原始神经外胚层肿瘤)、垂体腺瘤、浆细胞瘤形成、胸膜肺母细胞瘤、原发性中枢神经系统淋巴瘤、癌症、直肠癌、肾细胞癌、肾盂或输尿管癌症(如肾盂或输尿管移行细胞癌症、成视网膜细胞瘤、横纹肌肉瘤(如儿童横纹肌肉瘤)、唾液腺癌症、肉瘤(如尤文氏肿瘤家族中的肉瘤、卡堡氏、软组织或子宫肉瘤)、塞氏综合症、皮肤癌(如非黑素瘤、黑素瘤或默克氏细胞皮肤癌(merkel cell skin cancer)、小肠癌、鳞状细胞癌、幕上原始神经外胚层肿瘤(如儿童幕上原始神经外胚层肿瘤)、T细胞淋巴瘤(如皮肤T细胞淋巴瘤)、睾丸癌、喉癌、胸腺瘤(如儿童胸腺瘤)、胸腺瘤或胸腺癌、甲状腺癌(如儿童甲状腺癌)、滋养细胞肿瘤(如妊娠期滋养细胞肿瘤)、原发部位未知的癌瘤(如成年人或儿童原发部位未知的癌瘤)、尿道癌症(如子宫内膜子宫癌)、子宫肉瘤、阴道癌、视路或下丘脑神经胶质瘤(如儿童视路或下丘脑神经胶质瘤)、外阴癌、瓦尔登斯特伦巨球蛋白血症或威尔姆斯氏肿瘤(wilms tumor)(如儿童威尔姆斯氏肿瘤)。在各种实施例中,癌症已转移或尚未转移。
癌症可以是或可以不是激素相关或依赖性癌症(例如雌激素或雄激素相关癌症)。可以使用本发明的方法和/或组合物诊断、预后、稳定、治疗或预防良性肿瘤或恶性肿瘤。
在一些实施例中,个体患有癌症综合症。癌症综合症是一种基因病症,其中一种或多种基因中的基因突变使得患病个体易于发生癌症且也可以引起这些癌症的早发。癌症综合症通常不仅展示生存期内产生癌症的高风险,而且还产生多种独立的原发性肿瘤。许多这些综合症是由肿瘤抑制基因的突变引起,所述肿瘤抑制基因是涉及保护细胞避免变成癌性的基因。其它可能受影响的基因是DNA修复基因、致癌基因和涉及血管产生(血管生成)的基因。遗传性癌症综合症的常见实例是遗传性乳房-卵巢癌综合症和遗传性非息肉病结肠癌(林奇氏综合症(Lynch syndrome))。
在一些实施例中,分别向在K-ras、p53、BRA、EGFR或HER2中具有一种或多种多态现象或突变的个体给予靶向K-ras、p53、BRA、EGFR或HER2的治疗。
本发明的方法通常可以用于治疗任何细胞、组织或器官类型的恶性或良性肿瘤。
例示性治疗
视需要,可以向个体(例如使用本发明的任何方法鉴别为患有癌症或增加的癌症风险的个体)给予任何用于稳定、治疗或预防疾病或病症(如癌症)或增加的疾病或病症(如癌症)风险的治疗。在各种实施例中,治疗是已知的用于疾病或病症(如癌症)的治疗或治疗组合,包括(但不限于)细胞毒性剂、靶向疗法、免疫疗法、激素疗法、放射疗法、癌性细胞或可能变成癌性的细胞的手术去除、干细胞移植、骨髓移植、光动力疗法、姑息性治疗或其组合。在一些实施例中,使用治疗(如预防性药物)来预防、延缓具有增加的疾病或病症(如癌症)风险的个体中的疾病或病症(如癌症)或降低其严重程度。在一些实施例中,治疗是手术、一线化学疗法、辅助疗法或新辅助疗法。
在一些实施例中,靶向疗法是靶向癌症的特异性基因、蛋白质或有助于癌症生长和存活的组织环境的治疗。这种类型的治疗阻断癌细胞的生长和扩散,同时限制对正常细胞的损伤,与其它癌症药物相比通常引起较少的副作用。
一种较成功的方法靶向血管生成,肿瘤周围的新血管生长。靶向疗法(如贝伐珠单抗(bevacizumab)(阿瓦斯汀(Avastin))、来那度胺(lenalidomide)(雷利米得(Revlimid))、索拉非尼(sorafenib)(多吉美(Nexavar))、舒尼替尼(sunitinib)(舒癌特(Sutent))和沙力度胺(thalidomide)(撒利多迈(Thalomid)))干扰血管生成。另一实例是针对过表达HER2的癌症(如一些乳癌)使用靶向HER2的治疗,如曲妥珠单抗(trastuzumab)或拉帕替尼(lapatinib)。在一些实施例中,使用单克隆抗体阻断癌细胞外部上的特异性目标。实例包括阿仑单抗(alemtuzumab)(坎帕斯-1H(Campath-1H))、贝伐珠单抗、西妥昔单抗(cetuximab)(爱必妥(Erbitux))、帕尼单抗(panitumumab)(维克替比(Vectibix))、帕妥珠单抗(pertuzumab)(奥密塔克(Omnitarg))、利妥昔单抗(rituximab)(美罗华(Rituxan))和曲妥珠单抗。在一些实施例中,使用单克隆抗体托西莫单抗(tositumomab)(百克沙(Bexxar))向肿瘤递送辐射。在一些实施例中,口服小分子抑制癌细胞内部的癌症过程。实例包括达沙替尼(dasatinib)(斯普塞尔(Sprycel))、埃罗替尼(erlotinib)(特罗凯(Tarceva))、吉非替尼(gefitinib)(易瑞沙(Iressa))、伊马替尼(imatinib)(格列卫(Gleevec))、拉帕替尼(lapatinib)(泰克泊(泰克泊))、尼罗替尼(nilotinib)(塔西纳(Tasigna))、索拉非尼、舒尼替尼和坦罗莫司(temsirolimus)(托瑞斯(Torisel))。在一些实施例中,蛋白酶体抑制剂(如多发性骨髓瘤药物,硼替佐米(bortezomib)(万珂(Velcade)))干扰称为分解细胞中的其它蛋白质的酶的特殊蛋白质。
在一些实施例中,免疫疗法被设计成增强身体的天然抵抗力以对抗癌症。例示性类型的免疫疗法使用由身体或在实验室中产生的物质以支持、靶向或恢复免疫系统功能。
在一些实施例中,激素疗法通过降低身体中激素的量来治疗癌症。若干种类型的癌症(包括一些乳房和前列腺癌症)仅在身体中存在称为激素的天然化学物质的情况下生长和扩散。在各种实施例中,使用激素疗法治疗前列腺、乳房、甲状腺和生殖系统的癌症。
在一些实施例中,治疗包括干细胞移植,其中用称为造血干细胞的高度特化的细胞替换病变的骨髓。在血流和骨髓中发现造血干细胞。
在一些实施例中,治疗包括光动力疗法,其使用称为光敏剂的特殊药物和光来杀伤癌细胞。药物在其由某些类别的光活化之后起作用。
在一些实施例中,治疗包括癌性细胞或可能变成癌性的细胞的手术去除(如肿块切除术或乳房切除术)。举例来说,具有乳癌敏感性基因突变(BRCA1或BRCA2基因突变)的女性可以通过用于降低风险的输卵管-卵巢切除(去除输卵管和卵巢)和/或用于降低风险的双侧乳房切除术(去除两个乳房)来降低乳癌和卵巢癌的风险。可以使用激光(功率极强、极密集的光束)代替刀片(解剖刀)进行极谨慎的手术工作,包括治疗一些癌症。
除用于延缓、停止或消除癌症的治疗(也称为疾病定向治疗)以外,癌症护理的一个重要部分是缓解个体的症状和副作用,如疼痛和恶心。其包括支持个体的生理、情感和社交需求,一种称为姑息性或支持性护理的方法。人们通常同时接受疾病定向疗法和治疗以减轻症状。
例示性治疗包括放射菌素D(actinomycin D)、阿德曲斯(adcetris)、阿德力霉素(Adriamycin)、阿地介白素(aldesleukin)、阿仑单抗(alemtuzumab)、力比泰(alimta)、阿米西丁(amsidine)、安吖啶(amsacrine)、阿那曲唑(anastrozole)、阿可达(aredia)、阿纳托唑(arimidex)、阿诺新(aromasin)、天冬酰胺酶(asparaginase)、阿瓦斯汀(avastin)、贝伐珠单抗、比卡鲁胺(bicalutamide)、博莱霉素(bleomycin)、博德纳特(bondronat)、博尼弗斯(bonefos)、硼替佐米(bortezomib)、布西韦克(busilvex)、白消安(busulphan)、坎普托(campto)、卡培他滨(capecitabine)、卡铂(carboplatin)、卡莫司汀(carmustine)、康士得(casodex)、西妥昔单抗(cetuximab)、赤克斯(chimax)、苯丁酸氮芥(chlorambucil)、甲腈咪胍(cimetidine)、顺铂(cisplatin)、克拉屈滨(cladribine)、氯屈膦酸盐(clodronate)、氯法拉滨(clofarabine)、克立他酶(crisantaspase)、环磷酰胺(cyclophosphamide)、乙酸环丙孕酮(cyproterone acetate)、西普塔特(cyprostat)、阿糖胞苷(cytarabine)、环磷氮介(cytoxan)、达卡波秦(dacarbozine)、放线菌素D(dactinomycin)、达沙替尼(dasatinib)、道诺霉素(daunorubicin)、地塞米松(dexamethasone)、己烯雌酚(diethylstilbestrol)、多烯紫杉醇(docetaxel)、小红莓(doxorubicin)、多格尼尔(drogenil)、恩克依特(emcyt)、表柔比星(epirubicin)、艾普欣(eposin)、爱必妥(Erbitux)、埃罗替尼(erlotinib)、艾斯塔特(estracyte)、雌氮芥(estramustine)、艾托普斯(etopophos)、依托泊苷(etoposide)、艾弗特拉(evoltra)、依西美坦(exemestane)、法乐通(fareston)、富马乐(femara)、非格司亭(filgrastim)、氟达拉(fludara)、氟达拉滨(fludarabine)、氟尿嘧啶(fluorouracil)、氟他胺(flutamide)、格非尼布(gefinitib)、吉西他滨(gemcitabine)、健择(gemzar)、格列卫(gleevec)、格力卫(glivec)、长效格纳普特(gonapeptyl depot)、戈舍瑞林(goserelin)、哈拉维(halaven)、赫赛汀(herceptin)、赫卡汀(hycamptin)、羟基尿素(hydroxycarbamide)、伊班膦酸(ibandronic acid)、异贝莫单抗(ibritumomab)、伊达比星(idarubicin)、伊弗米德(ifosfomide)、干扰素、甲磺酸伊马替尼(imatinib mesylate)、易瑞沙(iressa)、伊立替康(irinotecan)、结塔纳(jevtana)、兰卫斯(lanvis)、拉帕替尼(lapatinib)、来曲唑(letrozole)、瘤可宁(leukeran)、亮丙瑞林(leuprorelin)、乐斯塔特(leustat)、洛莫司汀(lomustine)、玛卡斯(mabcampath)、玛瑟拉(mabthera)、美加西(megace)、甲地孕酮(megestrol)、甲胺喋呤(methotrexate)、米托蒽醌(mitozantrone)、丝裂霉素、木土兰(mutulane)、马利兰(myleran)、诺维本(navelbine)、尼拉斯塔(neulasta)、雷普根(neupogen)、多吉美(nexavar)、尼彭特(nipent)、诺瓦得士D(nolvadex D)、诺凡隆(novantron)、安可平(oncovin)、太平洋紫杉醇、帕米膦酸盐(pamidronate)、PCV、培美曲塞(pemetrexed)、喷司他汀(pentostatin)、帕杰它(perjeta)、丙卡巴肼(procarbazine)、普洛韦格(provenge)、泼尼松龙(prednisolone)、普洛斯普(prostrap)、雷替曲赛(raltitrexed)、利妥昔单抗(rituximab)、斯普塞尔(sprycel)、索拉非尼(sorafenib)、索塔莫西(soltamox)、链脲霉素(streptozocin)、己烯雌酚(stilboestrol)、斯迪木西(stimuvax)、舒尼替尼(sunitinib)、舒癌特(sutent)、他布伊德(tabloid)、他加米特(tagamet)、他莫芬(tamofen)、他莫昔芬(tamoxifen)、特罗凯(tarceva)、紫杉醇(taxol)、克癌易(taxotere)、喃氟啶(tegafur)和尿嘧啶、特莫达尔(temodal)、替莫唑胺(temozolomide)、沙力度胺(thalidomide)、噻利斯(thioplex)、噻替派(thiotepa)、硫鸟嘌呤(tioguanine)、拓优得(tomudex)、拓朴替康(topotecan)、托瑞米芬(toremifene)、曲妥珠单抗(trastuzumab)、维甲酸(tretinoin)、曲奥舒凡(treosulfan)、三亚乙基硫磷酰胺(triethylenethiophorsphoramide)、曲普瑞林(triptorelin)、特韦博(tyverb)、优弗拉尔(uftoral)、万珂(velcade)、维派德(vepesid)、凡善能(vesanoid)、长春新碱(vincristine)、长春瑞滨(vinorelbine)、夏克瑞(xalkori)、希罗达(xeloda)、益伏(yervoy)、扎克替玛(zactima)、扎诺沙(zanosar)、善唯达(zavedos)、泽韦林(zevelin)、诺雷德(zoladex)、唑来膦酸盐(zoledronate)、唑米他唑来膦酸(zometa zoledronic acid)和泽替加(zytiga)。
在一些实施例中,癌症是乳癌且给予个体的治疗或化合物是以下中的一个或多个:阿贝西利(Abemaciclib)、阿布拉生(Abraxane)(太平洋紫杉醇白蛋白稳定化纳米粒子配制物)、阿多-曲妥珠单抗恩他新(Ado-Trastuzumab Emtansine)、阿飞尼妥(Afinitor)(依维莫司(Everolimus))、阿那曲唑(Anastrozole)、阿可达(Aredia)(帕米膦酸二钠)、阿纳托唑(Arimidex)(阿那曲唑(Anastrozole))、阿诺新(Aromasin)(依西美坦(Exemestane))、卡培他滨(Capecitabine)、环磷酰胺、多烯紫杉醇、盐酸小红莓、艾伦斯(Ellence)(盐酸表柔比星(Epirubicin Hydrochloride))、盐酸表柔比星、甲磺酸艾日布林(Eribulin Mesylate)、依维莫司(依维莫司)、依西美坦(Exemestane)、5-FU(氟尿嘧啶注射剂)、法乐通(Fareston)(托瑞米芬(Toremifene))、芙仕得(Faslodex)(氟维司群(Fulvestrant))、富马乐(Femara)(来曲唑(Letrozole))、氟尿嘧啶注射剂、氟维司群(Fulvestrant)、盐酸吉西他滨、健择(盐酸吉西他滨)、乙酸戈舍瑞林(GoserelinAcetate)、哈拉维(Halaven)(甲磺酸艾日布林(Eribulin Mesylate)、赫赛汀(曲妥珠单抗)、伊布兰西(Ibrance)(帕博西里(Palbociclib))、伊沙匹隆(Ixabepilone)、艾克斯普拉(Ixempra)(伊沙匹隆(Ixabepilone))、卡德克拉(Kadcyla)(阿多-曲妥珠单抗恩他新)、克斯卡利(Kisqali)(瑞博西林(Ribociclib))、二甲苯磺酸拉帕替尼(Lapatinib Ditosylate)、来曲唑(Letrozole)、林帕拉扎(Lynparza)(奥拉帕尼(Olaparib))、乙酸甲地孕酮(MegestrolAcetate)、甲胺喋呤、顺丁烯二酸来那替尼(Neratinib Maleate)、尼尔克斯(Nerlynx)(顺丁烯二酸来那替尼)、奥拉帕尼(Olaparib)、太平洋紫杉醇、太平洋紫杉醇白蛋白稳定化纳米粒子配制物、帕博西里(Palbociclib)、帕米膦酸二钠、帕杰它(Perjeta)(帕妥珠单抗(Pertuzumab))、帕妥珠单抗、瑞博西林(Ribociclib)、柠檬酸他莫昔芬(TamoxifenCitrate)、紫杉醇(太平洋紫杉醇)、克癌易(多烯紫杉醇)、噻替派、托瑞米芬、曲妥珠单抗、特瑞夏尔(Trexall)(甲胺喋呤)、泰克泊(Tykerb)(二甲苯磺酸拉帕替尼(LapatinibDitosylate))、维泽尼奥(Verzenio)(阿贝西利(Abemaciclib))、硫酸长春碱、希罗达(Xeloda)(卡培他滨(Capecitabine))、诺雷德(Zoladex)(乙酸戈舍瑞林)、梯瓦(Evista)(盐酸雷诺昔酚(Raloxifene Hydrochloride))、盐酸雷诺昔酚、柠檬酸他莫昔芬(Tamoxifen Citrate)。在一些实施例中,癌症是乳癌且给予个体的治疗或化合物是选自以下的组合:盐酸小红莓(阿德力霉素)和环磷酰胺;盐酸小红莓(阿德力霉素)、环磷酰胺和太平洋紫杉醇(紫杉醇);盐酸小红莓(阿德力霉素)、环磷酰胺和氟尿嘧啶;甲胺喋呤、环磷酰胺和氟尿嘧啶;盐酸表柔比星、环磷酰胺和氟尿嘧啶;以及盐酸小红莓(阿德力霉素)、环磷酰胺和多烯紫杉醇(克癌易)。
对于表达mRNA或蛋白质的突变体形式(例如癌症相关形式)和野生型形式(例如与癌症不相关的形式)的个体,疗法对突变体形式的表达或活性的抑制优选是其对野生型形式的表达或活性的抑制的至少2、5、10或20倍。多种治疗剂的同时或依序使用可以显著降低癌症的发病率和降低变得对疗法具有抗性的所治疗的癌症的数目。此外,用作组合疗法的一部分的治疗剂与在所述治疗剂单独地使用时所需的相应剂量相比,可能需要较低的剂量便可治疗癌症。组合疗法中的每种化合物的低剂量降低由化合物引起的潜在不良副作用的严重程度。
在一些实施例中,由本发明或任何标准方法鉴别为具有增加的癌症风险的个体避免特定风险因子或改变生活方式以降低任何其它癌症风险。
在一些实施例中,使用多态现象、突变、风险因子或其任何组合来选择用于个体的治疗方案。在一些实施例中,选择较大的剂量或较大数目的治疗用于具有较大癌症风险或具有较差预后的个体。
其它用于包含在单独或组合疗法中的化合物
视需要,可以根据所属领域中已知的方法,从大型的天然产物或合成(或半合成)提取物的库或化学库鉴别其它用于稳定、治疗或预防疾病或病症(如癌症)或增加的疾病或病症(如癌症)风险的化合物。所属领域或熟悉药物研究和研发的技术人员将理解,检验提取物或化合物的确切来源对于本发明的方法来说不重要。因此,可以筛检几乎任何数目的化学提取物或化合物对来自具体类型的癌症或具体个体的细胞的作用,或筛检其对癌症相关分子(如已知在具体类型的癌症中具有改变的活性或表达的癌症相关分子)的活性或表达的作用。当发现粗提取物调节癌症相关分子的活性或表达时,可以使用所属领域中已知的方法进行阳性先导提取物的进一步分级分离以分离引起所观察的作用的化学成分。
用于检验疗法的例示性分析法和动物模型
视需要,可以使用细胞系(如具有使用本发明的方法在已诊断患有癌症或增加的癌症风险的个体中鉴别的突变中的一种或多种的细胞系)或疾病或病症的动物模型(如SCID小鼠模型)来检验本文中所公开的治疗中的一种或多种对疾病或病症(如癌症)的作用(Jain等人,《癌症研究中的肿瘤模型(Tumor Models In Cancer Research)》,Teicher编,Humana Press Inc.,Totowa,N.J.,第647-671页,2001,其以全文引用的方式并入本文中)。此外,存在大量可以用于确定具体疗法在稳定、治疗或预防疾病或病症(如癌症)或增加的疾病或病症(如癌症)风险方面的功效的标准分析法和动物模型。还可以在标准人类临床试验中检验疗法。
对于选择用于具体个体的优选疗法,可以检验化合物对个体中突变的一种或多种基因的表达或活性的作用。举例来说,可以使用标准RNA(Northern)、蛋白质(Western)或微阵列分析法来检测化合物调节具体mRNA分子或蛋白质的表达的能力。在一些实施例中,选择满足以下条件的一种或多种化合物:(i)抑制个体中(如来自个体的样品中)以高于正常水平表达的或具有高于正常活性水平的促进癌症的mRNA分子或蛋白质的表达或活性,或(ii)促进个体中以低于正常水平表达的或具有低于正常活性水平的抑制癌症的mRNA分子或蛋白质的表达或活性。满足以下条件的单独或组合疗法:(i)调节个体中最大数目的具有与癌症相关联的突变的mRNA分子或蛋白质,和(ii)调节个体中最少数目的不具有与癌症相关联的突变的mRNA分子或蛋白质。在一些实施例中,所选择的单独或组合疗法具有高药物功效且产生极少(如果存在)的不良副作用。
作为上文所描述的个体特异性分析的替代方案,DNA芯片可以用于比较具体类型的早期或晚期癌症(例如乳癌细胞)中mRNA分子的表达与正常组织中的表达(Marrack等人,《当前免疫学观点(Current Opinion in Immunology)》12,206-209,2000;Harkin,《肿瘤学(Oncologist.)》5:501-507,2000;Pelizzari等人,《核酸研究》28(22):4577-4581,2000,其各自以全文引用的方式并入本文中)。基于这一分析,可以选择用于患有这种类型的癌症的个体的单独或组合疗法以调节在这种类型的癌症中具有改变的表达的mRNA或蛋白质的表达。
除用于选择用于具体个体或个体组的疗法以外,表达谱可以用于监测在治疗期间发生的mRNA和/或蛋白质表达的变化。举例来说,表达谱可以用于确定癌症相关基因的表达是否恢复正常水平。如果未恢复正常水平,那么可以改变疗法中的一种或多种化合物的剂量以增加或降低疗法对相应的癌症相关基因的表达水平的作用。此外,这一分析可以用于确定疗法是否影响其它基因(例如与不良副作用相关联的基因)的表达。视需要,可以改变疗法的剂量或组成以防止或减少不合需要的副作用。
例示性配制物和给药方法
为了稳定、治疗或预防疾病或病症(如癌症)或增加的疾病或病症(如癌症)风险,可以使用所属领域的技术人员已知的任何方法配制和给予组合物(参见例如美国专利案第8,389,578号和第8,389,557号,其各自以全文引用的方式并入本文中)。用于配制和给药的一般技术见于《雷明顿:医药科学和实践(Remington:The Science and Practice ofPharmacy)》,第21版,David Troy编,2006,Lippincott Williams&Wilkins,Philadelphia,Pa.,其以全文引用的方式并入本文中。液体、浆料、片剂、胶囊、丸剂、粉末、颗粒、凝胶、软膏、栓剂、注射剂、吸入剂和气溶胶是这类配制物的实例。作为实例,可以使用所属领域中已知的其它方法制备被改性的或延长释放型口服配制物。举例来说,活性成分的合适的延长释放形式可以是骨架片或胶囊组合物。合适的骨架形成材料包括例如蜡(例如棕榈蜡、蜂蜡、石蜡、地蜡、虫胶蜡、脂肪酸和脂肪醇)、油、硬化油或脂肪(例如硬化菜籽油、蓖麻油、牛脂、棕榈油和大豆油)以及聚合物(例如羟基丙基纤维素、聚乙烯吡咯烷酮、羟基丙基甲基纤维素和聚乙二醇)。其它合适的骨架片材料是微晶纤维素、粉末纤维素、羟基丙基纤维素、乙基纤维素以及其它载剂和填充剂。片剂还可以含有颗粒、包衣粉末或丸粒。片剂还可以是多层的。任选地,成品片剂可以包覆包衣或未包覆包衣。
给予这类组合物的典型途径包括(但不限于)口服、舌下、颊内、局部、经皮、吸气、非经肠(例如皮下、静脉内、肌肉内、胸骨内注射或输注技术)、经直肠、经阴道和鼻内。在优选实施例中,使用延长释放型装置给予疗法。配制本发明的组合物以便使其中所含的活性成分在给予组合物时是生物可用的。组合物可以呈一种或多种剂量单位形式。组合物可以含有1、2、3、4或更多种活性成分且可以任选地含有1、2、3、4或更多种非活性成分。
替代性实施例
本文中所描述的任何方法可以包括呈实体格式的数据输出,如在计算机屏幕上或在打印纸上。本发明的任何方法可以与呈可以由医师使用的格式的可操作数据的输出组合。医学专业人员可以将文献中所描述的一些用于确定关于目标个体的基因数据的实施例与潜在染色体异常(如缺失或复制)或不具有潜在染色体异常的通知组合。本文中所描述的一些实施例可以与可操作数据的输出,以及产生临床治疗的临床决定的执行或不采取行动的临床决定的执行组合。
在一些实施例中,本文中公开用于产生公开本发明的任何方法的结果(如存在或不存在缺失或复制)的报告的方法。可以产生具有本发明的方法的结果的报告且其可以电子方式发送给医师、在输出装置上显示(如数字报告)或可以向医师递送书面报告(如报告的打印复印件)。此外,所描述的方法可以与产生临床治疗的临床决定的实际执行或不采取行动的临床决定的执行组合。
在某些实施例中,本发明提供用于进行这类方法、使用本文中所公开的多重PCR方法检测来自相同样品的CNV和SNV的试剂、试剂盒和方法以及计算机系统和具有编码指令的计算机介质。在某些优选实施例中,样品是怀疑含有循环肿瘤DNA的单细胞样品或血浆样品。这些实施例利用以下研究结果:与单独查询CNV或SNV相比,通过使用本文中所公开的高敏感性多重PCR方法查询来自单细胞或血浆的DNA样品中的CNV和SNV,可以获得改善的癌症检测,尤其对于呈现CNV的癌症,如乳癌、卵巢癌和肺癌。在某些说明性实施例中,用于分析CNV的方法查询50到100,000个或50到10,000个,或50到1,000个SNP,且对于SNV,查询50到1000个SNV或50到500个SNV或50到250个SNV。本文中所提供的用于检测怀疑患有癌症(包括例如已知呈现CNV和SNV的癌症,如乳癌、肺癌和卵巢癌)的个体的血浆中的CNV和/或SNV的方法提供以下优点:检测来自在基因组成方面通常由异源癌细胞群体构成的肿瘤的CNV和/或SNV。因此,集中于仅分析肿瘤的某些区域的传统方法通常会遗漏存在于肿瘤的其它区域中的细胞中的CNV或SNV。可以查询充当液体活检的血浆样品以检测仅存在于肿瘤细胞的亚群中的任何CNV和/或SNV。
提出以下实例以便提供所属领域的一般技术人员如何使用本文中所提供的实施例的完整公开内容和描述,并且并不打算限制本公开的范围,其也不打算表示以下实例是所进行的所有或仅有实验。已经致力于确保关于所用数量(例如量、温度等)的准确性,但是应该考虑一些实验误差和偏差。除非另外规定,否则份数都是体积份,并且温度用摄氏度表示。应理解,可以在不改变实例意图说明的基本方面的情况下,对所描述的方法进行改变。
实例
实例1.用于监测结肠直肠癌的个人化循环肿瘤DNA分析
已证实疾病复发的早期检测可以改善结肠直肠癌(CRC)患者的存活率。在手术后检测到循环肿瘤DNA(ctDNA)定义具有极高复发风险的CRC患者的子集。先前研究已使用小型基因集合测序或数字微滴式PCR进行ctDNA分析以监测早期CRC中的肿瘤负荷。
本实例的目的是对每名患者使用靶向16种肿瘤特异性突变的个人化多重PCR NGS平台,以评估手术后微小残留病和监测CRC中的治疗反应。
包括130名用治愈性手术和(任选的)辅助化学疗法治疗的I-IV期CRC患者(参见表1)。在手术之前的基线时和在手术之后的计划对照访视时纵向收集血浆样品(图20A)。全外显子组测序鉴别体细胞突变;根据实体瘤患者定制化监测技术标准工作流程,通过在手术前和手术后以及在辅助疗法期间收集的血浆样品中的大规模平行测序来分析靶向16种体细胞单核苷酸和插入缺失变异体的患者特异性多重PCR分析法(图20B)。
表1.患者特征和人口统计(N=130)
图22提供来自130名患者中的128名的超过800个血浆样品的ctDNA分布结果的示意性概述。图23A-B展示由手术后ctDNA状态进行分级的复发风险。基于第一手术后血液样品建立ctDNA状态,所述血液样品是在第6周和起始ACT之前抽取。图23(A)展示由ctDNA状态进行分级的无复发存活率的卡普兰迈耶分析。在随访结束时检查无事件患者。图23(B)展示根据ctDNA状态的复发率(未检查患者)。向58名患者给予辅助化学疗法,其可能影响ctDNA阳性患者的复发率。
图24(A)展示由辅助后ctDNA状态分级的在辅助化学疗法之后的复发风险。如果任何辅助后时间点是阳性,那么将患者评估为ctDNA阳性,且如果所有辅助后时间点都是阴性,那么评估为阴性。图24(B)展示在辅助治疗期间和辅助治疗后,代表性患者的ctDNA分布。
图25展示在用辅助化学疗法进行治疗之前,10名手术后ctDNA阳性患者的复发率,以及如何由ACT清除三名非复发性ctDNA阳性患者中的两名中的ctDNA。
图26A-B展示使用ctDNA和CT成像的复发时间(TTR)的比较:(A)12名复发患者的使用ctDNA和CT成像的TTR比较,其中由两种形态检测复发;(B)具有10.2个月ctDNA前置时间的代表性复发患者的连续ctDNA分布。
图27A-D展示四名代表性患者的连续ctDNA分布。
总之,用于靶向肿瘤特异性突变的个人化多重PCR分析法的大规模平行测序的实体瘤患者定制化监测技术RUO方法是用于检测和定量ctDNA的高敏感性和特异性平台。手术后ctDNA分析能够将CRC患者分级为具有极高或极低复发风险的子组(在ACT之前和之后)。在辅助化学疗法之前和之后。纵向ctDNA分析实现有效手术后治疗监测且复发ctDNA分析的早期检测在指导治疗决定方面具有显著效用(在辅助和辅助后情形下)。
实例2.用于监视和疗效监测的来自局部晚期膀胱癌患者的血浆cfDNA的测序
对不同癌症类型的研究表明循环肿瘤DNA(ctDNA)含量可以有效用于监测对新辅助疗法的治疗反应和/或比临床和放射性检测更早地检测疾病复发。在膀胱癌中,先前已使用血浆中的突变监测治疗期间的反应和鉴别转移性疾病的早期迹象。最近,描述NSCLC患者中的纵向ctDNA检测且研发个人化循环肿瘤DNA(ctDNA)检测分析法(Signatera
研究目标是使用在原发性肿瘤中鉴别的患者特异性突变,由纵向收集的血浆样品检测转移性复发、评估预后和监测ctDNA中的治疗反应。
临床方案.在2013与2017年之间前瞻性募集诊断患有局部晚期肌肉侵袭性膀胱癌(MIBC)且计划使用化学疗法的患者。所有患者在切除术(CX)之前用新辅助疗法或第一线化学疗法治疗且进行最多2年随访(图28)。在全身性疗法之前和之后以及在CX之后的计划对照访视时纵向收集血浆样品。
分子方案.由肿瘤和相匹配的正常样品的全外显子组测序(WES)鉴别患者特异性体细胞突变。使用来自纵向收集的血浆样品的cfDNA,使用个人化多重PCR分析法检测血浆中的患者特异性肿瘤DNA。对于每名患者,进行16种肿瘤特异性目标的测序且针对是否存在ctDNA以临床上盲式分析数据。当且仅当识别至少两种患者特异性目标且符合鉴定置信度评分阈值时,样品被视为ctDNA阳性。临床结果(放射性成像和治疗反应)是非盲的且直接与实体瘤患者定制化监测技术血浆识别结果进行比较。
研究中包括总共50名患者(表2)。
表2.患者特征和人口统计(N=50)
跨越5个HiSeq PE 2×50操作进行串联测序QC;每个目标读段深度的平均值和背景误差率展示于图29中。
复发的早期检测。针对CX之后的临床复发诊断十名患者且已在这些患者中的九名的血浆中鉴别ctDNA,其中中值是临床复发之前128天(表3)。对于一名患者,尚未分析在CX之后4个月时的最后一个对照样品。
表3.分子与临床复发之间的关系(n=9)
图30A-F描绘6名具有早期复发检测的患者。在分子复发时,在低到0.02%的VAF下检测到ctDNA,其在临床复发之前具有最多265天的前置时间(图30D、30E和30F)。在大部分患者中,对化学疗法的反应对应于ctDNA VAF降低(表4)。
表4.化学疗法之后的治疗反应的预测
图32A-B描绘使用ctDNA观察治疗反应的两名患者;在诊断时初始检测到的ctDNA由于新辅助治疗而下降且在CX之后保持检测不到。
总之,这些数据表明ctDNA分析(例如通过实体瘤患者定制化监测技术)可以帮助报告治疗反应且比放射性成像早最多265天鉴别疾病复发。存活率分析鉴别在诊断时或在切除术之后具有ctDNA的患者的显著较低的无复发存活率。最终,可以将ctDNA分析并入常规随访中以用于复发的早期检测且因此,有可能更早地开始替代性治疗,如免疫疗法。应在随机化临床试验中评估由ctDNA复发检测获得的总存活率益处。
实例3.基于高敏感性患者特异性多重PCR NGS的非侵袭性癌症复发检测和疗法监测分析法
循环游离DNA中肿瘤突变的鉴别具有用于临床表现之前的癌症复发的非侵袭性检测、在治愈性目的治疗之后的微小残留病的检测和治疗学上相关突变的检测的巨大潜力。评述和报告由当前版本的分析法进行的肿瘤特异性变异体检测的分析验证结果。
实体瘤患者定制化监测技术RUO.Signatera
分析验证.对两种乳癌细胞系(HCC2218、HCC1395)、一种肺癌细胞系(NCI-H1395)和其相匹配的正常对应物(分别是HCC2218-BL、HCC1395-BL和NCI-H1395-BL)来进行当前版本的实体瘤患者定制化监测技术(RUO)分析法的分析验证。将不同量的肿瘤细胞系DNA(0%、0.005%、0.01%、0.03%、0.05%、0.1%、0.3%、0.5%、1%)滴定到其各别相匹配的正常细胞系DNA中。使用来自相应的肿瘤细胞系DNA和其相匹配的正常细胞系DNA的全外显子组数据设计多重PCR分析法引物池(各自由16种对高置信度体细胞突变具有特异性的引物对分析法组成)。每个反应的用于库制备的起始全部输入物是20K基因组等效物;使用上文提及的多重PCR分析法引物池扩增来自添加有相应的肿瘤DNA的样品的SNV和插入缺失目标。将mPCR产物加注条形码,接着与其它mPCR条形码产物汇集在一起且接着使用IlluminaPaired End v2试剂盒,在50个双端读段循环下通过Illumina HiSeq 2500 Rapid Run进行测序,其中平均读段深度是约100,000个/分析法。
图34描绘跨越4个HiSeq PE 2×50操作的测序QC方法检验,包括背景转换和颠换误差率,且每个目标读段深度(DOR)的平均值是约100K(其中任何接受小于5K个读段的目标视为不合格且不考虑用于识别)。
在添加0.03%肿瘤DNA的情况下,由实体瘤患者定制化监测技术(RUO)获得SNV检测的分析敏感性是约60%(表5)。
表5.当前版本的实体瘤患者定制化监测技术(RUO)的分析敏感性结果
对于既定SNV集合,跨越6个目标展示在不同的所添加的突变体DNA浓度下,所预期的输入物百分比的数值与所检测的变异体等位基因出现率(VAF)百分比之间的关系,显示在高于0.03%肿瘤DNA浓度下的高敏感性(图36)。在≤0.01%SNV输入物(以<2个突变体拷贝为起始物质)下展示的假阴性包括由采样引起的突变体分子损失(图36)。
当由于具有16种高置信度SNV的集合识别至少两种SNV时,实体瘤患者定制化监测技术(RUO)的所估计的样品水平敏感性列举于表6中。
表6.所估计的样品水平敏感性
总结.实体瘤患者定制化监测技术RUO分析法提供在高敏感性、高特异性和低误差率下,通过定制的多重PCR分析法(由患者的肿瘤选择)的超深测序来以非侵袭方式检测血浆中的个人化癌症标签的复发的新型方法。基于分析验证结果,在SNV水平方面,实体瘤患者定制化监测技术RUO分析法具有99.9%特异性,以及在高于0.03%肿瘤分数下的大于65%敏感性和在高于0.1%肿瘤分数下的100%敏感性。在样品水平方面,实体瘤患者定制化监测技术RUO分析法在0.01%肿瘤分数下具有大于95%敏感性,在0.03%肿瘤分数下具有几乎100%敏感性且在0.05%和更高的肿瘤分数下具有100%敏感性。这些数据表明在单分子突变体水平方面的高检测率;其还暗示可以使用较小的血浆体积实现具有高特异性的相同单分子检测。实体瘤患者定制化监测技术分析法的性能表明其用于确定化学疗法治疗有效性的潜力。
实例.针对诊断、监视和复发的膀胱癌中多重患者特异性ctDNA生物标记物的纵向评估
背景.使用循环肿瘤DNA(ctDNA)作为用于诊断时的疾病分期(DX)、治疗反应和复发监测的生物标记物在许多癌症类型中是新兴的领域。在膀胱癌中,ctDNA的效用展示有前景的结果。本文中公开用于ctDNA监测的高敏感性和特异性的基于NGS的方法。
方法.前瞻性地包括50名用新辅助化学疗法治疗的局部晚期肌肉侵袭性膀胱癌患者的群组。对于每名患者,基于肿瘤和生殖系DNA的全外显子组测序来设计(SignateraTMRUO)16种肿瘤特异性突变的集合。总共,我们分析了在诊断时、在治疗期间、在切除术(Cx)时和在直到疾病复发的监测或最多2年随访期间,在386个时间点时纵向收集血浆样品的ctDNA。比较ctDNA分析的结果与放射性成像和临床结果。还可以针对治疗反应和疾病复发来分析来自纵向收集的尿液样品的ctDNA。
结果.在DX时,血浆ctDNA状态在无复发存活率方面具有强预后性。具体地说,62%(8/13)的在DX时呈ctDNA+的患者在新辅助治疗和Cx之后复发;相反,无(0/22)ctDNA-患者复发(对数秩;p<0.0001)。此外,还观察到在Cx之后存在ctDNA与疾病复发之间的强相关性。具体地说,在放射性成像之前约120天(0-245天),在100%(10/10)的ctDNA+患者中检测到在Cx之后的复发,而0%(0/38)的ctDNA-患者复发(对数秩;p<0.0001)。
结论.证明在DX时,ctDNA在膀胱癌中的强预后潜力,表明ctDNA在膀胱癌的分期中的作用。此外,证实在所有具有Cx之后的疾病复发的患者中检测到ctDNA。将ctDNA分析并入常规随访中以用于复发的早期检测可以实现更早地开始替代性治疗模式。
实例5.用于结肠直肠癌中的残留病检测、辅助疗法功效评估和早期复发检测的连续循环肿瘤DNA分析
背景.已证实疾病复发的早期检测可以改善结肠直肠癌(CRC)患者的存活率。先前研究已使用小型基因集合和ddPCR分析循环肿瘤DNA(ctDNA)以监测CRC中的肿瘤负荷。本文中,使用个人化多重PCR和NGS平台(Signatera
方法.分析根据护理标准治疗的130名I-IV期CRC患者的群组。对于每名患者,使用从WES获得的体细胞突变标签设计16种突变的肿瘤特异性集合。分析在手术前和手术后以及在ACT期间收集的血浆样品(n=829)。计算由手术后(n=91)和ACT后(n=58)的ctDNA状态分级的患者的无复发存活率。
结果.评估91名患者在手术之后,但在ACT之前的ctDNA状态。在75%(6/8)的ctDNA+且仅13%(11/83)的ctDNA-患者中观察到复发。在30%(3/10)的手术后ctDNA+患者中观察到有效ACT治疗。这些患者在ACT后连续收集的血液样品中始终呈ctDNA-,且在随访结束时始终未发生复发。评估58名患者的ACT后ctDNA状态。在77%(10/13)的ctDNA+和4%(2/45)的ctDNA-患者中观察到以放射方式证实的复发。平均起来,ctDNA比标准护理CT成像早9.13个月检测到复发。
结论.连续手术后ctDNA分析实现将患者分级为高或低复发风险子组、评估ACT治疗功效和复发的早期检测。重要的是,其还指示ACT可以消除最多30%的手术后ctDNA+患者中的残留病,且因此可以是ctDNA+患者的治疗选项。总之,ctDNA分析在指导治疗决定(在辅助和辅助后情形中)方面具有巨大潜力。
实例6.通过循环肿瘤DNA(ctDNA)的稳定、可扩展和个人化分析进行的残留乳癌(BC)的早期检测可以预测明显的转移性复发
背景.许多BC患者在主要治疗之后复发,但没有可靠的检验可以在远端癌转移变得明显之前便检测到。本文中展示通过个人化ctDNA分析实现复发患者的更早的鉴别。所述方法适用于所有患者且不限于通常由基因集合检测的热点突变。
方法.在手术和辅助疗法之后募集四十九名非转移性BC患者。在半年内连续收集血浆样品(n=208)。使用以分析方式验证的Signatera
结果.在18(89%)名临床复发患者中的16名中,在通过临床检查和生物化学(CA15-3)测量诊断转移性复发之前检测到ctDNA,且在整个随访期间保持ctDNA阳性。在2名未检测的患者中,一名具有局部复发且另一名具有两种原发性肿瘤。33名非复发性患者在任何时间点(n=142)都不呈ctDNA阳性。通过实体瘤患者定制化监测技术,以高准确性和最多2年(中值=9.5个月)的前置时间预测转移性复发。这些结果的概述提供于图37中。详细结果展示于图38-59中。
结论.使用可扩展的基于患者特异性ctDNA的经过验证的工作流程可以更早地鉴别将复发的患者。通过ctDNA分析实现的准确和更早的预测可以提供用于监测需要第二线补救性辅助疗法的乳癌患者的手段,以防止明显危及生命的转移性进展。
本文中呈现的结果表明,基于检测患者血液样品中的个人化癌症标记物/循环肿瘤DNA,所述方法在预测乳癌复发方面的敏感性极高。举例来说,正确地预测了18名复发病例中的16名且未鉴别到假阳性。所述方法还极其稳定。在作出阳性检测后,来自同一名患者的子序列血液样品始终保持阳性。
所述方法能够检测个人化癌症标记物/循环肿瘤DNA且进行复发的预测,例如在通过标准方法(例如成像)检测到复发之前27-610天。检测复发的中值时间是在通过标准方法检测到复发之前9个月。
实例7.I-SPY2试验中用于监测和预测对新辅助疗法(NAT)的反应的高风险早期乳癌患者中的个人化连续循环肿瘤DNA(ctDNA)分析和结果
背景.ctDNA分析提供用于监测对治疗的反应和抗性的非侵袭性方法。在NAT期间的连续ctDNA检验可以提供新出现的抗性和疾病进展的早期指示物。在本研究中,在I-SPY2试验(NCT01042379)中,分析来自接受NAT和确定性手术的高风险早期乳癌患者的ctDNA。在本实例中收集的数据将用于:(1)确定在早期治疗期间的ctDNA含量与pCR/残留癌症负荷/DRFS之间的关系;(2)比较ctDNA与MRI成像在预测对疗法的肿瘤反应方面的性能;和(3)检验在NAT之前和之后的ctDNA含量与3年无事件存活率(EFS)之间的关系。
方法.在84名高风险II期和III期乳癌患者中进行ctDNA分析,所述患者随机分配新辅助研究性药剂(n=57);AKT抑制剂MK-2206(M)与太平洋紫杉醇(T)的组合,接着给予小红莓和环磷酰胺(AC)(M+T->AC);或标准护理(T->AC)(n=27)。除T或M+T以外,HER2+患者还接受曲妥珠单抗(H)。
在手术之前,在NAT之前、在早期治疗(3周)时、在各方案(12周)之间和在NAT之后收集连续血浆。使用来源于预处理肿瘤活检和生殖系DNA全外显子组序列的突变分布来设计靶向16种对患者的肿瘤具有特异性的变异体的个人化分析法,以检测血浆中的ctDNA。在未实现pCR的患者子集(n=18-22)中,比较残留癌症中的突变与在预处理肿瘤中发现的突变。
分析:在这一分析中的84名患者中,15-25%的患者是HR-HER2-,40-60%的患者是HR+HER2-且35-35%的患者是HER2+。20-25%和30-42%的患者分别在对照和治疗臂中实现pCR。当前,收集数据以:(1)确定在早期治疗期间的ctDNA含量与pCR/残留癌症负荷/DRFS之间的关系;(2)比较ctDNA与MRI成像在预测对疗法的肿瘤反应方面的性能;和(3)检验在NAT之前和之后的ctDNA含量与3年无事件存活率(EFS)之间的关系。
结论.本研究提供用于评估ctDNA在连续监测对NAT的反应方面的临床显著性的平台。通过高敏感性ctDNA分析进行的准确和早期反应预测可以有助于及时和明智的治疗变化,以改善患者实现pCR的机率。最终,个人化ctDNA检验可以与成像和肿瘤反应的病理性评估互补以微调pCR作为替代终点,以获得改善的EFS。
实例8.通过循环肿瘤DNA(ctDNA)的可扩展和个人化分析进行的残留乳癌的早期检测可以预测明显的转移性复发
前言
乳癌是全世界最常诊断的癌症之一且是女性中癌症相关死亡的排名第二的起因。当前用于患有非转移性乳癌的女性的护理标准是手术,通常接着使用辅助疗法以消除可能引起复发或其它疾病进展的微观残留病。不幸的是,多达30%的在治愈性目的治疗之后未显示疾病迹象的女性最终复发且由于微小转移灶引起的转移性乳癌而死亡。当前用于疾病监测的工具(包括成像和/或生物化学方法,包括癌症抗原15-3(CA15-3)的血清含量)在检测微小转移灶方面具有有限的敏感性和准确性。较晚检测到转移与许多患者中的不良结果相关,强调研发微小残留病(MRD)的更早和更敏感的测量手段的需要。
已证实由细胞凋亡和坏死的癌细胞释放的循环肿瘤DNA(ctDNA)能够反映肿瘤的突变标签,且正在成为用于跨越不同癌症类型监测肿瘤进展的潜在非侵袭性生物标记。在乳癌中,ctDNA检测手术和/或辅助疗法之后的微小残留病和监测转移性疾病的效用展示有前景的结果。具体地说,已证实血浆ctDNA含量与肿瘤负荷的变化相关,由此提供更早的治疗反应的测量手段,且区分具有和不具有手术后最终临床复发的患者。尽管许多研究证明了ctDNA分析在乳癌中的潜在用途,但迄今为止,不存在能够可靠地检测所有患者中的微小残留病的可扩展的检验。
用于查询血浆cfDNA中的单核苷酸和INDEL变异体的个人化肿瘤特异性方法在临床检测之前预测非小细胞肺癌患者中的复发,表明这也可以适用于监测乳癌患者中的微小残留病。本文中,使用这一方法的增强和可扩展版本,我们寻求确定与常规监测方法相比,连续ctDNA分析在监测手术和辅助疗法后的乳癌复发方面的用途。主要目的是确定在原发性乳癌患者中,血浆中ctDNA的检测与明显转移性疾病的临床检测之间的“前置间隔”。
方法
患者和样品
EBLIS是由英国癌症研究所(Cancer Research UK)和国家健康研究所(NationalInstitute for health Research;NIHR)资助的多中心、前瞻性群组研究(NIHR REC编号13/LO/1152;IRAS:126462)。所有患者在参与试验之前提交书面知情同意书。试验方案由Riverside研究伦理委员会(Riverside Research Ethics Committee)批准,REC:13/LO/115;IRAS:126462。所有研究和技术人员对患者结果不知情。
由位于英国的3个中心募集总共197名患者。九名患者不满足试验准入条件;因此,对188名患者的群组进行随访,且进行6次每月一次的针对ctDNA的血液采样且同时进行临床检查和生物化学测量,包括CA153(图60)。符合条件的患者是18周岁或更大、不具有转移性疾病的临床迹象且因此视为在手术和辅助化学疗法之后不具有疾病。所有患者在参与研究的5年内完成辅助化学疗法。所有患者具有不良乳癌风险(在不接受疗法的情况下,在10年时的死亡率>50%的风险,对应于在不接受治疗的情况下,在10年时的复发率是65%)。
在研究的中点(2年)时,在期中分析且注意到发生50%的预测事件之后,我们选择在第一批50名患者中进行原发性肿瘤的全外显子组分析。
在K2-EDTA管中收集血液样品。在收集后2小时内通过血液的双重离心来处理样品,首先在1,000g下进行10分钟,接着血浆在2000g下进行10分钟。血浆以1ml等分试样形式储存在-80℃下。
Signatera
统计分析
所有数据以描述性方式呈现为平均值、中值或比例。卡普兰-迈耶方法确定从参与研究当天开始的无复发存活率。使用考克斯比例风险回归(Cox Proportional Hazardregression)建立疾病复发时间的模型。使用Stata,版本12.0(Stata Corp.,CollegeStation,Texas,USA)进行所有统计分析且使用R版本3.5.1(“survminer”程序包版本0.4.2.99)产生存活率图。
使用用于处理遗失连续数据的保守策略。如果遗失单一数据点,那么转入最后一个观察结果,或如果可以获得先前和后续数据,那么这两个值的平均值充当遗失数据的估计值。
全外显子组测序
通过目视检查来检验诊断性FFPE块状物,且使用具有最多的残留肿瘤的块状物进行DNA分离。由顾问组织病理学家审查单一H&E组织切片(D M)且使用1mm组织微阵列核心针巨视解剖肿瘤的最少2个区域。使用Gene Read试剂盒(Qiagen)根据制造商说明书从FFPE肿瘤核心提取DNA且如先前所描述测量DNA浓度。
使用Illumina HiSeq对200-500ng从每个FFPE原发性肿瘤块状物的核心的1-3个区域收集的肿瘤DNA进行全外显子组测序(测序由Novogene作为收费服务来进行),删除重复后的平均中靶读段深度是150x(对于所有49种肿瘤DNA)和50x(对于49个相匹配的生殖系样品)。所有测序数据已保存在欧洲基因组现象档案(European Genome-Phenome Archive)中。
定制集合设计.通过分析所有49名患者的原发性肿瘤和相匹配的正常WES来鉴别患者特异性体细胞变异体。基于所估计的具有变异体的癌细胞的比例来推断变异体的克隆性。使用所推断的变异体的克隆性和类型对每个肿瘤的所鉴别的体细胞SNV和短插入缺失进行优先级排序。使用标准实体瘤患者定制化监测技术分析法设计流水线产生用于既定变异体集合的PCR引物。对于每名患者,选择16种高排名相容分析法用于定制患者特异性集合。由整合式DNA技术将患者特异性16重PCR分析法排序。
cfDNA提取和定量.本研究中每种情况可以使用最多8ml血浆(范围,1-8ml;中值是5mL)。将全部体积的血浆用于cfDNA提取。使用QIAamp Circulating Nucleic Acid试剂盒(Qiagen)提取cfDNA且洗脱到50μL DNA悬浮缓冲液(Sigma)中。用Quant-iT高敏感性dsDNA分析法试剂盒(Invitrogen)对每个cfDNA样品进行定量。在49名患者中,从总共215个连续血浆样品分离cfDNA。
cfDNA库制备.使用来自每个血浆样品的最多66ng(20,000基因组当量)cfDNA作为用于定制库制备的输入物。对游离DNA进行末端修复,添加A尾部且与定制衔接子接合。将被纯化的接合产物扩增20个循环,使用Ampure XP珠粒(Agencourt/Beckman Coulter)纯化。
血浆多重PCR NGS工作流程.使用每个库的等分试样作为相关患者特异性16重PCR反应的输入物。样品使用实体瘤患者定制化监测技术肿瘤特异性分析法扩增且加注条形码,接着汇集在一起。由Illumina HiSeq 2500 Rapid Run进行测序,使用Illumina PairedEnd v2试剂盒进行50个双端读段循环,其中平均读段深度>100,000X/扩增子。
生物信息流水线.使用Pear软件合并所有双端读段。滤除在正向和后向读段中不匹配或具有低质量分数的碱基以最大限度地减少测序误差。用Novoalign版本2.3.4将合并的读段映射到hg19参考基因组。具有<5,000个高质量读段的扩增子视为QC不合格。针对每个样品广泛统计数据清单,使用内部程序检验来进行质量控制,所述清单包括读段总数、映射的读段、中靶读段、不合格目标数和平均误差率(图67A-D)。
血浆变异体识别.预先处理阴性对照样品(约1000个)的大型集合以构建变异体特异性背景误差模型。使用突变体和参考等位基因基于误差模型计算每个目标变异体的置信度评分,所述误差模型以全文引用的方式并入本文中。将具有高于预定义阈值(0.97)的置信度评分的具有至少2种变异体的血浆样品识别为ctDNA阳性。
分析验证.使用将来自三种癌细胞系的单核小体DNA滴定到其相匹配的正常对应物(ATCC)中来进行分析验证。使用两种乳癌细胞系(HCC2218和HCC1395)、一种肺癌细胞系(NCI-H1395)和其各自相匹配的正常B淋巴母细胞衍生的细胞系(HCC2218-BL、HCC1395-BL和NCI-BL1395)。对于每个细胞系对,对DNA进行外显子组测序,选择目标变异体,且使用标准实体瘤患者定制化监测技术流水线设计两个多重PCR引物池。在1%、0.5%、0.3%、0.1%、0.05%、0.03%、0.01%、0.005%、0%的平均VAF(基于DNA输入物)下进行将肿瘤滴定到正常单核小体DNA中(拷贝数目是二到九,随稀释因子而增加)。归因于肿瘤细胞株中异质性和非整倍性的可能性,单独目标的VAF可能与平均输入VAF不同。为了在每个滴定步骤中准确地计算每个目标的标称VAF,用10%VAF混合物进行独立实验。接着,使用来自这一实验的所观察的VAF计算输入校正因子(所观察的VAF/10%)。将校正因子应用于其在稀释系列中的各别目标。使用从16份人类血浆(每份约8mL)分离的cfDNA处理其它阴性样品。对于滴定系列,使用66ng cfDNA(对应于20,000单倍基因组当量)作为用于实体瘤患者定制化监测技术库制备的输入物;对于血浆cfDNA样品,使用所有所分离的DNA(在13-55ng范围内)作为输入物。接着,对这些库进行实体瘤患者定制化监测技术血浆工作流程(每个滴定样品使用两个引物池且每个cfDNA样品使用五个引物池)、测序且用实体瘤患者定制化监测技术分析流水线进行分析。图68A展示在各种浓度水平下,针对检测血浆中的目标所估计的敏感性。由阴性样品实现目标特异性>99.6%。假设定制集合具有10到16种克隆变异体,可以获得如图68B中报道的样品水平敏感性。估计样品水平特异性>99.8%。
结果
本文中,报道参与EBLIS研究的第一批50名患者的分析(图60)。一个肿瘤样品不适于进行外显子组测序且因此对49名患者进行。在报道普查日(2018年6月30日),十八名患者复发且31名保持无疾病。在49名患者中,除7名患者以外,其余所有患者都接受使用蒽环霉素/紫杉烷方案的辅助或NACT化学疗法(参见图69和表A)。四十一名患者在血液采样时始终接受辅助内分泌疗法(表B1-B3)。尽管在进入试验之前无需进行重复扫描,但除3名患者以外,所有其余患者在诊断时或在进入研究时都进行成像研究且都在正常限制内(表B1-B3)。
非复发患者是与复发患者在相同的时间框内募集的时序患者,对于非复发性患者,我们具有足够的从FFPE原发性肿瘤块状物分离的肿瘤DNA以用于外显子组分布分析且随访至少2年且进行连续血液采样。使用优化的实体瘤患者定制化监测技术工作流程以盲式方法分析连续血浆样品。
在18名复发患者中,由CT检测到10名,由骨扫描检测到3名,且由乳房X线照相术、MRI、肝酶升高和超声波各检测到一名。一名患者由于未知原因死亡。
循环肿瘤DNA检测和前置间隔
从所有患者收集FFPE肿瘤样品:39名患者在活检之前未接受全身性疗法;10名患者在其乳癌切除术之前接受新辅助化学疗法(NACT)(所有全身性疗法的细节,包括血液样品的时序提供于图60和表B1-B3中)。
为了针对每名患者评估是否存在循环肿瘤DNA,由每名患者的肿瘤的体细胞突变分布设计靶向16种体细胞SNV和INDEL变异体的患者特异性分析法(图64A-C)。接着,对来自49名患者的208个血浆样品中的每一个(范围:1-8个时间点)应用49种各别个人化分析法。
在89%(18名中的16名)的复发患者中检测到循环肿瘤DNA;HR+/HER2-、HER2+和三阴性乳癌(TNBC)中的检测分别是82%、100%和100%(图61A和B)。在未由ctDNA检测到的两名复发患者中,一名(1018)具有三种原发性癌症且另一名(1019)在胸骨中具有小型局部复发(随后切除)(图61A,表A)。应注意,一名患者(1072)最初具有正常血液分布,但在最后一次访视时呈ctDNA阳性;患者在血液分布分析时不具有疾病,但患者随后刚好在普查日期之前呈现远端癌转移(图61A和图65A)。我们在临床复发之前最多2年检测到复发性疾病,其中中值是266天(8.9个月;图61B)。当由亚型划分时,HR+/HER2-、HER2+和三阴性乳癌(TNBC)的中值前置时间分别是301、164和258天(图61B和C)。引人注目的是,在两名ER+PR+HER2-患者(1031和1051,图61A)中,在临床复发之前存在四个呈ctDNA阳性的时间点,转换成几乎2年的前置间隔。
在来自31名不具有疾病复发的患者的156个血浆样品中都未检测到ctDNA(图61A)。ctDNA的存在与显著不良预后相关联,由在第一个术后血浆样品(HR=11.8(95%CI4.3-32.5))和手术后随访血浆样品(HR=35.8(95%CI 8.0-161.3))中检测到ctDNA证实(图62A-B)。所有ctDNA阳性患者在手术之后50个月内复发。
对于所有患者,直到明显复发日,针对远端癌转移的放射性和扫描检查都呈阴性,在复发之前通常会出现患者症状。许多患者进行随访扫描以补充在呈现时进行的扫描且这些扫描还是呈阴性(表B1-B3)。七名患者(1004、1055、1072、1091、3018、3019、3048)在第一次变成阳性的ctDNA检验的4个月内进行扫描且都是阴性。
在整个研究过程期间,我们还监测49名患者中的43名中的CA15-3。这些患者中的三十九名在整个随访周期内具有正常结果。18名复发患者中的十二名在整个监测周期内具有正常CA15-3含量。两名患者(1051和1088)具有进行性CA15-3上升,但ctDNA与CA15-3相比敏感性更高,其中分别在CA15-3含量升高之前224和212天检测到ctDNA(图63A和图65B)。患者1111和1018具有在临床复发之前采集的单一血液样品且具有升高的CA15-3测量结果。患者1111具有阳性ctDNA检验,而患者1018具有阴性ctDNA检验。值得注意的是,六名其它患者(3名复发和3名非复发)具有CA15-3略微升高的偶然性血液样品,但这是波动的且并不反映疾病进展(表B1-B3)。
循环肿瘤DNA的表征
实体瘤患者定制化监测技术分析法被设计成靶向16种患者特异性体细胞SNV和INDEL变异体,所述变异体提供检测的最高似然性。由ctDNA检测的所有16名复发患者展示于图63和图65中。来自八名复发患者的十个血浆样品的所检测的变异体等位基因出现率(VAF)在0.01-0.02%内。最低的变异体等位基因出现率0.01%对应于在血浆样品中检测到单一突变体分子(图66)。可以在四名患者1004、01055、1072和1096中发现这一敏感性水平(图63、图65,表C)。检验的特异性大于99.5%需要通过测量到两种或更多种变异体超过识别算法的所选择的置信度阈值,以确定血浆中存在ctDNA来实现。特异性由所有非复发患者的血浆样品都未被识别为阳性的事实来强调。
根据由ctDNA检测的16例复发中的ctDNA分布随时间推移的变化,七名患者在所有所分析的时间点时具有阳性ctDNA检验且展示所检测的变异体数目和VAF百分比随时间推移而增加;六名患者最初是ctDNA阴性,随后变成阳性,且三名复发患者仅具有一个血浆时间点可用于分析,其都是ctDNA阳性(图63和图65)。
在图63中突出显示这些患者中的五名,其具有每种亚型中的至少一种,表示HR+/HER2-(1031和1051)、HER2+(1096)和TNBC(1055和1074)。患者1031、1051、1055最初是ctDNA阴性,但在稍后的时间点时变得可检测到ctDNA(图63A-C)。这些患者中的两名,都是HR+(1031和1051),在由ctDNA检测到的分子复发与临床复发之间的最大前置时间分别是721和611天(图63A-B)。患者1031、1055、1074和1096的所检测的变异体等位基因出现率在0.01到0.02%范围内且展示与疾病进展相关联的VAF的进行性上升(图63B-E)。所有患者在检测到ctDNA后,在整个随访期间保持阳性(图63和图65)。
整体上,可以通过变异体等位基因出现率和所检测的变异体的数目来监测疾病进展,如图63F中所示。因为患者的时间点数目不同,所以由来自同一名患者的一系列血浆样品中的第一个和最后一个时间点突出显示差异。中值变异体等位基因出现率从第一时间点的0.092%(范围:0.01%到9.2)增加到3.9%(范围:0.05到64.4%),然而在在第一时间点时检测到的变异体的中值数目是5(范围:2-12),相比之下,在最后一个时间点时是12个变异体(范围:5-15)。在早期时间点时检测到的较少变异体数目和这些变异体以极低拷贝数存在的事实指出检验患者肿瘤中的多种突变,以进行针对患者血浆中是否存在ctDNA的高敏感性检验的重要性。
讨论
本报告描述用于跨越主要子类型来监测乳癌患者中的微小残留病的可靠和可再现的方法。所述方法使用来自肿瘤的基因组数据设计患者特异性分析法。接着,以平均每个目标>100,000个读段的极高深度进行血浆cfDNA的测序,以实现低到单一突变体分子的敏感性检测。这种新的转化型技术是稳定的,并且可以扩展以用于现代医疗中的实施方案且已被生产用于研究用途。鉴于例如在英国,所有癌症从2018年秋季开始将接受基因组分布分析的当前通告,这是尤其及时的。实体瘤患者定制化监测技术平台能够提供检测微转移疾病的个体化方式,这由本研究的结果明确支持,因为除了一名以外,其余所有具有远端癌转移的复发性乳癌患者在明显复发之前都具有阳性血液检验且在一些情况下,显示几乎2年的前置间隔。
用这项技术的原型进行的先前研究展示在非小细胞肺癌患者中的前景(Abbosh等人2017,其以全文引用的方式并入本文中)且工作流程已被优化,因为实现了更高敏感性的ctDNA检测且成本更低。本文中,我们说明这一系统的极佳再现性和准确性。关注患者所特有的SNV而非已知的驱动基因代表了迄今为止,最准确和敏感的用于检测乳癌患者中的MRD的方式。
ctDNA测量结果在早期癌症的管理中的临床应用仍是极有争议的问题,且当前ASCO和ACP联合评审得出以下结论:在早期癌症治疗监测或早期残留病检测方面不存在临床效用证据和极少的临床有效性证据。这一结论可能部分归因于所有先前研究是在不存在与常规标记物的比较(如本文中所进行)的情况下进行的事实。
本研究产生若干要点。首先,现在有可能在成像时不具有疾病证据的情况下,以高度准确性预测患者中的复发。第二,大部分患者现正在美国和英国中心通过每年乳房X线照相术与(在一些情况下)CA15-3和肝脏功能检验的组合来进行随访。除了两个在不存在可检测的癌转移的情况下逐渐升高的CA15-3的病例以外,直到明显转移性复发,这些患者在我们的群组中都是正常的,表明由临床医生使用的当前方法的局限性。七名复发患者在第一次ctDNA检测的近期还进行了扫描且都是阴性。值得注意的是以下观察结果:一些患者具有短暂升高的CA15-3且保持无疾病,而实体瘤患者定制化监测技术检验在检测后始终是阳性且不存在假阳性。第三,尽管NACT确实减少原发性癌症(对原发性癌症进行外显子组测序),但残留癌症的突变标签反映这些患者中的残留转移性疾病,其表明基于原发性肿瘤突变分布设计的个人化检验不仅是可能的,而且是有效的。
然而,ctDNA散布到血浆中需要一定条件且因此检验可能限于在开始时具有侵袭性足够高的疾病的患者,且因此可能不适用于通常具有良好预后的具有侵袭性较小和较低的乳癌的患者。由于我们的分析法的侦测极限降到单分子,检测的缺陷可能与其中侵袭性较低的肿瘤可能释放较少的ctDNA分子的肿瘤生物学相关联。这由一名患者(1018)具有局部可切除的复发疾病,而ctDNA是阴性例示。此外,在具有多个原发性肿瘤的患者中,将需要对所有肿瘤进行分布分析以用于监测疾病进展。这在具有三个原发性肿瘤的患者1019中发生;不幸的是,由于可用的组织有限,仅一个肿瘤经历外显子组测序。在这种情况下,未由ctDNA检测到疾病复发且我们的假设是转移并非来源于所测序的肿瘤。最终,检验不适用于检测第二原发性乳癌,除非其是原始肿瘤的复发;这由患者1044例示,其中通过常规乳房X线照相发现第二对侧原发性癌症,但血浆始终保持ctDNA阴性(表A)。
本文所描述的分析平台并不意图鉴别来自血浆的易处理的目标。大部分所选择的用于肿瘤DNA检测的SNV是每名患者所特有的,且被选择作为肿瘤负荷的反映而非表示通常有助于癌症进展的驱动突变。然而,这也是实体瘤患者定制化监测技术分析法的优点,选择乘客和克隆突变是监测疾病负荷必不可少,因为驱动突变通常赋予选择性优点,引起改变肿瘤异质性。尽管16重分析法不提供可操作的目标,但肿瘤WES可以提供这类目标且还可以用其它鉴别易处理的突变的分析法检验血浆库。
我们的结果与其中在新辅助和辅助周期内始终监测可操作突变(例如在PIK3CA中)的方法的结果互补。尽管选择驱动突变可以监测一些肿瘤(REF)的进展,但研究表明,其不适用于检测所有患者中的早期转移性复发,因为并非所有患者在其肿瘤(REF)中都具有相同的靶向驱动突变。在一项先前研究中,仅78%(43/55)的病例鉴别到一种或多种体细胞突变且接着使用数字微滴式PCR(ddPCR)进行监测。我们还分析靶向10种乳癌基因中的>150个热点的“现成的”乳癌集合(Oncomine
我们的研究具有一些重要结果。迄今为止,已证实用靶向或细胞毒性疗法进行的全身性治疗仅在辅助情形中给予时是治愈性的;明显转移性疾病的治疗很少(如果存在)是治愈性的(REF)。本文所描述的方法提供替代方案:尝试用第二线疗法补救具有ctDNA的患者。另一种应用可能是有助于评估新的药物疗法,尤其具有增强免疫反应的机制的药物疗法。迄今为止,已成功地实现一项间接成功标准,即进展时间;实体瘤患者定制化监测技术ctDNA检测方法现将作为衡量准绳实现另一项成功标准,即ctDNA的降低或清除。
总之,实体瘤患者定制化监测技术平台能够以高度敏感性检测乳癌患者中的MRD。其优于常规随访手段且展示用于监测患者以进行精准医疗的前景。第一次,这提供了基于血液的检验,其使患者确定其疾病是可控的。
概述
许多BC患者在主要治疗之后复发,但没有可靠的检验可以在远端癌转移变得明显之前便检测到。本文中,我们展示通过可扩展的个人化循环肿瘤DNA(ctDNA)分析进行乳癌复发的更早的鉴别。所述方法适用于所有患者且不限于通常由基因集合检测的热点突变。
在手术和辅助疗法之后募集四十九名非转移性BC患者。在半年内连续收集血浆样品(n=208)。使用以分析方式验证的Signatera
在18名临床复发患者中的16名(89%)中,在通过临床检查、放射性成像和CA15-3测量来诊断转移性复发之前检测到ctDNA,且在随访期间保持ctDNA阳性。在2名未由ctDNA检测到的患者中,一名仅具有小型局部复发(现已切除)且另一名具有三个原发性肿瘤。31名非复发患者在任何时间点(n=156)都不是ctDNA阳性。由实体瘤患者定制化监测技术预测转移性复发,其中前置时间最多是2年(中值=8.9个月,HR:35.84(95%CI 7.9626-161.32))。
使用可扩展的基于患者特异性ctDNA的经过验证的工作流程跨越主要乳癌亚型在临床检测之前,在转移性乳癌复发之前检测微小残留病。由ctDNA分析实现的准确和更早的预测可以提供用于监测需要第二线补救辅助疗法,以试图防止明显危及生命的转移性复发的乳癌患者的手段。
表A1.所有49名患者的临床特征。标记有星号*的患者是已故的。
表B1.患者的血浆时间点
表B2.患者的CA15-3含量
表B3.患者的内分泌疗法
表C.样品水平和VAF的概述
实例9.尿道上皮膀胱癌患者中通过连续血浆游离DNA的超深测序进行的转移性复发的早期检测和疗效监测
前言
膀胱癌是最常见的尿道恶性疾病且约20-25%的新近诊断有尿道上皮癌的患者将发展肌肉侵袭性膀胱癌(MIBC),且10-30%的诊断患有非MIBC(NMIBC)的患者将进展成MIBC。用于治疗MIBC的当前标准方法是根治性切除术。不幸的是,20%的在手术时具有淋巴结阴性的患者和80%的在手术时具有淋巴结阳性疾病的患者将经历转移性复发,且5年内的总存活率(OS)平均是50%。
新辅助化学疗法(NAC)改善MIBC患者的存活率,且用吉西他滨和顺铂(GC)进行的治疗是MIBC的最常用的新辅助化学疗法(NAC)。当前,用吉西他滨和顺铂进行的治疗在约40-50%的患者中引起显著降期(在切除术时pT<2N0)。
膀胱癌患者中的转移性复发的早期检测可以提供新的治疗性方法以提高存活率。在切除术之后,在早期时间点时鉴别转移性复发(当复发不可由放射性成像检测到时)可以显著改善可能受益于早期/辅助治疗的患者的鉴别和改善这一患者群体的存活率结果。此外,复发和转移的早期确定可以帮助防止对治疗不起反应的患者的不必要的和可能有害的长期治疗。
当前,使用以预定间隔进行的标准计算机断层摄影(CT)成像来检测复发、转移和监测对治疗的反应。尽管成像技术提供肿瘤负荷的评估,但监测潜力受限于次最佳侦测极限和测量结果的固有可变性。因此,转移性复发和/或进展的早期检测以及治疗功效的评估仍是主要临床挑战。
使用循环肿瘤DNA(ctDNA)作为诊断时的疾病分期、肿瘤负荷、转移性复发的早期检测和治疗性治疗反应的生物标记物的完全可能性仍未得到满足。当前有前景的研究证实,游离DNA(cfDNA)可以用于监测早期肺癌演化和转移性疾病中的亚克隆发展(Abbosh等人,《自然》545,446-451(2017)(“Abbosh等人2017”),其以全文引用的方式并入本文中)。在膀胱癌中,已证实可以在血浆和尿液中检测到ctDNA且高含量ctDNA与随后检测到的临床疾病进展和转移性疾病相关联(
本文中,我们报道来自前瞻性研究的结果,所述研究涵盖68名在切除术之前用新辅助化学疗法(n=56)或第一线化学疗法(n=12)治疗的原发性肿瘤和相匹配的生殖系DNA的全外显子组测序(WES)。设计对各个患者的肿瘤突变标签具有特异性的敏感性、基于个人化多重PCR NGS的分析法且用于监测在化学疗法之前、在化学疗法期间和在化学疗法之后纵向获得的血浆样品中的体细胞突变。本研究的主要目标是研发实现使用ctDNA作为有效生物标记以用于转移性疾病的预后、早期检测和作为化学疗法反应的预测因子的ctDNA检测方法。
方法
患者和临床样品
诊断患有MIBC且在切除术之前接受新辅助化学疗法的患者和进行或未进行先验切除术的由于转移性疾病而接受化学疗法的患者在2013与2017年之间,在单一三级大学医院(Aarhus University Hospital,Denmark)参与研究。基于患者准则和机器人的可用性,以开放性切除术或机器人辅助方式进行根治性切除术。在所有患者中,进行扩大淋巴结清扫达到主动脉分叉水平。
根据丹麦国家指南(Danish national guidelines)治疗患者。以4个吉西他滨和顺铂(GC)系列方式给予新辅助化学疗法,以三周间隔给予。在诊断时具有转移或cT4b肿瘤的患者用最多6个GC系列治疗。在CX之前给予的化学疗法之后的病理学降期在治疗之后定义为
样品收集和DNA提取
分析来自血液、肿瘤活检和纵向收集的血浆样品的材料。在诊断时从TUR-B获得组织活检。如先前所描述,由在具有高癌瘤细胞百分比的最具代表性的位置进行打孔,从来自
外显子组测序和生物信息分析
使用100-500ng DNA制备肿瘤和相匹配的生殖系DNA的库且由
所有通过所用过滤器的变异体经历关于突变标签的活性的分析。最初将变异体装载于VRanges器具中且接着使用SomaticSignaturesR包提取序列上下文(Obenchain等人,《生物信息学(Bioinformatics)》30,2076-2078(2014);Gehring等人,《生物信息学》31,3673-3675(2015))。归因于群组的尺寸,未应用突变标签的重新提取。改为使用MutationalPatternsR包将在样品中鉴别的突变分布投影到已知的COSMIC标签上(参见cancer.sanger.ac.uk/cosmic/signatures和Blokzijl等人,《基因组医学(Genome Med.)》10,33(2018))。对Robertson等人,《细胞(Cell)》171,540-556.e25(2017)中鉴别的膀胱癌的突变标签1、2、5和13进行优先级排序以用于分析。
DNA损伤反应相关突变
所选择的DNA损伤反应基因描述于Teo等人,《临床癌症研究(Clin.Cancer Res.)》3610-3618(2017)中。针对受损或良性来分析在这些基因中鉴别的突变。所有功能丧失突变视为损伤。如Reva等人,《核酸研究》39,e118(2011)和Adzhubei等人,《自然方法(NatureMethods)》7,248-249(2010)中所描述,使用PolyPhen2和MutationAssessor进一步分析误义突变。在PolyPhen2和MutationAssessor中分别鉴别为可能的损伤/很可能的损伤或中/高的变异体视为损伤。
游离DNA库制备
如先前所描述制备游离DNA(cfDNA),使用来自每个血浆样品的最多66ng(20,000基因组当量)游离DNA(cfDNA)作为用于库制备的输入物。对cfDNA进行末端修复、添加A尾且与定制衔接子接合。将被纯化的接合产物扩增20个循环且使用
血浆多重PCR下一代测序(NGS)工作流程
使用每个库的等分试样作为相关患者特异性16重PCR反应的输入物。使用
血浆变异体识别
预先处理阴性对照样品(约1000个)的大型集合以构建背景误差模型。对于使用突变体和参考等位基因读段深度的每个目标变异体,基于误差模型计算置信度评分,如Abbosh等人2017中所描述。ctDNA阳性血浆样品定义为具有至少2个变异体且置信度评分高于预定算法阈值(0.97),如Abbosh等人2017中所描述。
血浆全外显子组测序
将每个血浆库的等分试样加注条形码且使用
RNA测序、数据处理和分析
使用
统计分析
使用程序包survminer和survival根据R统计学进行存活率分析。使用用于连续变量的威尔科克森秩和检验和用于类别变量的费舍尔精确检验进行统计显著性的评估。
结果
患者特征和原发性肿瘤分析
在Aarhus University Hospital,Denmark,在2014与2017年之间募集在切除术之前接受化学疗法的局部MIBC患者(图70)。总共68名患者满足所有纳入准则(参见图70、图71A-G和以下表IA)。
表IA:患者特征和人口统计
在平均目标覆盖率是104X(31X-251X,肿瘤样品)和66X(35X-120X,生殖系样品)的情况下进行肿瘤和相匹配的生殖系DNA的全外显子组测序(WES),每名患者鉴别平均488(11-3536)个突变。此外,进行46个肿瘤的RNA测序以确定膀胱癌子类型、免疫标签和细胞组成。所有患者的分子特征和临床数据的概述展示于图71A-G中。用于本研究的临床方案和采样方案的概述展示于图72中。
由基于超深多重PCR的下一代测序(NGS)进行的ctDNA监测
使用订制多重-PCR NGS方法进行ctDNA检测。基于组织和序列上下文中所观察的变异体等位基因出现率(VAF),由全外显子组测序(WES)数据对体细胞SNV和短INDEL进行优先级排序。设计独特的患者特异性分析法且合成十六中高等级体细胞突变,如图73中概述。对血浆cfDNA进行多重PCR NGS。仅当基于Abbosh等人2017中公开的先前研发的识别算法检测到至少两种目标变异体时才将样品识别为ctDNA阳性。在0.01%变异体等位基因出现率下,确定样品水平分析敏感性(其中检测到16种变异体中的2种或更多种)>95%。
使用这一方法,分析来自研究中所包括的68名患者的618个血浆样品中的ctDNA状态(图70)。在整个工作流程期间进行质量控制。从进一步分析中排除QC不合格的样品和扩增子。对于每名患者,在全外显子组测序(WES)和血浆测序中对45个SNP的集合进行基因分型以确保样品和谐性。中值目标覆盖率是120.000X。
用于预后和复发检测的ctDNA检测
在整个疾病病程期间,存在或不存在ctDNA与患者结果紧密相关(图74、图75A-C)。尤其关注以下三个时间点时的ctDNA状态。第一相关时间点是NAC给药之前的ctDNA状态,且发现这一时间点对结果具有强预后性。在膀胱癌中,第一介入是TURBT(膀胱肿瘤的经尿道切除术)且因此,发现此第一时间点充当代理以测量微小残留病。引人注目的是,94%的(34/36)的ctDNA阴性患者在此第一时间点时未复发,保持整个研究持续时间。相比之下,44%(11/25)的ctDNA阳性患者在这一时间点,在NAC之前,在切除术之后复发。因此,对于NAC和切除术(CX)之后的长期临床结果,这一早期时间点时的ctDNA的检测是极强的预后因子。
第二时间点是在NAC之后且在切除术之前,这一时间点时的ctDNA状态也对患者结果具有预后性。在ctDNA阴性患者中,仅7%(4/55;4名在切除术(CX)之后是阳性且复发)的患者复发,相比之下,70%(7/10)的ctDNA阳性患者经历复发。在切除术(CX)之前的ctDNA状态与CX时的病理相关联,因为100%的ctDNA阳性患者在这一时间点时具有残留T2+肿瘤和/或在切除术时鉴别的淋巴结癌转移(图75B)。
第三时间是在切除术之后。由在CX之后的ctDNA状态对患者进行分级显示ctDNA阳性患者的显著恶化结果(图75C)。大部分,96%的ctDNA阴性患者(50/52)不复发,然而,2/52名患者在CX之后超过1.5年时复发(未进行更接近复发的ctDNA分析)。相比之下,92%(12/13)的ctDNA阳性患者复发。值得注意的是,一名患者在临床评估之前死亡(图75D)。发现在CX之后的ctDNA状态对疾病复发具有高预后性,且与任何其它预测性因子(如切除术之前的N阶段和对化学疗法的反应)相比是更强的预测性因子(图75E)。
用于疾病监视的连续ctDNA测量结果
本文中公开ctDNA的连续测量结果可以用于监测疗法反应以及检测复发。在我们的研究中,在切除术之后,在患者不具有疾病时收集连续血浆时间点,以在监视情形中评估ctDNA的值。当包括疾病过程期间ctDNA的连续测量结果的分析时,我们观察到敏感性是92%且特异性是100%(图76)。平均起来,在通过放射性成像检测到之前96天(0-245天)观察到ctDNA的检测。举例来说,对于患者4265,观察到在NAC期间的ctDNA下降且接着在CX后第138天检测到ctDNA;然而在临床上,在186天后(在CX之后第324天)检测到复发。类似地,,患者4189在CX之前展示ctDNA阳性且接着是阴性,且接着在CX之后第273天又检测到ctDNA;然而,在第369天或96天后检测到临床复发(参见图76)。对于12名具有转移性复发的患者,我们发现ctDNA分析与常规成像相比具有103天的中值前置时间(范围;p=0.019)(图77)。前置时间可能由于与成像相比更频繁的血浆采样的分析而具有偏差。将我们的分析限于同时进行血浆和放射性成像的患者鉴别了八名患者,其中五名展示用于ctDNA分析的复发检测中的前置时间。其余三名患者展示同时复发检测(图77),且发现所有八名患者的所得平均前置时间是106天。
用于疗法反应监测的连续选择ctDNA测量结果
用NAC治疗膀胱癌,然而,先前在临床上适用的对治疗的反应的预测性生物标记物现在不可用,且使用在CX时的病理学降期作为治疗功效的代理(ref)。在我们的系列中,如所预期,疾病复发与化学疗法反应紧密相关联(图78A),然而,仅44%(×/y)的不具有反应的患者具有疾病复发,表明病理学降期对于评估治疗功效是次最佳的。本文中,我们发现在反应者和无反应者之间,在NAC期间的ctDNA的连续测量结果展示高度不同的分布(p=xx;图78F-G)。总共83%(34/41)的ctDNA阴性患者对化学疗法展示反应且53%(9/17)的从最初的阳性检验开始展示ctDNA清除的患者具有反应,表明ctDNA含量可以充当在治疗期间和之后的治疗功效的更好的指示物。在NAC之后的ctDNA阳性患者不展示对化学疗法的反应。整体上,ctDNA含量反映在群组中观察到的疾病病程特征(图78G)。
在对化学疗法起反应的患者中,未经治疗的肿瘤中的分子特征的分析展示突变标签5的显著更高的贡献(p=0.01)(图78B)。标签5的高贡献与ERCC2突变状态紧密相关联(图80),表明与DNA损伤反应(DDR)机制的相关性,如先前所报道的22。然而,反应者中的ERCC2突变并不显著更多(图78C)。整体上,DDR突变状态不是化学疗法反应的预测性生物标记物(图78D)。通过由分子亚型对肿瘤进行分级(图78E),我们发现与存活率的显著相关性(图78F),但子类型不预测对NAC的反应(图78G)。与其它子类型相比,分类为“浸润”的肿瘤展示更高的对化学疗法的反应率(图78G)。这与强调基底细胞型肿瘤与NAC治疗反应相关性最高的先前报告形成对比。
来自具有转移性疾病的患者的血浆ctDNA的全外显子组测序
在多重PCR NGS分析法中,对来自血浆样品的cfDNA进行全外显子组测序(WES),在10%或更高的等位基因出现率下测量ctDNA目标。对来自三名患者的四个样品进行测序达到平均目标覆盖率是307X(272X-340X)且鉴别到508-1294个突变。我们比较在血浆WES数据中鉴别的所有突变与来自原发性肿瘤的相关WES数据,以评估在转移性演化期间获得的突变变化(图79A-D)。发现原发性肿瘤的突变前景与转移性病灶之间的高类似性,表明在所选择的患者的疾病病程期间的有限克隆演化。平均起来,鉴别到在癌转移时,ctDNA中存在62个突变,其未在原发性肿瘤中检测到。有趣的是,在患者4119的血浆中鉴别到两个CYP2C19突变,其都影响密码子214。所得氨基酸位于涉及将化合物引导到蛋白质活性位点的通道中。这些突变可能在化学疗法期间发生且可以解释在这名患者中未观察到病理学降期。
讨论
本报告描述基于在治疗之前、在治疗期间和在治疗之后的期间进行的ctDNA检测和监测,用于转移性疾病的早期检测和改善的治疗的可靠和可再现的方法。在用治愈性目的手术治疗的患者中,ctDNA的检测充当隐性癌细胞且因此残留病的直接证据。
有趣的是,本文中发现ctDNA的检测通常是在通过成像技术检测到转移性疾病之前。具体地说,我们发现所有最终呈现转移性膀胱癌的患者在切除术之后都是ctDNA阳性,与成像相比平均前置时间是103天。因此,本文中所提供的分析ctDNA的方法提供用于在更早的时间点起始转移性复发的治疗的独特机会。重要的是,起始针对较小体积的转移性疾病的治疗可以提高反应率和有利地影响存活率。
本研究还描述用于鉴别具有低转移性复发风险的患者的可靠和可再现的方法。本文中发现所有保持不具有疾病的患者在切除术之后都是ctDNA阴性(100%特异性)。在切除术之后的检验的100%特异性可以用于鉴别具有低转移性复发风险的患者,且因此降低由昂贵的放射性成像进行持续监视的需要和相关患者焦虑。因此,与常规方法相比,本公开提供ctDNA状态与结果之间的优良关联性,由此使ctDNA分析在临床上更加有效。
本公开还发现ctDNA的动力学可以帮助鉴别在治疗期间已经对NAC起反应的患者。MIBC患者在切除术之前接受NAC以降低原发性肿瘤负荷且潜在地根除微转移,且大部分患者在NAC之后经历病理学降期。不具有病理学降期(即,残留原发性肿瘤或在切除术时的淋巴结浸润)或先验淋巴结浸润是与切除术之后的疾病复发相关联的风险因子。在我们的研究中,在NAC之前或期间,在37%(25/68)的患者中检测到ctDNA,且53%的具有低于我们的检测阈值的ctDNA降低的患者展示病理学降期。重要的是,具有持续可检测的ctDNA的患者都未呈现病理学降期。然而,观察到具有病理学降期的患者的子集具有疾病复发且反之亦然。这些结果表明尽管临床和组织病理学参数充当预后风险因子,但患者的基于病理的风险分级仍不理想。
本文中的公开内容还提供在化学疗法之前和期间的ctDNA状态可以作为新的有效的临床风险因子且可以潜在地帮助选择具有早期转移性扩散的患者,其可以受益于长期NAC和强化监视方案。在本文中,证明在化学疗法之前或期间呈ctDNA阴性的患者在切除术之后都未经历疾病复发,而44%(11/25)的在化学疗法之前或期间呈ctDNA阳性的患者具有疾病复发。因此,与当前可用的患者群组的风险因子相比,甚至在早期时间点时的ctDNA的存在可以指示转移性扩散且有助于这些患者的优良风险分级,据我们所知,其先前尚未以类似的一致性证明。具有初始可检测的ctDNA,但不具有最终疾病复发的迹象的患者的子集可以表示化学疗法或切除术有效地根除疾病的情况。因此,不具有早期转移性扩散迹象(ctDNA阴性)且在NAC之后具有病理学降期的患者可能符合膀胱保留方法的条件。
值得注意的是,对于许多患者,ctDNA出现率非常低(几乎没有突变拷贝),且需要基于NGS的超深测序方法以可靠地检测这些罕见变异体。基于原发性肿瘤的WES选择克隆突变使得有可能进行血浆ctDNA中的患者特异性突变的超深测序。更早的研究已证实原发性肿瘤与癌转移之间的基因异质性,表明可能需要频繁突变的基因的基因集合。通过应用来自血浆的cfDNA的WES,我们仍观察到原发性肿瘤与癌转移之间的异质性,但重要的是,在癌转移中检测到所有选自原发性肿瘤的克隆突变。
总之,本文中的公开内容证实检测膀胱癌患者中的ctDNA含量可以很好地预测转移性复发和治疗反应。具体地说,本发明人在本文中提供用于监测癌症患者的治疗反应的准确和可靠方法,以使患者确定疾病是可控的,以及比常规方法明显更早地检测到复发,其可以改善存活率结果。
实例10.I-III期结肠直肠癌患者中由超深测序进行的血浆游离DNA的纵向分析
本实例的目的是证实纵向手术后循环肿瘤DNA(ctDNA)分析实现不具有疾病的临床迹象的患者中的残留肿瘤负荷的鉴别和监测。具体地说,本实例证实ctDNA分析实现I-III期结肠直肠癌患者的个人化和风险分级手术后管理。
前言
结肠直肠癌(CRC)是全世界第三常见的癌症,每年新诊断130万病例,且是排名第二的癌症相关死亡原因。尽管改进了手术、实施筛检且治疗方案得到进步,但CRC患者的5年死亡率仍然较高,达到约40%,由此代表显著的全球健康负担。
用于CRC患者的当前护理标准包括肿瘤的手术切除,接着在所选择的患者中进行辅助化学疗法(ACT)。大部分II期患者未用ACT治疗,然而,约10-15%的患者在手术后具有残留病。如果可以鉴别残留病,那么用ACT进行治疗可以潜在地降低其复发风险。相比之下,大部分III期患者接受ACT。尽管,超过50%的患者已通过手术治愈。此外,约30%的用ACT治疗的III期患者经验复发,使其成为其它疗法的候选对象。因此,极需要改进的用于鉴别将受益于ACT的患者群体的工具。
复发性疾病的早期诊断是CRC中另一明显未满足的临床需求。在完成确定性治疗之后,建议进行复发监视以足够早的检测复发,从而可能进行治愈性手术。尽管进行监视,但过晚地检测到许多复发事件且仅10-20%的异时癌转移是出于治愈性目的来治疗。因此,需要更好的生物标记物,其可以更早地检测具有高复发风险的患者,由此实现适合的随访和治疗策略以改善患者存活率。
方法
患者
在2014-2018年之间,在奥尔胡斯大学医院(Aarhus University Hospital)、兰德斯医院(Randers Hospital)和赫宁医院(Herning Hospital)的手术部门募集I到III期CRC患者。在手术时收集肿瘤组织。在手术之前(最多事先14天,手术前)和在手术后第30天(允许最多在14天之前或之后抽取样品)收集血液样品,且接着每三个月一次收集血液样品直到死亡、患者退出研究或第36个月(以先达到者为准)。七十五名患者提供连续血液样品(每名患者3到14个样品),而其余50名患者仅提供两个血液样品(手术前和手术后第30天)。患者特征和人口统计展示于以下表12.1中。
收集所有患者的关于手术后临床介入的信息和其它临床病理学信息,如以下表12.2中所示。研究中的所有患者都经历原发性肿瘤的切除术。
所有患者按照丹麦国家指南接受治疗和随访。研究由丹麦中部地区(CentralRegion of Denmark)的生物医学研究伦理委员会批准(1-16-02-453-14)且根据赫尔辛基声明(Declaration of Helsinki)进行。所有参与者提供书面知情同意书。
癌胚抗原(CEA)分析
使用500μL血清,在Cobas e601平台(Roche)上根据制造商说明进行CEA分析。如分析医院建议,将非吸烟者和吸烟者的阈值水平分别设定为4.0μg/L和6.0μg/L。在样品收集之前8周未抽吸者视为曾经吸烟者。
组织收集和全外显子组测序
从所有患者以新鲜冷冻(n=102)或福尔马林固定和石蜡包埋组织(FFPE)(n=27)形式收集肿瘤组织。四名患者具有同步结肠直肠癌(CRC);从这些患者收集两种肿瘤的组织。从三名具有复发的患者收集转移性组织。从末梢血液白细胞提取匹配所有患者的组成DNA。
原发性新鲜冷冻或福尔马林固定石蜡包埋(FFPE)组织样品的中值病理学肿瘤细胞性是50%(范围是20-90%)(附录,表2)。使用
对相匹配的肿瘤DNA和白细胞层DNA进行全外显子组测序(WES)。样品和WES信息的概述展示于以下表12.5中。由在组织用于提取之前和之后切割的组织切片的H&E评估来评估癌症含量。同步CRC在表中标记有S1和S2。
如Lamy等人,《配对外显子组分析揭示尿道上皮癌中的克隆演化和潜在治疗目标(Paired Exome Analysis Reveals Clonal Evolution and Potential TherapeuticTargets in Urothelial Carcinoma)》,《癌症研究》76(19):5894-5906(2016)中所描述进行库制备、测序和数据分析。
血液收集和血浆分离
在奥尔胡斯大学医院,在K2-EDTA 10ml管状(BD 367525)中收集血液样品。在收集后2小时内,通过在室温下的血液双重离心来处理所有样品,首先在3000g下离心10分钟,接着在3000g下进行血浆离心保持10分钟。将血浆等分到5mL冷冻管中且在-80℃下储存。
游离DNA提取、定量和库制备
本研究中每种情况使用最多10ml血浆(范围:2-10mL;中值:8.5mL)且使用
使用来自每个血浆样品的最多66ng(20,000基因组当量)cfDNA作为用于库制备的输入物。对cfDNA进行末端修复、添加A尾且与定制衔接子接合,如Abbosh等人,《系统发生ctDNA分析描绘早期肺癌演化(Phylogenetic ctDNA analysis depicts early-stagelung cancer evolution)》,《自然》545(7655):446-451(2017)中所描述。将被纯化的接合产物扩增20个循环且使用
多重PCR分析法设计
通过分析所有患者的原发性肿瘤和相匹配的正常WES样品来鉴别患者特异性体细胞变异体。基于所估计的具有变异体的癌细胞的比例来推断变异体的克隆性。应注意,由于变异体等位基因出现率的极平坦分布,来自具有低肿瘤细胞分数的样品的克隆性推断是有限的。使用在变异体的组织和序列上下文中观察到的VAF将每个肿瘤的所鉴别的体细胞SNV和短INDEL按优先级排序。使用实体瘤患者定制化监测技术扩增子设计流水线产生用于既定变异体集合的PCR引物对。对于每名患者,选择16种高等级相容扩增子用于定制患者特异性集合。由整合式DNA技术将PCR引物排序。
血浆多重PCR下一代测序工作流程
使用每个库的等分试样作为相关患者特异性16重PCR反应的输入物。使用患者特异性分析法扩增样品且加注条形码,接着汇集产物。使用Illumina Paired End v2试剂盒,由
生物信息流水线
使用如Zhang,《生物信息学》30(5):614-620(2014)中所描述的Pear软件合并所有双端读段。滤除在正向和后向读段中不匹配或具有低质量分数的碱基以最大限度地减少测序误差。用Novoalign版本2.3.4(http://www.novocraft.com/)将合并的读段映射到hg19参考基因组。具有<5,000个高质量读段的扩增子视为测序质量控制(QC)不合格。针对每个样品的广泛统计数据清单,使用内部程序检验来进行QC,所述清单包括读段总数、映射的读段、中靶读段、不合格目标数和平均误差率。
血浆变异体识别
预先处理阴性对照样品(约1000个)的大型集合以构建背景误差模型。对于使用突变体和参考等位基因读段深度的每个目标变异体,基于误差模型计算置信度评分,如Abbosh等人,《系统发生ctDNA分析描绘早期肺癌演化》,《自然》545(7655):446-451(2017)中所描述。具有至少2个变异体且置信度评分高于预定算法阈值的血浆样品定义为ctDNA阳性,如Abbosh等人,2017,见上文中所描述。
统计分析
原发性结果测量值是由标准放射性准则评估的复发时间(TTR)。从手术日期到所记录的第一次放射性复发(局部或远端)或由结肠直肠癌引起的死亡测量TTR且在最后一次随访或非结肠直肠癌相关死亡时检查。使用卡普兰-迈耶方法进行存活率分析。使用考克斯比例风险回归评估ctDNA和CEA对TTR的影响。用在单变量分析中统计显著的临床参数进行多变量分析。AllP值是基于双侧检验且差异在P≤0.05时视为显著。使用用于Windows的STATA IC/12.1和R统计软件,版本2.4进行统计分析。
结果
在2014-2016年之间募集一百三十名UICC I到III期CRC患者。接着排除五名患者,因为其失访(n=3)或重新归类为IV期。使用肿瘤和相匹配的生殖系DNA的全外显子组测序(WES)鉴别体细胞突变,如图90A-B中所示。针对每名患者,设计靶向16种突变的肿瘤特异性多重PCR分析法集合。使用基于超深多重PCR的NGS分析和定量来自125名患者的795个血浆样品中的循环肿瘤DNA,其中中值随访是12.5个月(范围:1.4到38.5个月)。本研究的工作流程展示于图83A-E中。在工作流程中的每个步骤中进行质量控制。覆盖率质量控制合格的分析法的读段深度是>105,000X,如图91中所示。关于所有125名患者的ctDNA结果和动力学的详细信息列举于表12.6中且展示于图92中。在患者随访周期期间,24名(19.2%)患者经历放射性复发。
手术前检测ctDNA
在基线血浆样品(n=122)中,在89%的样品中检测到ctDNA,其中I、II和III期中的敏感性分别是40%、92%和90%,如图87A中所示。对相同样品进行癌胚抗原(CEA)分析,检测到43.3%的癌症,如图88中所示。
第30天时的手术后状态预测复发
为了评估检测残留病和预测未来复发的能力,进行手术后收集的血浆样品的ctDNA分析。在第30天(开始辅助化学疗法之前),可以收集94名患者血浆。有趣的是,绝大部分(89.4%)患者是ctDNA阴性,且仅10.6%的患者在手术之后是ctDNA阳性,如图89中所示。与在手术之后是ctDNA阴性的患者(11.9%,10/84)相比,这些ctDNA阳性患者具有显著更高的复发率(70%,7/10),如图87B中所示。ctDNA的存在与相比于ctDNA阴性患者显著缩短的复发时间(TTR)相关联(HR,7.2;95%CI,2.7-19;P<0.0000),如图87C中所示。与已知的预后因子(如阶段和淋巴管侵袭)相比,在多变量逻辑回归模型中,ctDNA状态是唯一的显著预后因子,如以下表12.7中所示。
用ACT治疗患者的子集(n=52),其可以改变ctDNA对结果的预后值。然而,即使对于这一子集,ctDNA阳性仍与高复发风险相关联(HR,7.1;95%CI 2.2-22;P=0.0008),如图90A-B中所示。ctDNA阴性患者的复发率是11.9%,与其用ACT(5/42)或未用ACT(5/42)治疗无关。总之,即使对于用ACT治疗的患者,手术后第30天时的ctDNA状态仍是未来复发的强预测因子。
辅助化学疗法消除第30天ctDNA阳性患者的子集中的ctDNA
尽管随机化研究已证实辅助化学疗法(ACT)可以降低III期CRC 21-24的整体复发率,但当前未知ACT是否可以特定地预防高风险ctDNA阳性子集中的复发。十名在第30天时呈ctDNA阳性的患者接着都用ACT治疗,如图87D中所示。在这些接着用ACT治疗的患者中,70%(n=7)复发性,而30%(n=3)在随访周期结束时仍不具有疾病。这一治疗功效与在向所有III期结肠癌症给予ACT时所估计的类似,如Upadhyay等人,《化学疗法在III期结肠癌中的使用:国家癌症数据库分析(Chemotherapy use in stage III colon cancer:aNational Cancer Database analysis)》,《医学肿瘤学的治疗进展(Ther Adv MedOncol.)》7(5):244-251(2015);André等人,《MOSAIC试验中使用奥沙利铂、氟尿嘧啶和甲酰四氢叶酸作为II期或III期结肠癌的辅助治疗实现改善的总存活率(Improved overallsurvival with oxaliplatin,fluorouracil,and leucovorin as adjuvant treatmentin stage II or III colon cancer in the MOSAIC trial)》,《临床肿瘤学杂志(J ClinOncol.)》27(19):3109-3116(2009);Gill等人,《用于II期和III期结肠癌的基于氟尿嘧啶的辅助疗法的综合分析:谁将受益和如何受益?(Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer:who benefits and byhow much?)》,《临床肿瘤学杂志》22(10):1797-1806(2004);Haller等人,《卡培他滨加奥沙利铂与氟尿嘧啶和亚叶酸相比作为III期结肠癌的辅助疗法(Capecitabine plusoxaliplatin compared with fluorouracil and folinic acid as adjuvant therapyfor stage III colon cancer)》,《临床肿瘤学杂志》29(11):1465-1471(2011)中所描述。因此,本文中呈现在图87D中呈现的结果,表明ACT可以消除高风险ctDNA阳性患者子集中的残留病。
对于三名未复发患者中的两名,可以纵向收集血浆样品。与ACT消除残疾病一致,这些患者在疗法期间显示ctDNA的完全清除且在研究持续时间内保持ctDNA阴性。相比之下,可以获得纵向血浆的六名复发患者在ACT期间保持ctDNA阳性或在ACT完成之后立即又变成ctDNA阳性。
纵向ctDNA监测可以测量ACT治疗的功效
在开始ACT之前,可以从8/10名ctDNA阳性患者纵向收集血液样品。分析这些纵向收集的血液样品以观察在治疗期间ctDNA含量的变化。在50%的患者(n=4)中,ctDNA状态变成阴性,如图87D中所示,而在另外四名患者中,ctDNA状态在治疗期间保持阳性。引人注目的是,所有四名未清除ctDNA的患者(100%)都经历疾病复发,表明残留ctDNA预示ACT未能消除残留病。在治疗期间清除ctDNA的四名患者中,两名在所有ACT后样品中保持ctDNA阴性且始终未复发,而另两名患者在治疗之后立即恢复ctDNA阳性且最终复发,如图87D中所示。
ACT后的ctDNA检测定义具有极高复发风险的患者子组
因为100%的在辅助化学疗法(ACT)期间未清除ctDNA的患者随后经历疾病复发,我们假设在ACT之后抽取的第一血液样品的ctDNA分析可以用于鉴别具有持续残留病的患者的子组,其将受益于进一步治疗。我们发现在58名具有ACT后血液样品的患者中,所有ctDNA阳性患者(7/7)都复发。相比之下,ctDNA阴性患者的复发率是13.7%(7/51)(费舍尔精确检验(Fisher exact test),P<0.0001),如图87E中所示。与其它预测性因子(如阶段、淋巴管侵袭、微观根治性切除术状态和CEA)相比,ACT后ctDNA状态是初始复发的更有效的预测因子,如以下表12.8中所示,且ctDNA状态复发时间(TTR)的高显著性预测因子(HR,18.0;95%CI,5.4-57;P<0.0000),如图87F中所示。
包括所有ACT后血液样品的纵向ctDNA分析是复发时间的更有效的预测因子(HR,29.0;95%CI,6.4-130;p<0.0000),且鉴别13名ctDNA阳性患者,其中92.3%(12/13)的患者复发,如图91中所示。尽管,纵向癌胚抗原(CEA)分析也是复发时间的显著预测因子,如图92中所示,但在多变量调节后,纵向ctDNA状态是复发时间的唯一的显著预测因子(HR,26.9;95%CI,5.11-142,;P=0.0001),如以下表12.9中所示。
纵向ctDNA分析预测患者结果且实现复发的早期检测
在75名具有纵向收集的血浆样品的患者中,在确定性治疗之后的监视期间的连续ctDNA分析鉴别转移性复发,其中敏感性是87.5%(14/16)且特异性是98.3%(58/59)。引人注目的是,93.3%(14/15)的ctDNA阳性患者复发,相比之下,ctDNA阴性患者的复发率仅是3.3%(2/60)(费舍尔精确检验,P<0.0001)。ctDNA阳性患者具有显著缩短的复发时间(TTR)(HR,44,0;95%CI,9.8-190;P<0.0000),如图93A-B中所示。所有75名患者的疾病病程和纵向ctDNA结果展示于图94中。连续ctDNA分析遗失两个复发事件(患者20和24,图94)。然而,两个遗失的癌转移的全外显子组测序证实存在用于血浆筛检的突变,如以下表12.10中所示。
对于两名复发患者(ID 20和24,表12.10),纵向分析未检测到手术后ctDNA,如图90A-B中所示。对于这两名患者,分析与其它患者相同量的血浆。归因于所有肿瘤和血浆样品中的45个通用SNP,可以拒绝可能的样品调换,其证实未进行样品调换。接着,进行两名患者的转移性复发病灶的全外显子组测序(WES)且证实癌转移中存在选择用于血浆分布分析的突变,表12.9。还对来自患者77的癌转移进行WES。对于这名患者,在通过放射性成像检测到复发之后,ctDNA纵向分析才检测到ctDNA,如图90A-B中所示。WES再次证实转移中存在选择用于血浆分布分析的突变。总之,阴性手术后结果是由ctDNA含量低于检测水平引起而不是由所选择的标记物是非信息性引起。
这一相同群体的纵向CEA分析鉴别复发,其中敏感性是68.8%(11/16)且特异性是64.4%(38/59),如图95中所示。在多变量分析中,ctDNA是复发时间(TTR)的唯一显著预测因子(HR,41;,95%CI,8.5-199;P<0.0000),如以下表12.11中所示。
对于具有转移性复发和可检测的ctDNA的患者,发现ctDNA分析与标准护理CT成像相比的平均前置时间是8.7个月(威尔科克森符号秩检验;P=0.0009),如图93C中所示;然而,使用癌胚抗原(CEA)分析不能建立前置时间,如图96中所示。从ctDNA检测开始且直到放射性复发检测,血浆样品保持ctDNA阳性,且观察到平均ctDNA VAF增加50倍,表明肿瘤负荷在患者等待复发的放射性检测时显著增加,如图93D中所示。
ctDNA分析显示临床上可操作的突变
已证实纵向ctDNA分析实现微转移疾病的早期检测,我们随后研究纵向ctDNA分析是否可以用于获得关于癌转移中存在的潜在地可操作突变的信息。
由可用的纵向样品鉴别11名具有转移性复发的患者,且通过进行原发性肿瘤全外显子组测序(WES)来鉴别临床上可操作突变,如以下表12.12中所示。
作为概念验证,设计其它靶向可操作突变的多重PCR集合且应用于纵向样品。在82%(9/11)的患者中检测到可操作突变,如图97A中所示。观察到平均ctDNA VAF与可操作突变的VAF之间的良好相关性,如图97B中所示。可操作变异体等位基因出现率(VAF)中的纵向变化通常展示与治疗的良好相关性极少的突变间变异,如图97C中所示。
讨论
本实例证明I-III期CRC患者中的纵向ctDNA分析可以有效地检测和监测整个临床疾病病程中的肿瘤负荷的变化。具体地说,其证明ctDNA充当稳定生物标记物以用于i)手术前CRC检测,ii)手术后和ACT后风险分级,iii)监测ACT功效,iv)检测临床可操作突变,和v)复发的早期检测。这些观察结果具有重要和潜在范例,其改变CRC患者的未来手术后管理的影响且成为用于研究ctDNA指导的管理的临床益处的未来介入试验的基础。
在手术前情形中,证明手术前ctDNA测量结果可以适用于疾病检测。
在患者分级方面,本文中使用的ctDNA分析将患者分为高复发风险组和低复发风险组,其潜在暗示用于辅助化学疗法(ACT)治疗和其它ACT后治疗决定的患者选择。先前,关于ACT治疗作出的决定是基于分期和临床风险因子。然而,本文中的公开内容证实与阶段、CEA和其它高风险特征相比,ctDNA状态是更有效的预后因子。因此,将来有可能基于ctDNA分析指定ctDNA阳性,但临床上低风险(I期和II期)患者(根据现在的标准护理,其将不接受ACT)进行ACT治疗。本发明人当前正在进行试验以在这种情形下,评估基于ctDNA的患者选择的临床受益(例如IMPROVE-IT ClinicalTrials.gov:NCT03748680和动态澳大利亚新西兰临床试验注册表(DYNAMIC Australian New Zealand Clinical Trials registry):ACTRN12615000381583)。
本公开还证明ctDNA阴性患者具有低复发风险,与给予ACT(11.9%)或不给予ACT(11.9%)无关。因此,将来有可能在对复发风险影响最小的情况下,取消ctDNA阴性,但临床上高风险(III期)患者的ACT。可以向这一患者组提供基于活性ctDNA的监视代替ACT,因此使许多仅由手术便治愈的患者避免化学疗法的毒性。此外,在其中不存在当前预后标记物的ACT后情形中,我们证明ctDNA分析鉴别仍具有残留病的患者。这一群体可以受益于强化治疗性治疗。
本公开还证明在ACT之前、期间和之后的纵向ctDNA监测可以提供ACT功效的患者水平测量结果。30%的清除ctDNA且在所有后续样品中保持阴性的患者在整个研究期间保持不具有疾病。因此,本实例提供ACT可以降低ctDNA阳性患者中的复发风险的第一线证据。本公开还证明,所有未清除ctDNA的患者在ACT完成后一年内复发,且所有仅具有短暂清除的患者也复发。在研究设计中合并有ctDNA清除的未来临床试验可以实现疗法功效的患者水平实时测量。
在手术后情形中,与标准护理放射性成像相比,ctDNA监测展示复发检测的显著改善,显示8.7个月的显著前置时间(P<0.001)。重要的是,在等待放射性检测时,ctDNA含量平均增加50倍,表明肿瘤负荷在8.7个月的前置时间期间显著增加。当前指南建议在治愈性CRC手术之后进行监视,但过晚地检测到大部分复发事件以至于不符合治愈性介入的条件。由ctDNA分析进行的残留病的早期检测可以提供更早的放射性检测的机会。除检测残留病以外,ctDNA分析还实现临床上可操作突变的鉴别。因此,ctDNA具有实现早期检测和指导治疗决定的潜力。
总之,本实例中的公开内容提供潜在范例,改变ctDNA在结肠直肠癌中的临床应用。如前所述,正在或已经设计其它临床试验以研究ctDNA指导的管理的临床益处。本文中所提供的结果能够将循环生物标记物用于个人化风险分级和疗法监测,以确保在正确的时间向正确的患者给予正确的治疗且保持正确的持续时间。
实例11.全外显子组血浆cfDNA分布分析捕获用于监测疾病演化的临床前复发的突变标签
本实例的目的是评估血浆游离DNA的全外显子组测序(cfDNA-WES)用于研究晚期癌症患者或具有高循环肿瘤DNA(ctDNA)负荷的患者中的突变标签和克隆演化的用途。具体地说,本文中说明cfDNA-WES分布分析用于检测原发性乳癌患者中的临床前转移的用途。
方法
在手术和辅助疗法后募集四十九名原发性乳癌患者。每六个月一次收集连续血浆样品以用于ctDNA分析,所述ctDNA分析通过超深测序和Natera实体瘤患者定制化监测技术工作流程使用靶向16种变异体的患者特异性分析法。在临床复发之前和临床复发时间附近对来自所有17名复发患者的血浆cfDNA进行全外显子组测序,以确定在血浆和肿瘤活检中鉴别的变异体之间的和谐性,且了解在疾病进展期间的肿瘤演化。
结果
来自3名复发患者的cfDNA-WES分布的基本分析展示在肿瘤活检和血浆中鉴别的患者特异性变异体之间的高度和谐性。还由血浆WES鉴别到35种实体瘤患者定制化监测技术检测的变异体中的34种且展示高度一致的变异体等位基因出现率(VAF)。一种未由cfDNA-WES检测到的变异体是先前由实体瘤患者定制化监测技术在0.2%VAF下检测到。
结论
本实例证实血浆WES可以检测原发性乳癌患者中的分子残留病。WES血浆的分析可能提供癌症演化的证据,其对于作出治疗决定来说可能是重要的。
实例12.使用循环肿瘤DNA(ctDNA)分子生物标记物以用于评估淋巴瘤中的治疗反应
本文中,在生物医药试点研究中,我们评估个人化、肿瘤特异性、基于多重PCR NGS的方法(Signatera
方法
从8名非霍奇金淋巴瘤(NHL)(6名弥漫性大型B细胞淋巴瘤和2名滤泡性淋巴瘤)患者(pts)收集的血液样品可以用于ctDNA分析。通过分析来自原发性肿瘤活检和相匹配的正常样品的全外显子组测序(WES)数据来鉴别患者特异性体细胞变异体。接着,使用实体瘤患者定制化监测技术工作流程,由相应的定制16重分析法以盲式分析血浆样品。如果至少两个患者特异性目标符合鉴定置信度评分阈值,那么样品视为ctDNA。
结果
对于非霍奇金淋巴瘤(NHL),从2.5mL(中值)血浆提取14.9ng(中值,范围:2.25-685ng)cfDNA。由4名患者在5个血浆时间点时检测ctDNA。在5个ctDNA+血浆样品中,4个血浆样品在血液收集时与临床进行性疾病或对疗法的部分反应相关。在任何时间点都不具有ctDNA检测的4名患者在血液收集时显示临床完全反应。
结论
可以将可扩展的患者特异性ctDNA监测分析法应用于基线检测、疗法监测和复发检测。实体瘤患者定制化监测技术的高敏感性ctDNA检测分析提供优于当前护理标准的非侵袭性监测手段。
- 用于借助于循环肿瘤DNA的个人化检测的癌症检测和监测的方法
- 一种用于评估先天肤质的五大基因组合的检测方法及根据检测结果制定的个人化护肤套盒