菲啶类衍生物及其制备方法和治疗白癜风药物
文献发布时间:2023-06-19 11:19:16
技术领域
本发明涉及医药技术领域,尤其涉及菲啶类衍生物及其制备方法和治疗白癜风药物。
背景技术
白癜风(vitiligo)是一种常见的皮肤色素脱失性疾病,由皮肤和/或毛囊的功能性黑素细胞的减少或丧失引起,主要症状为大面积的黑素缺失带来的局部或者泛发性皮肤白斑出现。世界卫生组织统计显示,白癜风在全球各国患病率为0.5~2%之间,在新疆、甘肃等省区发病率较高,患者多集中于青少年,无性别差异,春夏季高发。白癜风根据白斑分布方式可以分为寻常型与节段型,而寻常型又可进一步分为局限性、散在性、泛发性及肢端性四种,其中,散在性占白癜风患者总数的80%以上;此外,根据皮损区黑素细胞分布情况,白癜风又可分为完全性白斑与不完全性白斑两类,其中完全性白斑皮损处组织内黑素细胞消失,促黑色素生成药物对其无效。由于白癜风严重影响患者的外观和正常的社交活动,因此,对于白癜风治疗的研究具有十分重要的意义。
目前白癜风具体发病机制尚不清楚,可能致病机制的解释有多种,包括自身免疫学说、氧化应激学说、黑素细胞自身破坏学说、神经化学因子学说以及遗传学说等。由于潜在的发病机理是多方面的,因此应对白癜风的治疗手段也比较复杂多样,而如何有效增加皮损区黑素细胞的数量及黑色素合成是治疗白癜风的重要研究方向。目前,临床治疗白癜风并没有特效药物。
发明内容
本发明的目的在于提供一种菲啶类衍生物及其制备方法和治疗白癜风药物,本发明提供的菲啶类衍生物具有显著的抗白癜风作用。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种菲啶类衍生物,具有式I所示结构:
式I中,m=1、2或3;
n=1、2或3;
R
R
R
R
所述的取代是指基团中的一个或多个-H被下列至少一种取代基所取代:卤素基团、-OH、-CN、-NH
优选地,所述式I中,m=1或2,n=1或2;
R
R
R
优选地,所述式I中,m=1,n=1或2;
R
R
R
优选地,所述R
优选地,所述R
优选地,所述菲啶类衍生物为式I-1~式I-5所示结构化合物中的任意一种:
本发明提供了上述技术方案所述菲啶类衍生物的制备方法,包括以下步骤:
将化合物1、四氢呋喃、N,N-二甲基甲酰胺与氯化亚砜混合后进行氯化反应,去除所得氯化反应产物体系中的溶剂后与NH
在保护气氛条件下,将所述化合物2、碘苯类化合物、醋酸钯、6-四氟邻苯二甲酸、碳酸钾、乙腈、降冰片烯与水混合,进行偶联反应,得到化合物3;
将所述化合物3、四氢呋喃与硼烷混合,进行酰胺还原反应,得到化合物4;
将所述化合物4、二氯甲烷与BBr
将所述化合物5、NaH、化合物6与四氢呋喃混合,进行醚化反应,得到具有式I所示结构的菲啶类衍生物;
其中,所述化合物1的结构式为
所述碘苯类化合物的结构式为
所述化合物6的结构式为R
优选地,所述氯化反应的温度为45~55℃,时间为1.5~2.5h;
所述酰氯胺解反应的温度为3~7℃,时间为50~70min;
所述偶联反应的温度为80~90℃,时间为5~7h;
所述酰胺还原反应的温度为-70~-80℃,时间为1.5~2.5h;
所述脱保护基反应的温度为-70~-80℃,时间为3.5~4.5h;
所述醚化反应的温度为60~100℃,时间为6~18h。
本发明提供了一种治疗白癜风药物,包括药学上可接受的载体和有效成分,所述有效成分为上述技术方案所述菲啶类衍生物和/或其药学上可接受的盐。
优选地,所述有效成分的质量含量为0.1~99.5%。
本发明提供了一种菲啶类衍生物,本发明提供的菲啶类衍生物具有显著的抗白癜风作用。实施例的结果显示,本发明提供的菲啶类衍生物具有显著的体外黑色素生成上调活性,有进一步应用于制备治疗白癜风药物的潜力。
附图说明
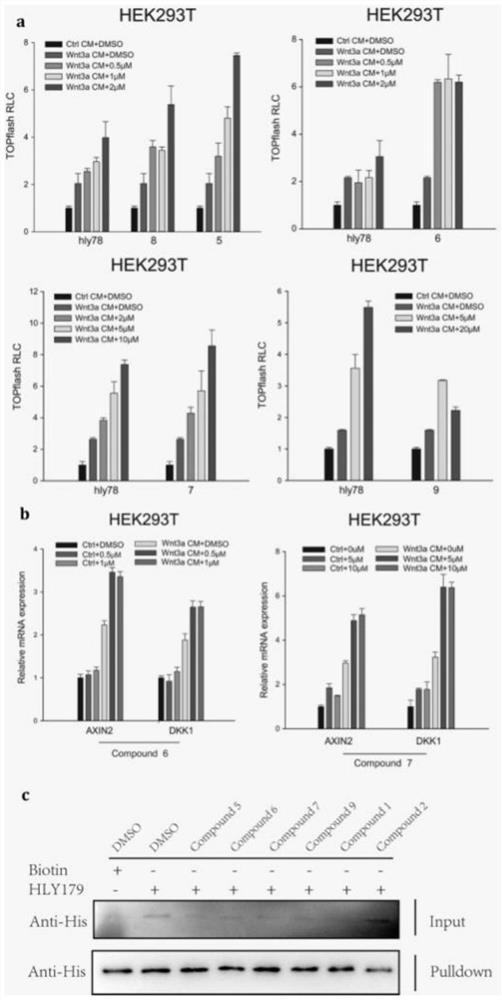
图1为实施例1~5制备的化合物的Wnt信号通路激动活性测试结果图。
具体实施方式
本发明提供了一种菲啶类衍生物,具有式I所示结构:
式I中,m=1、2或3;
n=1、2或3;
R
R
R
R
所述的取代是指基团中的一个或多个-H被下列至少一种取代基所取代:卤素基团、-OH、-CN、-NH
在本发明中,所述卤素基团优选包括-F、-Cl、-Br或-I。
在本发明中,所述式I中,m优选为1或2,更优选为1;n优选为1或2。
在本发明中,R
在本发明中,R
在本发明中,R
在本发明中,所述菲啶类衍生物优选为光学异构体或外消旋体。
在本发明中,所述菲啶类衍生物优选为式I-1~式I-5所示结构化合物中的任意一种,菲啶类衍生物中8、9位吡啶或氮唑环取代化合物具有显著抗白癜风活性:
本发明提供了上述技术方案所述菲啶类衍生物的制备方法,包括以下步骤:
将化合物1、四氢呋喃、N,N-二甲基甲酰胺与氯化亚砜混合后进行氯化反应,去除所得氯化反应产物体系中的溶剂后与NH
在保护气氛条件下,将所述化合物2、碘苯类化合物、醋酸钯、6-四氟邻苯二甲酸、碳酸钾、乙腈、降冰片烯与水混合,进行偶联反应,得到化合物3;
将所述化合物3、四氢呋喃与硼烷混合,进行酰胺还原反应,得到化合物4;
将所述化合物4、二氯甲烷与BBr
将所述化合物5、NaH、化合物6与四氢呋喃混合,进行醚化反应,得到具有式I所示结构的菲啶类衍生物;
其中,所述化合物1的结构式为
所述碘苯类化合物的结构式为
所述化合物6的结构式为R
本发明将化合物1、四氢呋喃、N,N-二甲基甲酰胺与氯化亚砜混合后进行氯化反应,去除所得氯化反应产物体系中的溶剂后与NH
在本发明中,所述化合物1、四氢呋喃(THF)、N,N-二甲基甲酰胺(DMF)与氯化亚砜的用量比优选为1mmol:(8~12)mL:(0.08~0.12)mL:(0.4~0.6)mL,更优选为1mmol:10mL:0.1mL:0.5mL。在本发明中,所述氯化反应的温度优选为45~55℃,更优选为50℃;时间优选为1.5~2.5h,更优选为2h;所述氯化反应优选在搅拌条件下进行。
氯化反应完成后,本发明优选通过减压蒸馏去除氯化反应产物体系中多余的溶剂,然后与NH
完成所述酰氯胺解反应后,本发明优选将所得酰氯胺解反应产物体系进行过滤,将滤饼用柱色谱纯化,得到化合物2。在本发明中,所述柱色谱纯化所用洗脱液优选为二氯甲烷-甲醇混合液,所述二氯甲烷和甲醇的体积比优选为100:1。
得到化合物2后,本发明在保护气氛条件下,将所述化合物2、碘苯类化合物、醋酸钯、6-四氟邻苯二甲酸(TFPA)、碳酸钾、乙腈、降冰片烯与水混合,进行偶联反应,得到化合物3;所述化合物3的结构式如下所示:
本发明对于提供所述保护气氛的保护气体种类没有特殊的限定,采用本领域技术人员熟知的保护气体即可,具体如氮气。在本发明中,所述化合物2、碘苯类化合物、醋酸钯、6-四氟邻苯二甲酸、碳酸钾和降冰片烯的用量比优选为0.1mmol:(0.24~0.28)mmol:(0.010~0.015)mmol:(0.025~0.030)mmol:(0.50~0.55)mmol:(0.28~0.29)mmol,更优选为0.1mmol:0.26mmol:0.013mmol:0.027mmol:0.52mmol:0.286mmol。本发明优选将化合物2、碘苯类化合物、醋酸钯、6-四氟邻苯二甲酸和碳酸钾混合,然后采用乙腈将所得混合物料溶解,之后再与降冰片烯的水溶液混合。本发明对于所述乙腈和水的用量没有特殊的限定,能够保证物料充分溶解并使偶联反应顺利进行即可。
在本发明中,所述偶联反应的温度优选为80~90℃,更优选为85℃;时间优选为5~7h,更优选为6h;所述偶联反应优选在搅拌条件下进行。
完成所述偶联反应后,本发明优选将所得偶联反应产物体系冷却至室温,加入饱和氯化铵溶液,之后用乙酸乙酯萃取,有机层利用无水硫酸钠干燥后进行浓缩,残余物用柱色谱纯化,得到化合物3。在本发明中,所述柱色谱纯化所用试剂优选为二氯甲烷-甲醇混合液,所述二氯甲烷和甲醇的体积比优选为100:1。
得到化合物3后,本发明将所述化合物3、四氢呋喃与硼烷混合,进行酰胺还原反应,得到化合物4;所述化合物4的结构式如下所示:
本发明优选将化合物3溶于四氢呋喃中,得到化合物3的四氢呋喃溶液,然后在-70~-80℃条件下将所述化合物3的四氢呋喃溶液与硼烷的四氢呋喃溶液混合。本发明对于所述四氢呋喃的用量没有特殊的限定,能够保证物料充分溶解并使酰胺还原反应顺利进行即可。
在本发明中,所述酰胺还原反应的温度优选为-70~-80℃,更优选为-78℃;时间优选为1.5~2.5h,更优选为2h;所述酰胺还原反应优选在搅拌条件下进行。
完成所述酰胺还原反应后,本发明优选向所得酰胺还原反应产物体系中加水淬灭,将所得混合物用乙醚萃取,有机层以饱和食盐水洗涤后进行浓缩,残余物用柱色谱纯化,得到化合物4。在本发明中,所述柱色谱纯化所用试剂优选为二氯甲烷-甲醇混合液,所述二氯甲烷和甲醇的体积比优选为100:1。
得到化合物4后,本发明将所述化合物4、二氯甲烷与BBr
在本发明中,所述化合物4、二氯甲烷与BBr
在本发明中,所述脱保护基反应的温度优选为-70~-80℃,更优选为-78℃;脱保护基反应的时间优选为3.5~4.5h,更优选为4h,本发明所述脱保护基反应的时间从BBr
完成所述脱保护基反应后,本发明优选向所得脱保护基反应产物体系中加入饱和碳酸氢钠溶液稀释,将所得混合物用二氯甲烷萃取,有机层浓缩后进行柱色谱纯化,得到化合物5。在本发明中,所述柱色谱纯化所用试剂优选为二氯甲烷-甲醇混合液,所述二氯甲烷和甲醇的体积比优选为100:1。
得到化合物5后,本发明将所述化合物5、NaH、化合物6与四氢呋喃混合,进行醚化反应,得到具有式I所示结构的菲啶类衍生物。在本发明中,所述化合物6中卤素基团优选包括-F、-Cl、-Br或-I。在本发明中,所述化合物5、NaH、化合物6与四氢呋喃的用量比优选为0.1mmol:(1.8~2.2)mmol:(0.8~1.2)mmol:(8~12)mL,更优选为0.1mmol:2mmol:1mmol:10mL。
在本发明中,所述醚化反应的温度优选为60~100℃,更优选为60~80℃;时间优选为6~18h,更优选为6~12h;所述醚化反应优选在搅拌条件下进行。
完成所述醚化反应后,本发明优选向所得醚化反应产物体系中加水淬灭反应,将所得混合物浓缩蒸去多余溶剂,之后用二氯甲烷萃取,有机层以饱和碳酸氢钠溶液与饱和食盐水依次洗涤后浓缩,残余物用柱色谱纯化,得到具有式I所示结构的菲啶类衍生物。在本发明中,所述柱色谱纯化所用试剂优选为二氯甲烷-甲醇混合液,所述二氯甲烷和甲醇的体积比优选为100:1。
在本发明中,制备具有式I所示结构的菲啶类衍生物的反应流程具体如下:
其中,(a)SOCl
本发明提供了一种治疗白癜风药物,包括药学上可接受的载体和有效成分,所述有效成分为上述技术方案所述菲啶类衍生物和/或其药学上可接受的盐。在本发明中,所述有效成分的质量含量优选为0.1~99.5%,更优选为0.5~90%。本发明对于所述药学上可接受的载体的种类没有特殊的限定,具体的,可以为乳糖、淀粉、低取代羟丙基纤维素、微晶纤维素、硬脂酸镁和羟甲基纤维素钠中的一种或几种。本发明对于所述治疗白癜风药物的剂型没有特殊的限定,采用制药和食品领域公认的方法制备成各种剂型均可,如液体制剂(包括注射剂、混悬剂、乳剂、溶液剂或糖浆剂等)、固体制剂(包括片剂、胶囊剂、颗粒剂或冲剂等)、喷剂、气雾剂等。本发明提供的治疗白癜风药物可经注射(包括静脉注射、静脉滴注、肌肉注射、腹腔注射或皮下注射)、口服、舌下给药、粘膜透析等给药途径治疗白癜风。
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
(1)将化合物1(具体为2-溴-5-甲氧基苯甲酸,260mg,1mmol)溶于四氢呋喃(10mL)中,然后加入N,N-二甲基甲酰胺(0.1mL)与氯化亚砜(0.5mL,4mmol),将所得反应液在搅拌、50℃条件下反应2h;反应完成后减压蒸去多余的溶剂,在5℃条件下,将残余物逐滴加入到质量浓度为30%的乙胺水溶液(乙胺含量为10mmol)中,继续搅拌1h后过滤,将滤饼用柱色谱纯化(洗脱液为二氯甲烷-甲醇混合液,所述二氯甲烷和甲醇的体积比为100:1),得到化合物2(收率75%);
(2)在氮气保护条件下,将醋酸钯(3.0mg,0.013mmol)、6-四氟邻苯二甲酸(6.2mg,0.027mmol)、无水碳酸钾(72.3mg,0.52mmol),化合物2(0.1mmol)及2-乙基碘苯(0.26mmol)加入到圆底烧瓶中,加入无水乙腈溶解后,再加入降冰片烯水溶液(含降冰片烯0.286mmol),将所得反应液在搅拌、85℃条件下反应6h;反应完成后冷却至室温,加入饱和氯化铵溶液(30mL),之后用乙酸乙酯(3×15mL)萃取,有机层利用无水硫酸钠干燥后进行浓缩,残余物用柱色谱纯化(洗脱液为二氯甲烷-甲醇混合液,所述二氯甲烷和甲醇的体积比为100:1),得到化合物3(收率75%);
(3)将化合物3(30mg,0.1mmol)溶于四氢呋喃(5mL)中,在-78℃条件下加入硼烷的四氢呋喃溶液(20mg,硼烷的浓度为1mol/L),将所得反应液在搅拌、-78℃条件下反应2h;反应完成后加水(50mL)淬灭,将所得混合物用乙醚(2×20mL)萃取,有机层以饱和食盐水洗涤后进行浓缩,残余物用柱色谱纯化(洗脱液为二氯甲烷-甲醇混合液,所述二氯甲烷和甲醇的体积比为100:1),得到化合物4(收率75%);
(4)将化合物4(29mg,0.1mmol)溶于二氯甲烷中,冷却至-78℃后滴加BBr
(5)将化合物5(0.1mmol)溶解于四氢呋喃(10mL)中,加入NaH(50mg,2mmol)与化合物6(R
化合物6中R
目标化合物的英文名称为:
8-((1,3-dimethyl-1H-pyrazol-5-yl)methoxy)-5-ethyl-4-methyl-5,6-dihydro-phenanthridine,结构及表征数据具体如下:
实施例2
按照实施例1的方法制备菲啶类衍生物,不同之处在于,将化合物1替换为
最终得到无色无定形粉末,即为具有式I-2所示结构的化合物(收率75%);英文名称为:
8,9-bis((1,3-dimethyl-1H-pyrazol-5-yl)methoxy)-5-ethyl-4-methyl-5,6-dihydro-phenanthridine,结构及表征数据具体如下:
实施例3
按照实施例1的方法制备菲啶类衍生物,不同之处在于,将化合物6中R
最终得到无色无定形粉末,即为具有式I-3所示结构的化合物(收率87%);英文名称为:
8-((1,5-dimethyl-1H-pyrazol-3-yl)methoxy)-5-ethyl-4-methyl-5,6-dihydro-phenanthridine,结构及表征数据具体如下:
实施例4
按照实施例1的方法制备菲啶类衍生物,不同之处在于,将化合物6中R
最终得到无色无定形粉末,即为具有式I-4所示结构的化合物(收率85%);英文名称为:
5-ethyl-4-methyl-8-(pyridin-3-ylmethoxy)-5,6-dihydrophenanthridine,结构及表征数据具体如下:
实施例5
按照实施例1的方法制备菲啶类衍生物,不同之处在于,将化合物1替换为
最终得到无色无定形粉末,即为具有式I-5所示结构的化合物(收率76%);英文名称为:
5-ethyl-4-methyl-8,9-bis(pyridin-3-ylmethoxy)-5,6-dihydrophenanthridine,结构及表征数据具体如下:
应用例1
本发明提供的菲啶类衍生物的抗白癜风活性:
一、黑色素含量的测定
1、B16小鼠黑色素瘤细胞的准备
离心收集约10
2、除杂
向所得细胞沉淀中加入200μL纯水重悬细胞,再加入1mL乙醇/乙醚混合液(体积比为1:1),充分摇匀并于室温下静置15min;静置完成后,将所得混合物于3000rpm、5min条件下继续离心,弃上清液,细胞沉淀备用。
3、黑色素颗粒溶解与检测
向所得细胞沉淀中加入1mL质量浓度为10%的二甲基亚砜溶液(由二甲基亚砜与浓度为1mol/L的NaOH水溶液配制而成),充分振荡后于80℃条件下封口静置30min,之后于470nm波长处检测吸光度值(A);其中,黑色素含量计算具体如下:
黑色素含量=A/细胞数。
结果见表1。
表1实施例1~5制备的化合物在细胞内抗白癜风活性数据
由表1可知,实施例1~5制备的化合物具有显著的体外黑色素生成上调活性。
二、生物活性
对实施例1~5制备的化合物的Wnt信号通路激动活性进行测试,具体是采用NaOH溶解法:取对数期的B16黑素瘤细胞,弃去培养液,用PBS液清洗2次,加入培养液,加入96孔板中,每孔加入100μL。过夜后(12h),细胞贴壁,使用同MTT法同浓度同体积的含药培养基加入,并设置对照组(细胞+培养基)和空白对照组(培养基),每个浓度组设置5个复孔,培养基中均含59%胎牛血清,于37℃的59&CO
实施例6
将实施例1~5制备的化合物作为有效成分,按照表2中配方制备药物组合物片剂,具体是将实施例1~5制备的化合物与乳糖、淀粉、低取代羟丙基纤维素和微晶纤维素混合,加入1%羟甲基纤维素钠溶液适量制成软料,然后依次进行过筛制粒、湿粒烘干和过筛整粒,最后加入硬脂酸镁和滑石粉混合均匀后压片即得;每个片剂中含有实施例1~5制备的化合物成分5~60mg,其中,配方1~5分别采用实施例1~5制备的化合物,配方6采用实施例1制备的化合物,配方7采用实施例2制备的化合物。
表2药物组合物片剂的原料药和辅料配方
实施例7
将实施例1~5制备的化合物作为有效成分,按照表3中配方制备药物组合物胶囊制剂,具体是将实施例1~5制备的化合物与乳糖、淀粉和微晶纤维素混合,加入1%羟甲基纤维素钠溶液适量制成软料,然后依次进行过筛制粒、湿粒烘干和过筛整粒,最后加入硬脂酸镁混合均匀后装入胶囊壳内即得;每个胶囊制剂中含有实施例1~5制备的化合物成分5~50mg,其中,配方1~5分别采用实施例1~5制备的化合物,配方6采用实施例1制备的化合物。
表3药物组合物胶囊制剂的原料药和辅料配方
以实施例6和实施例7得到的药物组合物为药物活性成分制成制剂,每日用药剂量在5~200mg范围内,所述用药剂量具体是以制剂中药物活性成分(即实施例1~5制备的化合物)的含量计。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 菲啶类衍生物及其制备方法和治疗白癜风药物
- 菲啶类衍生物及其药物组合物和其制备方法与应用