乳腺癌治疗剂
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及一种乳腺癌治疗剂,其中组合使用具有成纤维细胞生长因子受体(Fibroblast growth factors receptor,FGFR)抑制作用的单环吡啶衍生物或其药理学上可接受的盐、及雌激素受体拮抗剂或芳香化酶抑制剂。更具体而言,本发明涉及一种乳腺癌治疗剂,其与雌激素受体拮抗剂或芳香化酶抑制剂组合施用,其中该乳腺癌治疗剂包含5-((2-(4-(1-(2-羟基乙基)哌啶-4-基)苯甲酰胺)吡啶-4-基)氧基)-6-(2-甲氧基乙氧基)-N-甲基-1H-吲哚-1-甲酰胺或其药理学上可接受的盐。
背景技术
式(I)所表示的5-((2-(4-(1-(2-羟基乙基)哌啶-4-基)苯甲酰胺)吡啶-4-基)氧基)-6-(2-甲氧基乙氧基)-N-甲基-1H-吲哚-1-甲酰胺作为对FGFR1、FGFR2、及FGFR3的抑制剂为人所知,并且报告称该化合物具有胃癌、肺癌、膀胱癌及子宫内膜癌的细胞增生抑制作用(专利文献1)。据报告,上述化合物对胆管癌(专利文献2)、乳腺癌(专利文献3)及肝细胞癌(专利文献4)表现出高治疗效果。作为上述化合物的药理学上可接受的盐,已知有琥珀酸盐及马来酸盐(专利文献5)。
雌激素受体拮抗剂如他莫昔芬(tamoxifen)和氟维司群(fulvestrant)等用于治疗雌激素受体阳性的乳腺癌(非专利文献1)。
芳香化酶抑制剂如依西美坦(exemestane)和阿那曲唑(anastrozole)等抑制芳香化酶,该芳香化酶将肾上腺皮质分泌的雄激素转化为雌激素(非专利文献2)。因此,与雌激素受体拮抗剂同样地用于治疗雌激素受体阳性的乳腺癌。一般而言,芳香化酶抑制剂用于绝经后的乳腺癌患者,而雌激素受体拮抗剂用于绝经前的乳腺癌患者。
乳腺癌根据雌激素受体、黄体酮受体及人类表皮生长因子受体2型(Humanepidermal growth factor receptor Type2,HER2)表达的有无进行分类,并结合病变部分的手术切除,根据分类进行药物疗法。
引证文献清单
专利文献
[专利文献1]美国专利公开号2014-0235614
[专利文献2]美国专利公开号2018-0015079
[专利文献3]美国专利公开号2018-0303817
[专利文献4]WO 2019/189241
[专利文献5]美国专利公开号2017-0217935
非专利文献
[非专利文献1]Howell et al.,“Comparison of Fulvestrant VersusTamoxifen for the Treatment of Advanced Breast Cancer in Postmenopausal WomenPreviously Untreated With Endocrine Therapy:A Multinational,Double-Blind,Randomized Trial”,Journal of Clinical Oncology,2004,22(9),1605-1613.
[非专利文献2]Deeks et al.,“Exemestane A Review of its Use inPostmenopausal Women with Breast Cancer”Drugs,2009,69(7),889-918
发明内容
技术问题
本发明的目的是提供一种乳腺癌治疗剂,其涉及组合施用多种药物。
问题的解决方案
鉴于这种情况,本发明的诸位发明人进行了大量研究,结果发现,将上述式(I)所表示的化合物与雌激素拮抗剂或芳香化酶抑制剂组合施用会对乳腺癌表现出高治疗效果,从而促成了本发明的实现。
即,本发明提供以下[1]至[18]。
[1]一种乳腺癌治疗剂,其包含:式(I)所表示的5-((2-(4-(1-(2-羟基乙基)哌啶-4-基)苯甲酰胺)吡啶-4-基)氧基)-6-(2-甲氧基乙氧基)-N-甲基-1H-吲哚-1-甲酰胺、
或其药理学上可接受的盐,该化合物或其药理学上可接受的盐与雌激素受体拮抗剂或芳香化酶抑制剂组合施用。
[2]一种乳腺癌治疗用药物组合物,其包含:式(I)所表示的5-((2-(4-(1-(2-羟基乙基)哌啶-4-基)苯甲酰胺)吡啶-4-基)氧基)-6-(2-甲氧基乙氧基)-N-甲基-1H-吲哚-1-甲酰胺或其药理学上可接受的盐,该化合物或其药理学上可接受的盐与雌激素受体拮抗剂或芳香化酶抑制剂组合施用。
[3]一种乳腺癌治疗用药物组合物,其包含:式(I)所表示的5-((2-(4-(1-(2-羟基乙基)哌啶-4-基)苯甲酰胺)吡啶-4-基)氧基)-6-(2-甲氧基乙氧基)-N-甲基-1H-吲哚-1-甲酰胺或其药理学上可接受的盐、及雌激素受体拮抗剂或芳香化酶抑制剂。
[4]一种乳腺癌治疗用试剂盒,其包含:包含式(I)所表示的5-((2-(4-(1-(2-羟基乙基)哌啶-4-基)苯甲酰胺)吡啶-4-基)氧基)-6-(2-甲氧基乙氧基)-N-甲基-1H-吲哚-1-甲酰胺或其药理学上可接受的盐的制剂、及包含雌激素受体拮抗剂或芳香化酶抑制剂的制剂。
[5]一种用于治疗乳腺癌的方法,该方法包括:将式(I)所表示的5-((2-(4-(1-(2-羟基乙基)哌啶-4-基)苯甲酰胺)吡啶-4-基)氧基)-6-(2-甲氧基乙氧基)-N-甲基-1H-吲哚-1-甲酰胺或其药理学上可接受的盐、及雌激素受体拮抗剂或芳香化酶抑制剂施用于有需要的患者。
[6]一种式(I)所表示的5-((2-(4-(1-(2-羟基乙基)哌啶-4-基)苯甲酰胺)吡啶-4-基)氧基)-6-(2-甲氧基乙氧基)-N-甲基-1H-吲哚-1-甲酰胺或其药理学上可接受的盐,该化合物或其药理学上可接受的盐用于治疗乳腺癌,与雌激素受体拮抗剂或芳香化酶抑制剂组合施用。
[7]一种用于治疗乳腺癌的组合,其包含:式(I)所表示的5-((2-(4-(1-(2-羟基乙基)哌啶-4-基)苯甲酰胺)吡啶-4-基)氧基)-6-(2-甲氧基乙氧基)-N-甲基-1H-吲哚-1-甲酰胺或其药理学上可接受的盐、及雌激素受体拮抗剂或芳香化酶抑制剂。
[8]式(I)所表示的5-((2-(4-(1-(2-羟基乙基)哌啶-4-基)苯甲酰胺)吡啶-4-基)氧基)-6-(2-甲氧基乙氧基)-N-甲基-1H-吲哚-1-甲酰胺或其药理学上可接受的盐用于制造乳腺癌治疗剂的用途,该化合物或其药理学上可接受的盐与雌激素受体拮抗剂或芳香化酶抑制剂组合施用。
[9]所述治疗剂、组合物、试剂盒、方法、化合物、组合或用途,其中式(I)所表示的5-((2-(4-(1-(2-羟基乙基)哌啶-4-基)苯甲酰胺)吡啶-4-基)氧基)-6-(2-甲氧基乙氧基)-N-甲基-1H-吲哚-1-甲酰胺或其药理学上可接受的盐、及该雌激素受体拮抗剂或该芳香化酶抑制剂同时或分开施用。
[10]所述治疗剂、组合物、试剂盒、方法、化合物、组合或用途,其中该药理学上可接受的盐是1.5琥珀酸盐。
[11]所述治疗剂、组合物、试剂盒、方法、化合物、组合或用途,其中该雌激素受体拮抗剂是氟维司群、他莫昔芬或其药理学上可接受的盐、或者美雄烷(mepitiostane)。
[12]所述治疗剂、组合物、试剂盒、方法、化合物、组合或用途,其中该雌激素受体拮抗剂是氟维司群。
[13]所述治疗剂、组合物、试剂盒、方法、化合物、组合或用途,其中该芳香化酶抑制剂是依西美坦、阿那曲唑或来曲唑(letrozole)。
[14]所述治疗剂、组合物、试剂盒、方法、化合物、组合或用途,其中该芳香化酶抑制剂是依西美坦。
[14a]所述治疗剂、组合物、试剂盒、方法、化合物、组合或用途,其中该芳香化酶抑制剂是来曲唑。
[15]所述治疗剂、组合物、试剂盒、方法、化合物、组合或用途,其中该乳腺癌是雌激素受体阳性的。
[16]所述治疗剂、组合物、试剂盒、方法、化合物、组合或用途,其中该乳腺癌是局部晚期乳腺癌、转移性乳腺癌、复发性乳腺癌或无法切除的乳腺癌。
[17]所述治疗剂、组合物、试剂盒、方法、化合物、组合或用途,其用于治疗表达成纤维细胞生长因子受体(FGFR)的乳腺癌。
[18]所述治疗剂、组合物、试剂盒、方法、化合物、组合或用途,其中FGFR是FGFR1、FGFR2或FGFR3。
本发明的有益效果
通过将式(I)所表示的化合物与雌激素拮抗剂或芳香化酶抑制剂组合施用,有可能对乳腺癌发挥缩小肿瘤体积的效果。
附图说明
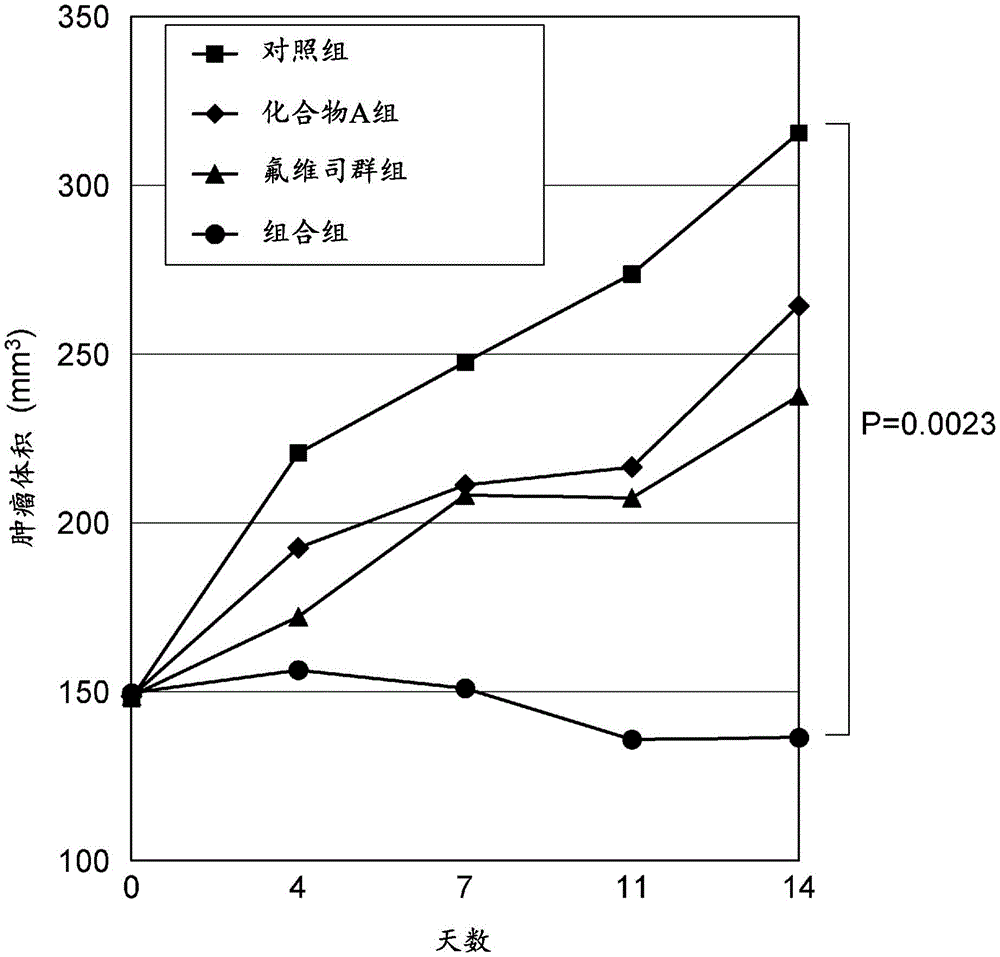
图1是表示实例1中药物施用开始后各组的平均肿瘤体积的变化的曲线图。
图2是表示实例4中药物施用开始后各组的平均肿瘤体积的变化的曲线图。
具体实施方式
根据本发明的式(I)所表示的化合物或其药理学上可接受的盐可通过专利文献1中披露的方法生产。
在本说明书中,药理学上可接受的盐的实例包括与无机酸的盐、与有机酸的盐、以及与酸性氨基酸的盐。
与无机酸的盐的合适实例包括与盐酸、氢溴酸、硫酸、硝酸、和磷酸的盐。
与有机酸的盐的合适实例包括与乙酸、琥珀酸、富马酸、马来酸、酒石酸、柠檬酸、乳酸、硬脂酸、苯甲酸、甲磺酸、乙磺酸、和对甲苯磺酸的盐。
与酸性氨基酸的盐的合适实例包括与天冬氨酸和谷氨酸的盐。
优选的药理学上可接受的盐是琥珀酸盐或马来酸盐,更优选的盐是琥珀酸盐。特别优选为1.5琥珀酸盐(在下文中,将式(I)所表示的化合物的1.5琥珀酸盐表示为化合物A)。
根据本发明的乳腺癌治疗剂能以例如片剂、颗粒剂、细粒剂、粉剂、或胶囊剂等固体制剂,液剂,冻胶剂,糖浆剂等形式经口施用。另外,根据本发明的乳腺癌治疗剂也能以注射剂、栓剂、软膏剂、糊剂等形式肠胃外施用。
根据本发明的乳腺癌治疗剂可通过日本药典(JP)、欧州药典(EP)、或美国药典(USP)中描述的方法配制。
式(I)所表示的化合物或其药理学上可接受的盐的剂量可根据症状的严重程度,患者的年龄、性别、体重、差异性敏感度,施用方法、施用时期、施用间隔、剂型等适当选择。当对成人(体重60kg)经口施用时,剂量为每日0.5mg至5g,优选为1mg至1g,进一步优选为1mg至500mg。可将该剂量每日分1至3次施用。
在本说明书中,雌激素受体拮抗剂意指与在乳腺癌细胞中表达的雌激素受体结合的药物。通过这种机制,雌激素受体拮抗剂可抑制雌激素受体与雌激素的结合,并阻碍乳腺癌细胞的增生。
雌激素受体拮抗剂的合适实例包括氟维司群、他莫昔芬或其药理学上可接受的盐(如柠檬酸盐)、以及美雄烷等。优选为氟维司群。
另外,雌激素受体拮抗剂也可为依拉司群(Elacestrant)、H3B-6545、托瑞米芬柠檬酸盐、SAR439859、AZD9833、rintodestrant、ZN-c5、LSZ102、D-0502、LY3484356、SHR9549、布利司群(brilanestrant)、和giredestrant。
当例如雌激素受体拮抗剂为氟维司群时,雌激素受体拮抗剂的剂量及施用方法为第一次、2周后、4周后、之后每4周,每剂500mg进行肌内施用。
当雌激素受体拮抗剂为他莫昔芬或其药理学上可接受的盐时,每日分1至2次经口施用20至40mg(以他莫昔芬计)。
当雌激素受体拮抗剂为美雄烷时,每日分2次经口施用20mg。
另外,也可使用抑制雌激素的合成的芳香化酶抑制剂代替雌激素受体拮抗剂。芳香化酶抑制剂的合适实例包括依西美坦、阿那曲唑、和来曲唑。优选为依西美坦。
/>
当例如芳香化酶抑制剂为依西美坦时,芳香化酶抑制剂的剂量及施用途径为每日1次、在饭后经口施用25mg。
当芳香化酶抑制剂为阿那曲唑时,每日1次、经口施用1mg。
当芳香化酶抑制剂为来曲唑时,每日1次、经口施用2.5mg。
在本说明书中,组合施用意指将包含式(I)所表示的化合物或其药理学上可接受的盐的制剂与包含雌激素受体拮抗剂或芳香化酶抑制剂的制剂同时或分开施用于患者。另外,也可施用一个制剂形式的包含式(I)所表示的化合物或其药理学上可接受的盐、及雌激素受体拮抗剂或芳香化酶抑制剂的组合物。
在本说明书中,乳腺癌意指在乳腺(乳腺管和小叶)发生的良性或恶性的肿瘤。乳腺癌包括局部晚期乳腺癌、转移性乳腺癌、复发性乳腺癌或无法切除的乳腺癌。
实例
以下实例对本发明更详细地进行说明。
实例1组合使用化合物A与氟维司群(商品名:芙仕得(Faslodex),肌内注射250mg,阿斯利康股份有限公司(AstraZeneca PLC))对来源于人乳腺癌患者的肿瘤(OD-BRE-0438)的增生抑制作用
各组使用五例NOD-SCID小鼠(NOD.CB17-Prkdcscid/J,雌性,日本查尔斯河实验室股份有限公司(Charles River Laboratories Japan,Inc.)),对施用化合物A及氟维司群时的抗肿瘤效果进行评价。
OD-BRE-0438是由Oncodesign公司(Oncodesign SA)建立的来源于激素受体阳性乳腺癌患者的肿瘤。通过将肿瘤组织皮下移植到NOD-SCID小鼠,进行继代。将从小鼠摘除的上述各个肿瘤切碎成约5mm方块,使用套管针(Φ3.5mm)将其移植到各小鼠的右侧腹皮下部,供抗肿瘤效果评价使用。
将β-雌二醇(富士胶片和光纯药株式会社(FUJIFILM Wako Pure ChemicalCorporation))用99.5%乙醇(富士胶片和光纯药株式会社)制成浓度为1mg/mL的溶液后,用供水用杀菌水制备成最终浓度2.5μg/mL。从肿瘤移植日到试验结束日,将该溶液通过饮水施用给小鼠。
使用电子数显卡尺(Digimatic
肿瘤体积(mm
将化合物A溶解在纯化水中至2.5mg/mL的浓度。作为氟维司群,直接使用市售的制剂(50mg/mL)。
肿瘤移植后30天,将小鼠按各自相同的平均肿瘤体积分配至各组。
将施用开始日设定为第0日,在以下条件下施用药物。
以25mg/kg(10mL/kg)的剂量,1日1次、连续14日对各组的小鼠经口施用化合物A。另外,以250mg/kg(5mL/kg)的用量,在第0日及第7日皮下注射氟维司群。对照组未经处理。
在第0日、第4日、第7日、第11日及第14日,对各小鼠的肿瘤体积进行测量。结果(平均值)如表1和图1中所示。此外,对于第14日的对照组及各施用组的肿瘤体积,进行Dunnett多重检验。
[表1]
药物施用开始后平均肿瘤体积的变化(mm
参考实例1人CYP19A1表达载体的构建
用Xba I及Not I切割PB-CMV-MCS-EF1α-Puro载体(系统生物科学有限责任公司(System Bioscience,LLC)),并向其中插入多克隆位点以制备PB510B2载体。接着,用Xba I及Cla I切割PB510B2载体,并向其中插入人CYP19A1(NM_001347249.2)的ORF,以获得PB510B2_hCYP19A1载体。
参考实例2pC3-PBase载体的构建
构建载体pC3-载体,该载体是去除了pcDNA3 1(-)哺乳动物表达载体(赛默飞世尔科技公司(Thermo Fisher Scientific Inc.))的新霉素表达模块(SV40启动子新霉素ORF-SV40聚A)而获得的载体。接着,通过将Super PiggyBac转座酶表达载体(系统生物科学有限责任公司)的Super PiggyBac转座酶的ORF插入pC3-载体的CMV启动子的下游,获得pC3-PBase载体。
参考实例3来源于人的乳腺癌细胞系ZR-75-1-hCYP19A1的建立
将来源于人的乳腺癌细胞系ZR-75-1(ECACC)接种在6孔的微量板(FALCON)上。使用包含10% FBS(西格玛公司(SIGMA))、及青霉素/链霉素(富士胶片和光纯药株式会社)的RPMI-1640培养基(含有4,500mg/L葡萄糖、L-谷氨酰胺、酚红、HEPES、和丙酮酸钠,富士胶片和光纯药株式会社)作为培养基。对于该接种的细胞,使用培养箱在5% CO
制备A液,其通过将Lipofectamine
参考实例4来源于人的乳腺癌细胞系MCF-7-hCYP19A1的建立
将来源于人的乳腺癌细胞系MCF-7(ECACC)接种在6孔的微量板(FALCON)上。使用包含10% FBS(西格玛公司)、青霉素/链霉素(富士胶片和光纯药株式会社)的E-MEM培养基(含有L-谷氨酰胺、酚红、丙酮酸钠、非必须氨基酸、和1500mg/L的碳酸氢钠,富士胶片和光纯药株式会社)作为培养基。对于该接种的细胞,使用培养箱在5% CO2和37℃的条件下培养过夜。
制备A液,其通过将Lipofectamine
实例2组合使用化合物A与依西美坦对来源于人的乳腺癌细胞系ZR-75-1-hCYP19A1的皮下移植肿瘤的增生抑制作用
各组使用五例BALB/c裸小鼠(CAnN.Cg-Foxn1/CrlCrlj,雌性,日本查尔斯河实验室股份有限公司),对施用化合物A及依西美坦时的抗肿瘤效果进行评价。
对小鼠皮下注射0.3mg/kg的盐酸美托咪定、4mg/kg的咪达唑仑(midazolam)及5mg/kg的酒石酸布托啡诺的混合麻醉液。切开各小鼠的背部皮肤及腹膜,切除左右卵巢,并缝合切开部分。之后,皮下施用65mg/kg(13.0mg/mL、100μL/小鼠)的氨苄青霉素钠注射液(明治制果制药株式会社(Meiji Seika Pharma Co.,Ltd.))、1mg/kg(0.2mg/mL、100μL/小鼠)的安醒顺(Antisedan)(日本全药工业株式会社(Nippon Zenyaku Kogyo Co.,Ltd.))、及5mg/kg(1.0mg/mL、100μL/动物)的卡洛芬注射液(日本Zoetis株式会社(ZoetisJapan))。
7日后到试验结束日,以0.1mg/小鼠(200μL/小鼠)对小鼠经口施用雄烯二酮(西格玛奥德里奇公司(Sigma-Aldrich))。
在开始施用雄烯二酮的第二日,将1×10
将化合物A溶解在纯化水中至2.5mg/mL的浓度。另外,将Tween(注册商标)80与0.5%甲基纤维素溶液混合,至0.4%的浓度。将依西美坦溶解在该混合液中,至5mg/mL的浓度。
将施用开始日设定为第0日,在以下条件下施用药物。对于各组的小鼠,每日1次、连续11日经口施用50mg/kg(20mL/kg)剂量的化合物A、1mg/小鼠(200μL/小鼠)剂量的依西美坦、或两剂化合物A及依西美坦(各自为与上述相同的剂量)。对照组未经处理。
在第0日、第4日、第7日、及第11日,对各小鼠的肿瘤体积进行测量。结果(平均值)如表2所示。使用电子数显卡尺(Digimatic
肿瘤体积(mm
[表2]
药物施用开始后平均肿瘤体积的变化(mm
实例3化合物A和来曲唑、阿那曲唑或依西美坦组合使用对表达人芳香化酶的来源于人的乳腺癌细胞系MCF-7-hCYP19A1的作用
使用包含10% FBS(西格玛公司)、青霉素/链霉素(富士胶片和光纯药株式会社)、及1.5μg/mL的嘌呤霉素的E-MEM培养基(含有L-谷氨酰胺、酚红、丙酮酸钠、非必须氨基酸、和1500mg/L碳酸氢钠,富士胶片和光纯药株式会社)(以下称为培养基),在5% CO
向细胞培养板的各孔中各加入200μL的在含有10%FBS的培养基中制备成0.25×10
第二日,去除培养基,并向其中添加100μL的包含10%FBS(活性炭吸附FBS,吉布科公司(GIBCO))、青霉素/链霉素、及1.5μg/mL的嘌呤霉素的不含酚红的RPMI-1640培养基(富士胶片和光纯药株式会社)(以下称为测定培养基),在培养箱中进行培养。
在第二日,去除培养基,各加入100μL测定培养基进行培养基替换,并使细胞在培养箱中培养2日。之后,去除培养基,添加25μL的在测定培养基中制备成最终浓度为10nmol/L的雄烯二酮、各25μL的在测定培养基中分别制备成最终浓度为25ng/mL和100ng/mL的FGF2(吉布科公司)和FGF10(R&D系统公司(R&Dsystems))后,向其中添加以下描述的各溶液各25μL,并在培养箱中培养7日。在添加药物后3日及5日去除培养基,并添加雄烯二酮、FGF2和FGF10溶液、以及以下描述的各溶液各25μL,进行培养基替换。
(1)含有在测定培养基中制备成最终浓度为2,000nmol/L的来曲唑(西格玛公司)、制备成最终浓度为10,000或2,000nmol/L的阿那曲唑(西格玛公司)、制备成最终浓度为2,000nmol/L或400nmol/L的依西美坦(西格玛公司)或最终浓度为0.05%的DMSO的测定培养基(在对照组或化合物A单剂的情况下使用)
(2)含有在测定培养基中制备成最终浓度为1,000nmol/L的化合物A和最终浓度为0.005%的DMSO的测定培养基(在对照组或(1)中所述的单药的情况下使用)
关于培养后各孔中的细胞数,使用CellTiter Glo
从板去除培养基后,向各孔中添加50μL的测量试剂与培养基1:1混合所得的溶液。在使用板混合器对板进行5分钟混合后,使用读板仪(EnVision
[表3]
[表4]
[表5]
[表6]
[表7]
实例4组合使用化合物A与来曲唑对来源于人的乳腺癌细胞系MCF-7-hCYP19A1的皮下移植肿瘤的增生抑制作用各组使用五例BALB/c裸小鼠(CAnN.Cg-Foxn1/CrlCrlj,雌性,日本查尔斯河实验室股份有限公司),对施用化合物A及来曲唑时的抗肿瘤效果进行评价。
对小鼠皮下注射0.3mg/kg的盐酸美托咪定、4mg/kg的咪达唑仑(midazolam)及5mg/kg的酒石酸布托啡诺的混合麻醉液。切开各小鼠的背部皮肤及腹膜,切除左右卵巢,并缝合切开部分。之后,将用生理盐水(大塚制药厂株式会社(Otsuka PharmaceuticalFactory,Inc.))稀释至1mg/mL最终浓度的拜有利(Baytril)2.5%注射液(拜耳公司(Bayer))作为恩诺沙星(enrofloxacin)和用生理盐水(大塚制药厂株式会社)稀释至0.2mg/mL最终浓度的安醒顺(日本全药工业株式会社)作为盐酸阿替美唑分别以相当于5mg/kg(100μL/小鼠)和1mg/kg(100μL/小鼠)的量皮下施用。
7日后,以0.1mL/小鼠(1.56mg/小鼠)的浓度肌内施用利用芝麻油稀释庚酸睾酮(testosterone enanthate)肌内注射液(富士制药工业股份有限公司(Fuji Pharma Co.,Ltd.))至15.6mg/mL浓度所得溶液。之后,1周1次向小鼠肌内施用该溶液,直到试验结束日为止。
在开始施用庚酸睾酮的第二日,将1×10
将化合物A溶解在纯化水中至2.5mg/mL的浓度。另外,将来曲唑溶解于无菌0.3%羟丙基纤维素水溶液中,至0.05mg/mL的浓度。
将施用开始日设定为第0日,在以下条件下施用药物。对于各组的小鼠,每日1次、连续14日经口施用25mg/kg(10mL/kg)剂量的化合物A、0.01mg/小鼠(200μL/小鼠)剂量的来曲唑、或两剂化合物A及来曲唑(各自为与上述相同的剂量)。对照组未经处理。
在第0日、第4日、第7日、第11日及第14日,对各小鼠的肿瘤体积进行测量。结果(平均值)如表8和图2所示。使用电子数显卡尺(Digimatic
肿瘤体积(mm
此外,对于第14日的对照组及各施用组的肿瘤体积,进行Dunnett多重检验。
[表8]
药物施用开始后平均肿瘤体积的变化(mm
/>
- 预测对乳腺癌治疗剂的反应的方法和治疗乳腺癌的方法
- DPPE与其它化学治疗剂联合治疗乳腺癌的用途