一种6,8-二溴咪唑并1,2-b哒嗪的制备工艺
文献发布时间:2024-04-18 19:52:40
技术领域
本发明属于化工技术领域,具体涉及一种6,8-二溴咪唑并[1,2-b]哒嗪的制备工艺。
背景技术
咪唑并哒嗪也是典型的含氮杂环化合物咪唑并哒嗪其衍生物,被广泛应用于新药研发领域,包括众多抗癌药物、肿瘤、神经药物、内分泌疾病、消化系统疾病等,目前已有多种咪唑并吡嗪骨架的药物被应用于临床。咪唑并[1,2-b]哒嗪是头孢唑兰的3位侧链,是合成头孢唑兰的重要中间体,而6,8-二溴咪唑并[1,2-b]哒嗪化合物是重要的有机合成重要的中间体,特别是在药物化学的研究和应用中,它的多功能团或潜在的多功能团性是其被用于有机合成的多种药物中。
现有技术中咪唑并[1,2-b]哒嗪的工艺合成路线繁琐,耗时长,大部分以重结晶和柱层析的方式得到产品,并不易实现规模化生产。如专利CN201510630627.X公开了一种6-氯-8-溴咪唑并[1,2-b]哒嗪的合成方法,GIA方法的反应温度较高,达到了40-100℃,反应时间过长,达到了5-30h,且产率和产品纯度也较低,需经重结晶得到复合纯度要求的目标产品。因此,为了满足日益增长的市场需求,开发一种操作简便,反应条件温和,生产成本低,产率高,适合工业化生产的6,8-二溴咪唑并[1,2-b]哒嗪的制备工艺十分必要。
发明内容
针对上述问题,本发明提出了一种6,8-二溴咪唑并[1,2-b]哒嗪的制备工艺,该工艺的溴化反应和合环反应在同一个反应釜中进行,操作简便,相对现有技术缩短了反应时间、反应条件更温和,所制备产品的品质更佳,显著提高了产品收率。
具体的,如下所示,本发明6,8-二溴咪唑并[1,2-b]哒嗪制备工艺包括以下步骤:
(1)将3-氨基-6-溴哒嗪溶解于溶剂后与溴化试剂发生溴化反应得到第一混合物料;
(2)向第一混合物料中加入催化剂及合环试剂进行反应,得到第二物料混合;其中,所述催化剂为单用或复配的镧系路易斯酸。
在上述技术方案中,本发明研发团队在大量实验研究的基础上,意外地发现在现有技术路线中的合环阶段加入催化剂,尤其是加入单用或复配的镧系路易斯酸可显著缩短反应的时间,且适用的反应条件更温和,更重要的是,所制备的6,8-二溴咪唑并[1,2-b]哒嗪的纯度和产品状态更佳,即产品质量更佳,同时产品收率得到了显著提高。本发明实施例验证加入镧系路易斯酸催化剂后,产品的收率提高了34%以上。
基于以上发现,研发团队开展了大量单用或复配镧系路易斯酸催化剂的选择性实验,发现当所述单用的镧系路易斯酸为氯化镧,氯化钕或氯化铒,以及当所述复配的镧系路易斯酸为复配的氯化镧和氯化铷、氯化镧和氯化铒、氯化铒和氯化铷中的一种复配时,制备工艺所得产品纯度和产品收率高,且副反应发生少,反应条件温和,反应效率高。
进一步的,研发团队选择单用的镧系路易斯酸催化剂对制备工艺中所加入催化剂的用量进行了大量的探索实验。可选的,催化剂的加入量为3-氨基-6-溴哒嗪摩尔量的3‰-10‰,在此催化剂加入范围内所制备6,8-二溴咪唑并[1,2-b]哒嗪产品的纯度高,收率达到了70.2%以上,远高于对比例中的45%;优选的,催化剂的加入量为3-氨基-6-溴哒嗪摩尔量的5‰。
更进一步的研究发现,在催化剂的加入量为3-氨基-6-溴哒嗪摩尔量5‰的条件下,当所用催化剂为复配的氯化镧和氯化铷,且氯化镧和氯化铷的摩尔比为1:1时,所制备产品的品质和产品收率更佳。
在上述技术方案中,本发明研发团队对步骤(1)中所用溶剂类型及溶剂的加入量,以及两者的优选进行了大量选择性实验。可选的,所述溶剂为DMF(N,N-二甲基甲酰胺),二氯甲烷,乙醇或甲醇中的一种。可选的,所述溶剂与3-氨基-6-溴哒嗪的质量比为2-10:1,优选为6-10:1,进一步优选为6:1。本发明实施例中给出了不同种类溶剂以及不同溶剂用量的制备结果。
在上述技术方案中,本发明研发团队对步骤(1)中所用溴化试剂的种类及加入量,以及优选方案进行了大量选择性实验。可选的,所述溴化试剂为NBS(N-溴代琥珀酰亚胺)或者二溴海因(DBDMH)中的一种。可选的,当所述溴化试剂为NBS时,溴化试剂与3-氨基-6-溴哒嗪的摩尔比为1.1-1.8:1,优选为1.3-1.5:1;当所述溴化试剂为二溴海因时,溴化试剂与3-氨基-6-溴哒嗪的摩尔比为0.6-1.1:1,优选为0.7-0.9:1。本发明实施例中给出了不同种类溴化试剂以及不同溴化试剂用量的制备结果。
在上述技术方案中,本发明研发团队对步骤(2)中所用合环试剂的种类及合环试剂的加入量,以及相关优选方案进行了大量选择性实验。可选的,所述合环试剂为氯乙醛或溴乙醛缩二甲醛,优选为氯乙醛。可选的,所述合环试剂与3-氨基-6-溴哒嗪的摩尔比为1.1-2.5:1,优选为1.1-1.5:1。本发明实施例中给出了不同种类合环试剂以及不同合环试剂用量的制备结果。
在上述技术方案中,所述步骤(1)的反应温度为0-5℃,反应时间为3-5h。
在上述技术方案中,本发明研发团队对步骤(2)中的反应温度进行了探索和优化。可选的,所述步骤(2)的反应温度为20-60℃,优选为30-40℃;反应时间为2-3h。本发明实施例中给出了步骤(2)在不同温度条件下的制备结果。
值得注意的是,上述技术方案中所述第一混合物中主要包括了中间体3-氨基-4,6-二溴哒嗪。实际操作过程中,待第一混合物中起始原料3-氨基-6-溴哒嗪HPLC检测<0.5%,则说明溴化反应结束。上述技术方案中所述第二混合物中主要包括了目标产品6,8-二溴咪唑并[1,2-b]哒嗪,待检测中间体3-氨基-4,6-二溴哒嗪HPLC<0.5%,则说明合环反应结束。
此外,上述技术方案中还包括了步骤(3):将所述第二混合物料的pH值调节至7-8,随后经萃取得到所述6,8-二溴咪唑并[1,2-b]哒嗪产品。本发明技术方案调节所述第二混合物料的pH后只需将产品萃取到有机溶剂,再将溶剂浓缩即可得到纯度较高的产品,避免了现有工艺中采用柱层析和重结晶的后续处理方式,因而本发明工艺流程更简化,适用于工业化生产。进一步的,所述步骤(3)的还包括对萃取后的有机相进行旋蒸;更进一步的,还包括在旋蒸前对萃取所得溶剂进行水洗和/或饱和食盐水洗。
在上述技术方案中,本发明研发团队对步骤(3)中调节pH值所用的碱液进行了探索和优化。可选的,可采用碳酸氢钠,碳酸钠或氢氧化钠溶液调节所述第二混合物的pH值。实施例中给出了碱液调节第二混合物pH值的制备结果。
在上述技术方案中,本发明研发团队对步骤(3)中所用萃取剂进行了探索和优化。可选的,萃取用的溶剂为二氯甲烷,二氯乙烷,乙酸乙酯中的一种,优选为二氯甲烷。实施例中给出了采用不同萃取剂进行萃取的制备结果。
相对于现有技术,本发明在同一个反应釜中进行溴化反应和合环反应制备6,8-二溴咪唑并[1,2-b]哒嗪的工艺,该工艺操作简便,反应条件温和,缩短了反应时间、提高了生产效率,所制备产品的品质更佳,产品收率提高了34%以上,适合工业化放大生产。
附图说明
构成本申请的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
在附图中:
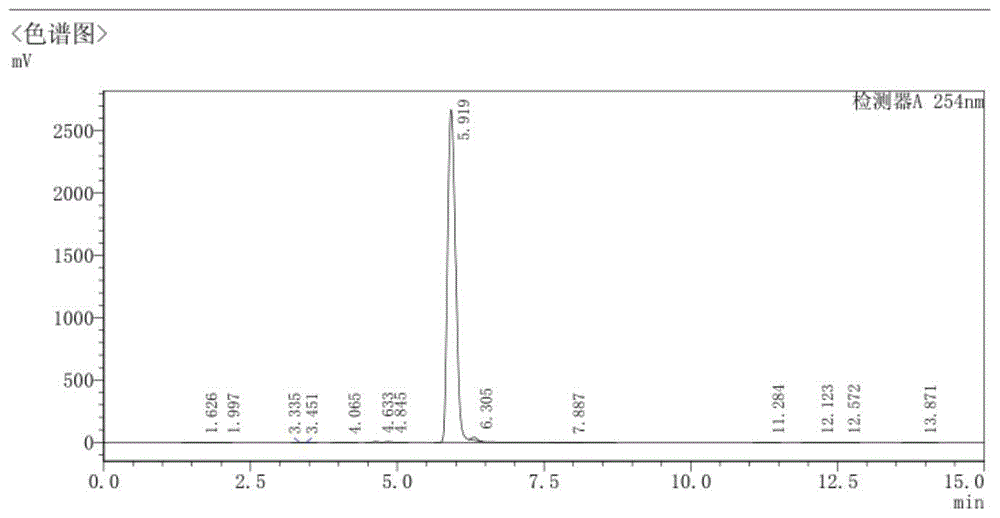
图1为实施例1所制备6,8-二溴咪唑并[1,2-b]哒嗪产品的液相图谱;
图2为实施例1中第二混合物的状态图;
图3为对比例1所制备6,8-二溴咪唑并[1,2-b]哒嗪产品的液相图谱;
图4为对比例1中第二混合物的状态图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
为了便于理解本发明,下面将对本发明进行更全面的描述,给出了本发明的较佳实施例。但应当理解为这些实施例仅仅是用于更详细说明之用,而不应理解为用以任何形式限制本发明,即并不意于限制本发明的保护范围。
需说明的是,除有定义外,以下实施例中所用的技术术语具有与本发明创造所属领域技术人员普遍理解的相同含义。本实施例中诸如“第一”“第二”等之类的关系术语仅仅用来将一个与另一个具有相同名称的部件区分开来,而不一定要求或者暗示这些部件之间存在任何这种实际的关系或者顺序。限定有“第一”“第二”等的特征可以明示或者隐含地包括一个或者更多个该特征。
下列实施例中的试剂来源:
3-氨基-6溴哒嗪:自制
催化剂载体:江西慧骅科技有限公司
催化剂:山东德胜新材料有限公司
氯乙醛:江苏雷恩环保科技有限公司
乙醇:淄博齐星化学科技有限公司
二氯甲烷:山东华盛新材料有限公司
HPLC检测条件:色谱柱型号:液相柱型号:lunaC18,5um,250*4.6;柱温:25℃;流动相:乙腈:水=80:20;流速:1ml/min;吸收波长:254nm;进样量:0.2ul;
本发明采用HPLC检测产品纯度;产品收率为烘干之后重量转为摩尔数与原料的摩尔比。
实施例1
本实施例展示了一种工况下6,8-二溴咪唑并[1,2-b]哒嗪的制备工艺,该工艺包括以下步骤:
(1)在反应釜中投入1.74kg3-氨基-6-溴哒嗪(10mol,1eq),加入17kgDMF搅拌30min,搅拌溶解,温度降至5-10℃分批加入2.67kgNBS(15mol,1.5eq)保持温度不变,搅拌反应3-5h;(2)HPLC检测反应中原料残留3-氨基-6-溴哒嗪<0.5%,视为反应完全,反应中先加入催化剂0.25kg氯化镧(0.1mol,10‰),将2.56kg氯乙醛(40%水溶液,13mol,1.3eq)缓慢滴加进反应釜,滴加完升温至30-40℃,反应搅拌2-3小时;(3)HPLC检测3-氨基-4,6-二溴哒嗪反应完全<0.5%,冷却至室温,将第二混合物用饱和碳酸氢钠中和至pH在7-8之间,直接加入二氯甲烷50L*3萃取、水洗,然后饱和食盐水洗,旋蒸合并固体得到浅棕色的6,8-二溴咪唑并[1,2-b]哒嗪产品2.08kg,产品的纯度99.3%,收率74.8%;产品的液相图谱如图1所示;步骤(2)反应完全后第二混合物的状态如图2所示,可看出第二混合物澄清,说明其杂质含量少,进一步证实本实施例所制备6,8-二溴咪唑并[1,2-b]哒嗪产品的纯度高。
对比例1
对比例1所示6,8-二溴咪唑并[1,2-b]哒嗪的制备工艺的操作条件和工艺参数与实施例1所示工艺相同,但在步骤(2)中未加入催化剂;具体的,该对比例包括以下步骤:
(1)在反应釜中投入1.74kg3-氨基-6-溴哒嗪(10mol,1eq),加入17kgDMF搅拌30min,搅拌溶解,温度降至5-10℃分批加入2.67kgNBS(15mol,1.5eq)保持温度不变,搅拌反应3-5h;(2)HPLC检测反应中原料残留3-氨基-6-溴哒嗪<0.5%,视为反应完全,直接将2.56kg氯乙醛(40%水溶液,13mol,1.3eq)缓慢滴加进反应釜,滴加完毕,升温至30-40℃,反应搅拌6~8小时;HPLC检测反应原料中3-氨基-4,6-二溴哒嗪未反应完全,再升温至85-100℃,反应5h;(3)HPLC检测3-氨基-4,6-二溴哒嗪无残留,视为反应完全,反应体系为红棕色油状,冷却至室温,溶液用饱和碳酸氢钠中和,反应液直接加入二氯甲烷50L*3萃取、水洗,然后饱和食盐水洗,旋蒸合并固体得到浅棕色产品1.25kg,纯度93.8%,收率45.0%;产品的液相图谱如图3所示,步骤(2)反应完全后第二混合物的状态如图4所示,可看出本对比例所制得第二混合物的较为浑浊,说明其内含有一定量的杂质。
随后,对实施例1和对比例1的相关反应条件和产品参数进行对比,结果如表1所示。
表1
由表1可证实,对比例1中不加催化剂反应使得产品纯度降低,且反应完全后第二混合物的状态从澄清的淡棕色变为红棕色油状,说明了对比例1中副产物增加,所得产品需进一步提纯;因而实施例1制备工艺整体上提高了产品质量。此外,实施例1相对于对比例1,产品收率提高了66%;值得注意的是,加入了催化剂的实施例1的整体反应时间和反应温度均低于对比例1,可提高生产效率及节约能耗。
实施例2
一种6,8-二溴咪唑并[1,2-b]哒嗪的制备工艺,具体的:(1)在反应釜中投入1.74kg3-氨基-6-溴哒嗪(10mol,1eq),加入17kg不同溶剂搅拌30min,搅拌溶解,温度降至5-10℃分批加入2.67kgNBS(15mol,1.5eq)保持温度不变。(2)搅拌反应一段时间,检测反应中3-氨基-6-溴哒嗪<0.5%原料剩余,检测反应完全,反应中先加入催化剂0.25kg氯化镧(0.1mol,10‰),直接将2.56kg氯乙醛(40%水溶液,13mol,1.3eq)缓慢滴加进反应釜,滴加完升温至30-40℃,反应搅拌2-3小时;(3)检测反应原料中中间态3-氨基-4,6-二溴哒嗪反应完全<0.5%,冷却至室温,将第二混合物用饱和碳酸氢钠中和至pH为7-8之间,直接加入二氯甲烷50L*3萃取、水洗,然后饱和食盐水洗,旋蒸合并固体得到浅棕色的6,8-二溴咪唑并[1,2-b]哒嗪产品。
确定其他条件和参数不变,唯一变量为步骤(1)所用溶剂,实施例2.1-实施例2.4的具体工艺参数和条件,产品参数如表2。
表2
如表2可验证,步骤(1)中所用溶剂可选为DMF,二氯甲烷,乙醇或甲醇中的一种。研发团队发现当其他工艺条件和参数相同时,采用DMF为反应溶剂的反应时间更短,此外,采用DMF为反应溶剂的实施例1得到的收率以及纯度最好,产品收率高出采用其他溶剂实施例20%左右。需注意,相对于对比例1的产品收率45%,即使当步骤(1)所用溶剂为甲醇时的产品收率亦高于对比例1收率34%以上。
进一步的,以DMF为溶剂设置实施例2.5-2.9以验证步骤(1)中溶剂用量对制备工艺的影响,实施例2.5-实施例2.9具体工艺参数及产品参数如表3。
表3
由表3可验证,步骤(1)中加入DMF的量与原料3-氨基-6-溴哒嗪的质量比可选为2-10:1,进一步可选为6-10:1,更进一步可选为6:1。表3证实当步骤(1)所用溶剂DMF的量过少时影响产品收率以及纯度,当DMF用量增加至一定量后将不再提高产品收率,但溶剂用量大将增加反应废水量,不利于后期处理。
实施例3
一种6,8-二溴咪唑并[1,2-b]哒嗪的制备工艺,包括以下步骤:(1)在反应釜中投入1.74kg3-氨基-6-溴哒嗪(10mol,1eq),加入10.5kgDMF搅拌30min,搅拌溶解,温度降至5-10℃分批分别加入不同量NBS(11mol,1.1eq;13mol,1.3eq;15mol,1.5eq;18mol,1.8eq)或者二溴海因(6mol,0.6eq;7mol,0.7eq;9mol,0.9eq;11mol,1.1eq)进行溴化反应,保持温度不变,搅拌反应3-5h;(2)检测反应中3-氨基-6-溴哒嗪<0.5%原料剩余,检测反应完全,反应中先加入催化剂0.25kg氯化镧(0.1mol,10‰),直接将2.56kg氯乙醛(40%水溶液,13mol,1.3eq)缓慢滴加进反应釜,滴加完升温至30-40℃,反应搅拌2-3小时;(3)检测反应原料中中间态3-氨基-4,6-二溴哒嗪反应完全<0.5%,冷却至室温,将第二混合物用饱和碳酸氢钠中和至pH为7-8之间,直接加入二氯甲烷50L*3萃取、水洗,然后饱和食盐水洗,旋蒸合并固体得到浅棕色6,8-二溴咪唑并[1,2-b]哒嗪产品。实施例3.1-实施例3.8的溴化试剂及添加量,以及产品参数如表4。
表4
由表4可验证,当所述溴化试剂为NBS时,溴化试剂与3-氨基-6-溴哒嗪的摩尔比可选为1.1-1.8:1,进一步可选为1.3-1.5:1,更进一步可选为1.3:1;当所述溴化试剂为二溴海因时,溴化试剂与3-氨基-6-溴哒嗪的摩尔比可选为0.6-1.1:1,进一步可选为0.7-0.9:1。
实施例4
一种6,8-二溴咪唑并[1,2-b]哒嗪的制备工艺,该工艺包括以下步骤:(1)在反应釜中投入1.74kg3-氨基-6-溴哒嗪(10mol,1eq),加入10.5kgDMF搅拌30min,搅拌溶解,温度降至5-10℃分批分别加入2.31kgNBS(13mol,1.3eq)保持温度不变,搅拌反应3-5h;(2)检测反应中3-氨基-6-溴哒嗪<0.5%原料剩余,检测反应完全,反应中先加入催化剂0.25kg氯化镧(0.1mol,10‰),直接将氯乙醛(40%水溶液,11mol,1.1eq;13mol,1.3eq;15mol,1.5eq;等)或者溴乙醛缩二甲醛(11mol,1.1eq;13mol,1.3eq;15mol,1.5eq;等)缓慢滴加进反应釜,滴加完升温至30-40℃反应搅拌2-3小时;(3)检测反应原料中中间态3-氨基-4,6-二溴哒嗪反应完全<0.5%,冷却至室温,将第二混合物用饱和碳酸氢钠中和至pH在7-8之间,直接加入二氯甲烷50L*3萃取、水洗,然后饱和食盐水洗,旋蒸合并固体得到浅棕色6,8-二溴咪唑并[1,2-b]哒嗪产品。本实施例中的合环试剂及添加量,以及产品参数如表5。
表5
由表5可验证,所述合环试剂可选为氯乙醛或溴乙醛缩二甲醛,进一步可选为氯乙醛。此外,通过对比所得6,8-二溴咪唑并[1,2-b]哒嗪产品的纯度及产品收率可证实,合环试剂与3-氨基-6-溴哒嗪的摩尔比可选为1.1-2.5:1,进一步可选为1.1-1.5:1,更进一步可选为1.3:1。
实施例5
一种化合物6.8-二溴咪唑并(1.2b)哒嗪制备工艺,该工艺包括以下步骤:(1)在反应釜中投入1.74kg3-氨基-6-溴哒嗪(10mol,1eq),加入10.5kgDMF搅拌30min,搅拌溶解,温度降至5-10℃分批分别加入2.31kgNBS(13mol,1.3eq)保持温度不变,搅拌反应3-5h;(2)检测反应中3-氨基-6-溴哒嗪<0.5%原料剩余,检测反应完全,反应中加入不同用量的不同催化剂(0.03mol,3‰;0.05mol,5‰;0.07mol,7‰;0.010mol,10‰),直接将氯乙醛(40%13mol,1.3eq)缓慢滴加进反应釜,滴加完升温至30-40℃,反应搅拌2-3小时;(3)检测反应原料中中间态3-氨基-4,6-二溴哒嗪反应完全<0.5%,冷却至室温,将第二混合物用饱和碳酸氢钠中和至pH在7-8之间,直接加入二氯甲烷50L*3萃取、水洗,然后饱和食盐水洗,旋蒸合并固体得到浅棕色的6,8-二溴咪唑并[1,2-b]哒嗪产品。
本实施例中确定其他条件和参数不变,变量为步骤(2)中加入催化剂的用量,以及催化剂的种类为变量,催化剂的种类及添加量,以及产品参数如表6所示。
表6
由表6中实施例5.11-5.14、实施例5.21-5.24、实施例5.31-5.34可验证,所述单用的镧系路易斯酸可选为氯化镧,氯化钕和氯化铒,进一步可选为氯化镧;通过对比所得6,8-二溴咪唑并[1,2-b]哒嗪产品的纯度及产品收率可证实,当使用单用的镧系路易斯酸作为催化剂时,催化剂的用量可选为3-氨基-6-溴哒嗪摩尔量的3‰-10‰,进一步可选为5‰。
此外,本实施例在催化剂用量为3-氨基-6-溴哒嗪摩尔量的5‰条件下,进一步验证了不同种类复配的镧系路易斯酸对产品纯度和收率的影响。由实施例5.4-5.6可验证,所述复配的镧系路易斯酸可选为复配的氯化镧和氯化铷,氯化镧和氯化铒,氯化铒和氯化铷;通过对比本实施例的产品纯度和产品收率数据可知,本发明所用催化剂更进一步可选为复配的氯化镧和氯化铷,其中氯化镧和氯化铷的摩尔比为1:1。
实施例6
一种化合物6,8-二溴咪唑并[1,2-b]哒嗪的制备工艺,包括以下步骤:
(1)在反应釜中投入1.74kg3-氨基-6-溴哒嗪(10mol,1eq),加入10.5kgDMF搅拌30min,搅拌溶解,温度降至5-10℃分批分别加入2.31kgNBS(13mol,1.3eq)保持温度不变,搅拌反应3-5h;(2)检测反应中3-氨基-6-溴哒嗪<0.5%原料剩余,检测反应完全,反应中先加入催化剂123g氯化镧(0.05mol,5‰)直接将氯乙醛(40%13mol,1.3eq)缓慢滴加进反应釜,滴加完后将反应液升温至4个温度梯度20-30℃,30-40℃,40-50℃,50-60℃反应搅拌2-3小时;(3)检测反应原料中中间态3-氨基-4,6-二溴哒嗪反应完全<0.5%,冷却至室温,将第二混合物用饱和碳酸氢钠中和至pH在7-8之间,直接加入二氯甲烷50L*3萃取、水洗,然后饱和食盐水洗,旋蒸合并固体得到浅棕色的6,8-二溴咪唑并[1,2-b]哒嗪产品。如表7所示,实施例6.1-实施例6.4示出了步骤(2)中反应温度对工艺的影响。
表7
由表7可证实,所述步骤(2)中的反应温度为可选为20-60℃。此外,(2)中的反应温度较低时,如20-30℃,将可能导致出现反应缓慢、反应周期长并进而导致收率下降,当步骤(2)中的反应温度上升至30-40℃时,产品的纯度和收率表现较佳,相对于更高温度的操作条件,无需额外升温消耗更多的能源或额外进行副产物的处理;当步骤(2)中的反应温度上升较高的温度,如40-50℃或温度更高的50-60℃,则可能生成少量的油状副产物。因此,所述步骤(2)的反应温度进一步可选为30-40℃。
实施例7
一种6,8-二溴咪唑并[1,2-b]哒嗪的制备工艺,该工艺包括以下步骤:(1)在反应釜中投入1.74kg3-氨基-6-溴哒嗪(10mol,1eq),加入10.5kgDMF搅拌30min,搅拌溶解,温度降至5-10℃分批分别加入2.31kgNBS(13mol,1.3eq)保持温度不变,搅拌反应3-5h;(2)检测反应中3-氨基-6-溴哒嗪<0.5%原料剩余,检测反应完全,反应中先加入催化剂123g氯化镧(0.05mol,5‰)直接将氯乙醛(40%13mol,1.3eq)缓慢滴加进反应釜,滴加完升温30-40℃反应搅拌2-3小时;(3)检测反应原料中中间态3-氨基-4,6-二溴哒嗪反应完全<0.5%,冷却至室温,将第二混合物中用不同碱液中和至pH在7-8之间,加入二氯甲烷50L*3萃取、水洗,然后饱和食盐水洗,旋蒸合并固体得到浅棕色的6,8-二溴咪唑并[1,2-b]哒嗪产品。表8示出了步骤(3)中不同碱液对工艺的影响。
表8
由表8可证实,可采用碳酸氢钠,碳酸钠或氢氧化钠溶液调节所述第二混合物的pH值,进一步可选为采用碳酸氢钠溶液来调节所述第二混合物的pH值。
实施例8
一种化合物6,8-二溴咪唑并[1,2-b]哒嗪的制备工艺,该工艺包括以下步骤:(1)在反应釜中投入1.74kg3-氨基-6-溴哒嗪(10mol,1eq),加入10.5kgDMF搅拌30min,搅拌溶解,温度降至5-10℃分批分别加入2.31kgNBS(13mol,1.3eq)保持温度不变,搅拌反应3-5h;(2)检测反应中3-氨基-6-溴哒嗪<0.5%原料剩余,检测反应完全,反应中先加入催化剂123g氯化镧(0.05mol,5‰)直接将氯乙醛(40%13mol,1.3eq)缓慢滴加进反应釜,滴加完升温30-40℃反应搅拌2-3小时;(3)检测反应原料中中间态3-氨基-4,6-二溴哒嗪反应完全<0.5%,冷却至室温,溶液用碳酸氢钠中和至pH在7-8之间,直接加入不同萃取溶剂50L*3萃取、水洗,然后饱和食盐水洗,旋蒸合并固体得到浅棕色的6,8-二溴咪唑并[1,2-b]哒嗪产品。表9示出了步骤(3)中不同萃取剂对工艺的影响。
表9
由表9可验证,步骤(3)中萃取用的溶剂可选为二氯甲烷,二氯乙烷,乙酸乙酯中的一种。
需注意的是,以上内容是结合具体的实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明;本实施例尺寸数据并不定限定本技术方案,只是展示其中一种具体的工况。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单改进和润饰,都应当视为属于本发明保护的范围。
- 一种旋耕机用安全防具
- 一种防刺伤的安全防具