用于沉淀铂族金属颗粒的方法
文献发布时间:2024-04-18 19:58:53
根据第一项权利要求的前序部分,本发明涉及一种从含有一种或多种铂族金属离子的一种或多种前体化合物的进料回收铂族金属的方法。
近年来,金属基纳米颗粒受到了大量关注,因为金属处于纳米尺度时经常具有完全不同的性质。铂族金属诸如Pt、Pd、Rh的纳米颗粒由于它们独特的催化性质获得了特别的关注。铂族金属,尽管昂贵且地质来源有限,但对于质子交换膜燃料电池(PEMFC)或直接醇类燃料电池(DAFC)仍是最佳的催化剂。目前,铂族金属纳米颗粒的合成策略旨在优化纳米颗粒的性质和减少所需的金属量。另外,能够实现铂族金属的二次回收策略备受推崇,优选以纳米颗粒的形式。
铂族金属纳米颗粒合成的常用方法包括用氢气作为还原剂的化学还原,例如用于胶体Pt纳米颗粒的受控形状合成。当H
至今,能够从稀释的前体化合物液体进料流有效地回收单质形式铂族金属的方法尚未公开,所述前体化合物含有溶解、分散或悬浮于其中的相应铂族金属离子。
合成或回收Pt族金属纳米颗粒的已知方法的劣势在于它们既不是选择性的,也不是可持续的:有些方法需要添加在金属回收后需去除的危险化学试剂,使用辅助材料;或使用剧烈鼓泡以供应H
EP3242963B1公开了电化学合成金属氧化物或金属氢氧化物的纳米颗粒,如氧化铁、铜和氢氯化锌(自旋过渡材料)、臭葱石和混合金属(氢)氧化物库(例如水钠锰矿、层状双氢氧化物和尖晶石),通过一种称为气体扩散电结晶(GDEx)的方法。根据EP3242963B1的方法,将氧气供应到含有溶解于其中的所述金属化合物的溶液中并在气体扩散阴极上被还原,从而生成羟基离子(OH
因此需要一种方法,其允许从复杂的进料流分离单质形式的铂族金属,所述进料流含有一种或多种铂族金属离子的一种或多种前体化合物。
因此,本发明的目的在于提供一种方法,用于从含有一种或多种铂族金属的一种或多种前体化合物的进料中回收单质形式的铂族金属颗粒。
根据本发明,用一种显示出第一权利要求的特征部分的技术特点的方法使其得以实现。
此外,本发明的方法是从含有一种或多种铂族金属离子的一种或多种前体化合物的进料回收铂族金属的工艺,包括以下步骤:
(i)向配备有阴极的电化学槽的阴极隔室供应含有一种或多种前体化合物的进料,以在阴极隔室中形成液相,所述阴极包括具有多孔电化学活性材料的气体扩散电极,
(ii)将含有CO
(iii)向阴极施加电势,以例如导致CO
(iv)并且从液相回收一种或多种单质形式的铂族金属的沉淀颗粒。
所述进料可源于多种多样的来源,而且经常是工业原料。进料尤其可源于从报废产品例如从可回收材料如电路板、电子设备、催化剂、燃料电池、玻璃、陶瓷、颜料、医用牙科用品、药品、光伏材料、超合金等回收铂族金属或贵金属的工艺。此类工业废弃物进料的一个问题是铂族金属离子的浓度可能相当低,大约为每吨废弃物几百克。进料通常是水基的,尽管也可能存在一种或多种有机溶剂。
含有一种或多种铂族金属离子的一种或多种前体化合物的进料,可作为含有一种或多种前体化合物的固体或液体进料,供应到阴极隔室中。液体进料可以是溶液,所述溶液包含溶解于其中的一种或多种铂族金属离子的一种或多种前体化合物,抑或是在液体分散剂中一种或多种前体化合物的分散体或悬浮体。
技术人员应该清楚阴极隔室中的液相包含所述进料。
已令人惊讶地发现,本发明的方法使得从进料沉淀相应单质形式的铂族金属的颗粒或颗粒团簇成为可能,所述进料含有一种或多种铂族金属离子的一种或多种前体化合物。这些颗粒适合回收用于进一步利用。已发现单质形式的铂族金属颗粒沉淀主要出现在阴极电解质中,且允许从液相简化的回收金属颗粒。
沉淀颗粒可采取纳米颗粒的形式,其粒度最大为100nm或者更小。沉淀颗粒也可采取金属颗粒的团聚体或团簇的形式,其中所述团簇或团聚体具有大于100nm的粒度。有利地,本发明的方法允许通过操控一个或多个工艺参数来控制颗粒的尺寸。例如为了控制沉淀颗粒的粒度,可以改变一种或多种前体化合物的浓度、供应到阴极隔室的电解质的流量、气体流量、电解质的电导率等。
颗粒可包括单一类型的铂族金属,抑或两种以上不同铂族金属的混合物,取决于进料的组成。
因此,可使铂族金属颗粒以尤其适合进一步处理的形式回收。在一个实例中,单质的铂族金属沉淀颗粒可重新溶解于适合的溶剂中,用于在特定的应用中使用,它们可分散于分散剂中以提供悬浮液或浆液,或者它们可被制成干粒料。
有利地,本发明的方法为电化学工艺且所消耗的电荷主要涉及CO
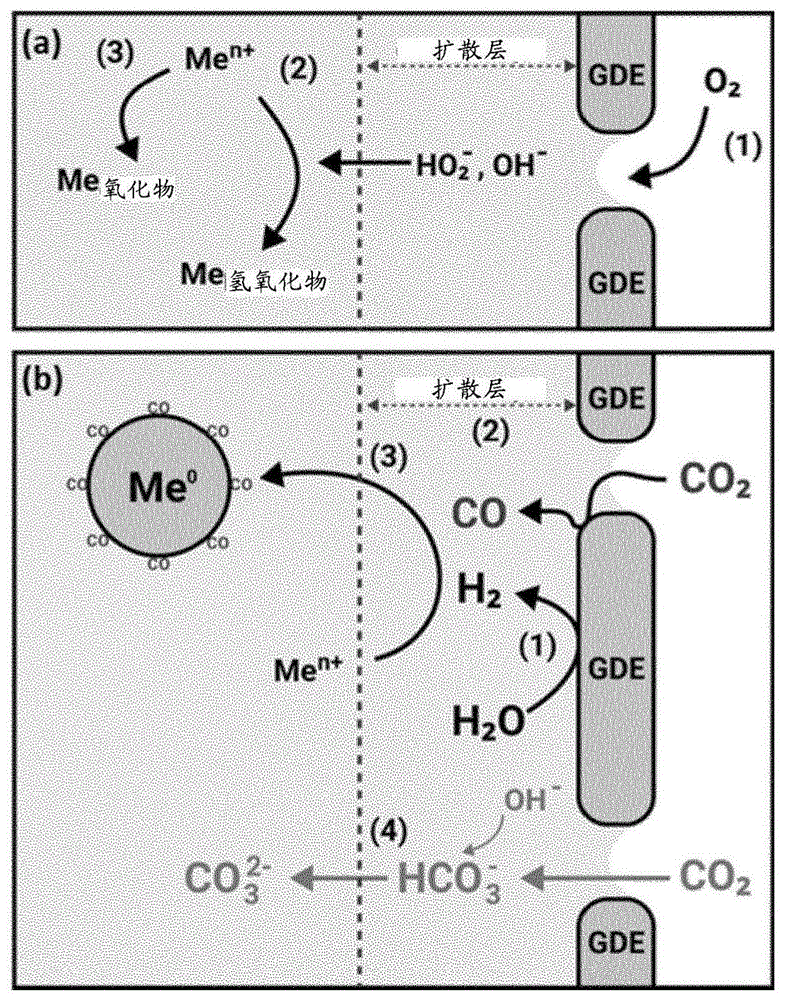
发明人假定,不想被该理论所约束,在本发明的条件下CO是CO
尽管某些铂族金属离子的还原可能发生于气体扩散电极的表面处,往往是电化学还原,但铂族金属离子的还原主要发生于阴极隔室中,特别是在阴极电解质中。不想被该假设所约束,发明人认为,铂族金属的还原主要出现在电极附近的位置,其中CO
本发明的方法可以作为连续工艺、分批工艺进行,且阴极隔室中所含的液相可以再循环利用。
在一个优选实施方案中,液相可包含一种或多种溶剂,所述溶剂选自包括水、可生成H
在阴极隔室内的液相中存在水的情况下,要么源自进料、要么源自阴极电解质,假定水还原反应生成H
使用气体扩散电极允许供应足够数量的CO
CO
E
E
E
E
E
E
E
E
注意到这些反应主要在电催化剂存在的情况下发生,且在电催化剂不存在的情况下是可以忽略不计的。
在有机溶剂存在的情况下,除CO外还可形成其他封盖剂,例如柠檬酸和/或草酸,其可有助于控制沉淀的单质铂族金属颗粒的粒度。
在一个优选的实施方案中,将CO
含有一种或多种铂族金属离子的液体进料可以是含水的液体进料。除水外,液体进料还可包含一种或多种的各类常用的工业溶剂,在上文公开了其实例。更优选地,含有一种或多种铂族金属离子的液体进料是该类金属化合物在水中的液体进料。
如果阴极电解质中存在水,假定CO
E
E
根据本发明,尽管CO
CO
含有一种或多种铂族金属离子的进料可包含单一类型的铂族金属离子或者两种以上不同铂族金属离子的混合物。本发明的方法使得获得单质形式铂族金属的沉淀物成为可能,不依赖于铂族金属的性质。
本发明的方法通常在低于液相沸点的温度进行。在液相包含水的优选实施方案中,本发明的工艺优选在最高为100℃的温度下进行,优选10℃-100℃的温度,更优选15-75℃,最优选15-50℃,尤其是大约室温的温度。与此相反,现有技术的工艺需要使用高于100℃的高温,往往高达210℃,因此是非常耗能的。
有利地,本发明的工艺可在单一步骤、单一反应器、在大气压力和适中的温度下进行。
液相的pH在本发明的工艺开始时可在宽pH范围内变化,但是优选地,液相的pH在工艺开始时最大为5.0,优选地最大为4.0,更优选地最大为3.0。在这些pH值下,铂族金属沉淀为氢氧化物的风险可降至最低。发明人观察到在反应过程中,阴极电解质的pH可逐渐向碱性pH发展。在本发明的一个实施方案中,随着铂族金属以其金属形式沉淀的进行,液相的pH可发生演变。实际上,如上文所描述的CO
液相的pH在本发明的工艺开始时可至少为7.0,优选地至少为8.0,更优选地约为9.0,或者例如在目的在于单质Pt沉淀的情况下甚至为11.0。pH可在反应过程中进行演变,通常这意味着下降,例如pH到8.0或9.0。
优选地,考虑到存在于液相中的铂族金属离子的性质,将液相的pH在本发明的工艺开始时作调整。有利地,如果液相只含有Pt离子作为铂族金属离子,pH可演变至11,因为只有高于该pH,预计才会出现Pt氢氧化物的形成。
在本发明的范围内铂族金属是指选自Pd、Pt、Rh、Ru、Os、Ir的一种或多种金属,包括两种以上所述金属的混合物。各金属离子可以相近的浓度存在于进料中,但各铂族金属离子的浓度也可不同。金属离子可以单一氧化态存在于进料中,例如Pd
本发明的方法适合用于从来源广泛的溶液中回收铂族金属。本发明的工艺适合用于从报废应用回收铂族金属,例如从来源于催化转化器、印刷电路板、电子元件等的进料回收铂族金属离子。本发明的工艺同样适合用于从废料流例如废渣、尾矿、废水、沥滤液等回收铂族金属。
在本发明的方法主要用于从工业废料流中回收铂族金属的情形中,这些废料流中的铂族金属离子的浓度可能相当低(例如ppm至ppb范围)。不过,本发明的方法也适合从较高浓度的溶液中回收金属。
CO
当使用气体混合物时,技术人员能够调整CO
有利地,将CO
技术人员应该明确,阴极隔室可包含阴极电解质,且在本发明的工艺过程中,电解质可以适当的流量供应到阴极隔室,例如辅助实现所需的质量流量。
在一个优选的实施方案中,气体扩散阴极所经受的电化学势是相对于参比电极的还原电势,优选低于CO
有利地,施加到电极的电荷为-10至-50mAcm
附图列表
-图1.1显示当(a)使用O
-图1.2是适合于本发明使用的电化学反应器的示意图。
-图2.1实施例2中,在-1.4V vs Ag/AgCl下的CO
-图2.2在实施例2中在GDEx过程期间,作为Rh洗提液pH的函数的Rh浓度演变。每个柱体上方的数据表示金属去除的百分率。
-图3.1显示了实施例3中在-1.4V vs Ag/AgCl下CO
-图3.2显示了实施例3中所得产物的XRD图样。
-图4.1显示了在-1.4V vs Ag/AgCl下CO
-图4.2显示了在-1.4V vs Ag/AgCl下还原CO
-图4.3显示了使用CO
-图5.1显示了在-1.4V vs Ag/AgCl下CO
-图5.2显示了在-1.4V vs Ag/AgCl下CO
-图5.3显示了实施例5中使用CO
-图6.a)当CO
-图7.左:当Ar在气相处流动且在阴极电解质容器中鼓泡CO
在下文的实施例中将进一步说明本发明。
材料和方法
化学品
六氯铂(IV)酸(H2PtCl6,Pt 39.93wt%)(Johnson Matthey)、氯化钯(II)(PdCl2,99.9%)(Sigma-Aldrich)、氯化铑(III)水合物(RhCl3·xH2O,99.98%)(Sigma-Aldrich)、氯化钠(NaCl,99.5%)(Acros Organics)、盐酸(HCl,37wt%)(Sigma-Aldrich)、二氧化碳(CO
电化学反应器
电化学反应器(图1.2)由三隔室电化学槽构成。经过第一隔室,具有设定过压的气体(即CO
合成步骤
在NaCl 0.5M中制备三种铂族金属离子(PGM)前体化合物(H2PtCl6、PdCl2、RhCl3)储备溶液0.1M,并用于制备相应的工作溶液。GDEx过程的背景电解质是由用浓HCl调节的pH为3的NaCl0.5M溶液组成的。阴极电解质溶液是通过混合背景电解质和PGM储备溶液制备的,使得Pt
气体(即CO
用配备有Metrohm Unitrode pH电极的Metrohm 781pH/离子计每5秒测量pH。在不同时间从阴极电解质中取出1ml等分试样,加入100μl的HCL 0.1M以淬灭反应并避免未反应的金属离子沉淀。将等分试样离心,并用0.3μm孔径过滤器过滤。
将过滤后的溶液用电感耦合等离子体-光发射光谱仪(ICP-OES)(Varian 750ES)分析,以监测液相中的金属浓度。由清液变为深色浑浊溶液是形成纳米颗粒的标志。在将阴极电解质等分试样离心之后上清液变透明时,将实验停止。将纳米颗粒留置整夜以沉降。倒出上清液,用去离子水使纳米颗粒重新悬浮并用Hettich Rotina 35离心机以10000rpm离心操作以去除剩余的NaCl。重复洗涤步骤直至上清液的电导率与去离子水相似。将产物在Ar气氛下室温干燥并存放用于进一步表征。
表征
X射线衍射(XRD):通过粉末X射线衍射在以40kV电压和40mA电流的Seifert3003T/T衍射仪中将干燥后的样品进行分析,该衍射仪采用Cu的Kα射线
D=(κλ)/(βcosθ)(1)
其中D为晶畴的平均微晶尺寸,κ为Scherrer常数,对于球形颗粒通常被认为是0.89,λ为X射线波长,β为强度最大值的一半处的线展宽(FWHM),为弧度制,θ(°)为反射面上的Bragg角。
电子扫描显微镜(SEM):用Philips XL30 FEG扫描电子显微镜得到干燥样品的显微图像,图像由二次电子成像且加速电压为30kV。通过将粉末分散于乙醇中并超声30分钟制备样品。然后,将10μL倒入安装在样品支架上的铝箔中。通过使用软件ImageJ(NIH)对至少100个颗粒计数来评估平均粒度及分布。此后,将数据拟合为对数正态分布以获得平均粒度和标准差。
实施例1-空白实验.
图2中(灰线)显示了在没有PGM时(空白),随CRR进行,作为单位体积所消耗电荷的函数的pH演变。从酸性条件(即pH~3)开始,当仅500C L
实施例2.
以1M HCL作为基质的洗提溶液被用作初始溶液,该洗提溶液包含1.035M的氯离子,0.7M的硫酸根离子,以及pH为-0.02,下列元素的浓度以mg L
本发明方法对象的操作条件与实施例1中所呈现的相同。
在GDEx期间在阴极(在该阴极上发生CO
表2.1.
金属去除百分率
实施例3
以1M HCl为基质制备旨在部分地模拟实施例2中洗提溶液的合成溶液,以mg L
图3.1显示了pH和Rh浓度随时间的演变。在处理12小时且pH达到~1.5之后,大于98%的Rh已从溶液中去除,在实验结束时,98.1%的Rh已从含有100mg L
表3.1在实验结束时在Rh模拟洗提中所述金属的金属去除百分率
实施例4.
沥滤溶液从用过的催化转化器的预处理获得。沥滤液被负载(supported)于HCl6M+H
本发明的方法对象的操作条件与先前的实施例中所呈现的相同。
图4.1显示了实验过程中pH的演变。在用GDEx处理沥滤液的过程中出现的不同的颜色变化被突出显示。处理约22小时后,pH达到0.5(图5.a)且~99%的PGM已从沥滤液中去除(图4.2)。表4.1显示了沥滤液的不同成分的浓度演变,其中从溶液中最少量除去非PGM元素是值得注意的,尤其是处理高达7小时,同时已出现最高的PGM去除率。如图4.3所示,从该处理中回收的产物由纯PGM的混合物组成。
表4.1.在不同取样时间从沥滤液去除金属的百分率。
实施例5
沥滤溶液从废催化转化器的预处理获得。沥滤液被负载于HCl 6M+H
本发明方法对象的操作条件与先前的实施例中所呈现的相同。
图5.1显示了实验过程中pH的演变。在用GDEx处理沥滤液过程中出现的颜色的不同变化被突出显示。如实施例1所示,通过22小时的处理,大部分PGM被去除,用GDEx对该沥滤液的处理时间停止于22小时(即,见图5.1中对两个实验的pH演变的比较)。达到的最大pH为~0.75,此时99.9%的PGM已从沥滤液中去除(图5.2)。不过,表5.1中显示,最大PGM回收率出现在处理最初7小时内。如图5.3所示,从处理回收的产物由纯PGM的混合物组成。
实施例6在气体隔室用Ar替代CO
为了说明在GDEx过程中CO
为了说明生成的CO在GDEx过程中在CO
参考文献列表.
1.P.Hervés,M.Pérez-Lorenzo,L.M.Liz-Marzán,J.Dzubiella,Y.Lu andM.Ballauff,Chem.Soc.Rev.,2012,41,5577–5587.
2.D.Banham and S.Ye,ACS Energy Lett.,2017,2,629–638.
3.E.Antolini,J.Power Sources,2007,170,1–12.
4.S.Sui,X.Wang,X.Zhou,Y.Su,S.Riffat and C.Liu,J.Mater.Chem.A,2017,5,1808–1825.
5.A.P.Reverberi,N.T.Kuznetsov,V.P.Meshalkin,M.Salernoand B.Fabiano,Theor.Found.Chem.Eng.,2016,50,59–66.
6.L.D.Rampino and F.F.Nord,J.Am.Chem.Soc.,1941,63,2745–2749.
7.T.S.Ahmadi,Z.L.Wang,T.C.Green,A.Henglein and M.A.El-Sayed,Science(80-.).,1996,272,1924 LP–1925.
8.J.M.Petroski,Z.L.Wang,T.C.Green and M.A.El-Sayed,J.Phys.Chem.B,1998,102,3316–3320.
9.B.Wu,N.Zheng and G.Fu,Chem.Commun.,2011,47,1039–1041.
10.G.Chen,Y.Tan,B.Wu,G.Fu and N.Zheng,Chem.Commun.,2012,48,2758–2760.
11.S.-H.Chang,M.-H.Yeh,C.-J.Pan,K.-J.Chen,H.Ishii,D.-G.Liu,J.-F.Lee,C.-C.Liu,J.Rick,M.-Y.Cheng and B.-J.Hwang,Chem.Commun.,2011,47,3864–3866.
12.C.Zhang,S.Y.Hwang and Z.Peng,J.Mater.Chem.A,2013,1,14402–14408.
13.C.F.Zinola,J.Electrochem.Soc.,2017,164,H170–H182.
14.C.Delacourt,P.L.Ridgway,J.B.Kerr and J.Newman,J.Electrochem.Soc.,2008,155,B42.
15.T.Burdyny and W.A.Smith,Energy Environ.Sci.,2019,12,1442–1453.
16.R.A.Prato,V.Van Vught,S.Eggermont,G.Pozo,P.Marin,J.Fransaer andX.Dominguez-Benetton,Sci.Rep.,2019,9,15370.
17.G.Pozo,P.de la Presa,R.Prato,I.Morales,P.Marin,J.Fransaer andX.Dominguez-Benetton,Nanoscale,2020,12,5412–5421.
18.G.Pozo,D.van Houtven,J.Fransaer and X.Dominguez-Benetton,React.Chem.Eng.,2020,5,1118–1128.
19.R.A.Prato M.,V.Van Vught,K.Chayambuka,G.Pozo,S.Eggermont,J.Fransaer and X.Dominguez-Benetton,J.Mater.Chem.A,2020,8,11674–11686.
20.N.Yang,S.R.Waldvogel and X.Jiang,ACS Appl.Mater.Interfaces,2016,8,28357–28371.
21.J.Albo,M.Alvarez-Guerra,P.
22.H.Ooka,M.C.Figueiredo and M.T.M.Koper,Langmuir,2017,33,9307–9313.
23.W.Zhou,J.Wu and H.Yang,Nano Lett.,2013,13,2870–2874.
24.N.Gupta,M.Gattrell and B.MacDougall,J.Appl.Electrochem.,2006,36,161–172.
25.R.E.Cameron and A.B.Bocarsly,Inorg.Chem.,1986,25,2910–2913.
26.C.F.Holder and R.E.Schaak,ACS Nano,2019,13,7359–7365.
27.H.Einaga and M.Harada,Langmuir,2005,21,2578–2584.
28.A.Henglein and M.Giersig,J.Phys.Chem.B,2000,104,6767–6772.
29.P.Jeanty,C.Scherer,E.Magori,K.Wiesner-Fleischer,O.Hinrichsen andM.Fleischer,J.CO2 Util.,2018,24,454–462.
- 用于从含有铂族金属的工业废弃物回收铂族金属的方法
- 一种处理铂族金属氯化铵沉淀渣的方法