1,3-二芳巯基中氮茚衍生物的电化学制备方法和应用
文献发布时间:2024-04-18 19:58:53
技术领域
本发明属于有机合成技术领域,具体涉及一种1,3-二芳巯基中氮茚衍生物的电化学制备方法和应用。
背景技术
中氮茚,又称为吲哚嗪,是通常用于各种小分子药物发现计划的特权结构。功能化的吲哚嗪在天然产物和合成药物中得到了广泛的应用,这些药物与广泛的生物活性有关,如抗真菌、抗癌、抗氧化剂、磷酸酶抑制和拮抗剂。特别是,3-亚磺酰吲哚嗪是CRTH2受体的配体,在治疗各种呼吸系统疾病中很有价值
现有技术中合成吲哚嗪衍生物主要有两条路线。路线一,通过吡啶衍生物和卤代化合物制成吡啶鎓盐,之后和炔烃反应制成多取代中氮茚,该路线的主要问题是炔烃的价格、炔烃结构本身的限制以及炔烃的可得性存在较大的问题,使得使用该路线制取多取代中氮茚存在目标产物的结构多样性和可调性较差,同时成本非常高昂。路线二,也是通过吡啶衍生物和卤代化合物制成吡啶鎓盐,然后和缺电子烯烃在氧化剂的存在下,制取多取代吡啶。相对于路线一,路线二具有很大的优势,如烯烃结构相对炔烃丰富、价格低廉且易获取。但该路线使用的各种氧化剂均存在不同的问题。考虑到吲哚嗪在各种生物活性和物质化学中的普及,仍然需要开发出强大的吲哚嗪合成方法。
电催化电合成是一种高效、环保、可控性强的有机合成方法,具有广泛的应用前景。随着电催化剂的不断发展和改进,电催化电合成在各个领域发挥越来越重要的作用。近十多年来,电催化与电合成在基础理论和应用研究方面都展现了很多的新颖、前沿的热点研究方向,持续吸引着科研人员的关注和投入,不断融合材料、能源、环境、信息、AI智能等学科知识,迎来一个黄金发展期。利用电催化合成化学近年来引起大多数有机合成研究者的兴趣。电催化转化被认为是在温和条件下进行有机合成的一种有前景的替代方法。它们提供了独特的直接途径,可以使通常难以使用其他反应类型合成的分子结构。电催化反应的发展是合成化学中一个有吸引力的合成策略。与过渡金属催化剂相比,电解质具有成本低、合成通用性强、无毒环保等优点。
目前,没有通过电催化合成吲哚嗪衍生物的相关现有技术的公开。
[1]Yang DT,Radtke J,Mellerup SK,et al.One-Pot Synthesis of BrightlyFluorescent Mes2B-Functionalized Indolizine Derivatives via CycloadditionReactions[J].Org Lett.2015May 15;17(10):2486-9.
[2]Prasad ChD,Kumar S,Sattar M,Adhikary A,et al.Metal freesulfenylation and bis-sulfenylation of indoles:persulfate mediated synthesis[J].Org Biomol Chem.2013Dec 14;11(46):8036-40.
[3]Nalbandian CJ,Miller EM,Toenjes ST,et al.A conjugate Lewis base-
acid catalyst for the sulfenylation of nitrogen containingheterocycles under mild conditions[J].Chem Commun(Camb).2017Jan 26;53(9):1494-1497.
发明内容
基于现有技术中存在的问题和不足,本发明旨在提供1,3-二芳巯基中氮茚衍生物的电化学制备方法和应用,提供一种原料来源安全无毒、以电为能源、对环境友好反应条件绿色温和高效的制备1,3-二芳巯基中氮茚衍生物的新途径。本发明制备得到的1,3-二芳巯基中氮茚衍生物具有一定的抗癌细胞增殖活性,且在克级反应中具有良好的产率,在工业生产方面具有潜在的应用前景。
术语:
1、中氮茚:又称吲哚嗪,是通常用于各种小分子药物发现计划的特权结构,功能化的吲哚嗪在天然产物和合成药物中得到了广泛的应用,这些药物与广泛的生物活性有关。
2、电解质:电解质是溶于水溶液中或在熔融状态下自身能够导电的化合物。根据其电离程度可分为强电解质和弱电解质,几乎全部电离的是强电解质,只有少部分电离的是弱电解质。
3、溶剂:是一种可以溶化固体、液体或气体溶质的液体,继而成为溶液,溶剂通常拥有比较低的沸点和容易挥发,或是可以由蒸馏来去除,从而留下被溶物,因此,溶剂不可以对溶质产生化学反应。
4、四丁基碘化铵:白色结晶粉末。微溶于水(10g/100mL)及甲醇。
5、溴化钠:无色立方晶系晶体或白色颗粒状粉末,溴化钠微溶于醇,可与稀硫酸反应生成溴化氢。在酸性条件下,溴化钠能被氧化,游离出溴。
6、碘化钾:是一种无机化合物,化学式为KI,为无色或白色晶体,无臭易溶于水和乙醇。水溶液见光变暗,并游离出碘。
7、IC50:又称为半抑制浓度,可以作为药物抗肿瘤活性的指标,能指示某一药物或者抑制剂在抑制某些物质如酶、细胞受体或者微生物的半量,该值可以用来衡量药物诱导凋亡的能力,即诱导能力越强,该数值越低。在细胞凋亡实验中可以通过计算出IC50值,来研究药物对肿瘤细胞的抑制作用。
一方面,本发明提供了一种1,3-二芳巯基中氮茚衍生物的制备方法,所述制备方法包括如下步骤:以中氮茚为原料,以烷基或芳基硫醇为硫源,在电催化体系作用下,制备得到1,3-二芳巯基中氮茚衍生物;
所述的中氮茚衍生物的结构如式1所示:
其中,所述R
具体地,所述电催化体系包括电源、电解质和溶剂。
进一步具体地,所述电源为直流电源。
优选地,所述电源直流电源的条件为:电流为5-15mA,电压为10-20V。
进一步优选地,所述电源直流电源的条件为:电流为10mA,电压为15V。
具体地,所述电解质为四丁基碘化铵、溴化钠、碘化钾和碘化钠中的一种或几种。
进一步具体地,所述电解质为四丁基碘化铵、溴化钠中的一种或几种。
优选地,所述电解质为四丁基碘化铵。
具体地,所述溶剂选自1,2-二氯乙烷、二甲基亚砜、乙腈、N,N-二甲基甲酰胺、二氯甲烷和水中的一种或几种。
进一步具体地,所述溶剂选自1,2-二氯乙烷、乙腈、N,N-二甲基甲酰胺、二氯甲烷和水中的一种或几种。
优选地,所述溶剂选自乙腈、N,N-二甲基甲酰胺、二氯甲烷和水中的一种或几种。
优选地,所述溶剂选自乙腈、二氯甲烷和水中的一种或几种。
进一步优选地,所述溶剂选自乙腈和水,所述溶剂为混合溶剂。
具体地,所述混合溶剂水:乙腈的比例选自1:1-6。
进一步具体地,所述混合溶剂水:乙腈比例选自1:1-5
优选地,所述混合溶剂水:乙腈比例选自1:2-5
优选地,所述混合溶剂水:乙腈比例选自1:3-5
进一步优选地,所述混合溶剂水:乙腈比例为1:5
具体地,所述产物1,3-二芳巯基中氮茚衍生物结构如式2所示:
其中,所述R
进一步具体地,所述的1,3-二芳巯基中氮茚衍生物的结构式中的R
所述R
所述R
优选地,所述的1,3-二芳巯基中氮茚衍生物的结构式包括但不限于以下结构:
具体地,所述中氮茚与硫醇的摩尔比选自1:1.2-6。
进一步具体地,所述中氮茚与硫醇的摩尔比选自1:1.4-5.8。
优选地,所述中氮茚与硫醇的摩尔比选自1:1.6-5.6;进一步优选为1:
1.8-5.4;再进一步优选为1:2-5.2;更进一步优选为1:2.2-5;更进一步优选为1:2.4-4.8;更进一步优选为1:2.6-4.6;更进一步优选为1:2.8-4.4;更进一步优选为1:3-4.2;更进一步优选为1:3.2-4.0;更进一步优选为1:3.4-3.8;更进一步优选为1:3.4-3.6。
具体地,所述电解质与中氮茚的摩尔比选自0.001-7:1。
进一步具体地,所述电解质与中氮茚的摩尔比选自1-5:1。
优选地,所述电解质与中氮茚的摩尔比选自2-7:1;进一步优选为为3-7:1;再进一步优选为4-7:1。
具体地,上述制备方法的合成路线为:
其中,所述R
进一步具体地,反应物1与反应物2摩尔比选自1:1.2-6。
优选地,所述反应物1与反应物2摩尔比选自1:1.4-5.8;进一步优选为1:1.6-5.6;再进一步优选为1:1.8-5.4;更进一步优选为1:2-5.2;更进一步优选为1:2.2-5;更进一步优选为1:2.4-4.8;更进一步优选为1:2.6-4.6;更进一步优选为1:2.8-4.4;更进一步优选为1:3-4.2;更进一步优选为1:3.2-4.0;更进一步优选为1:3.4-3.8;更进一步优选为1:3.4-3.6。
又一方面,本申请提供了一种上述制备方法所制备的化合物,所述化合物选自以下结构:
又一方面,本申请提供了上述制备方法和/或上述化合物的应用,所述应用包括在农用化学品、荧光材料、制药、医疗和/或化工领域的应用。
具体地,所述应用包括在制备抗癌药物中的应用。
进一步具体地,所述应用包括在制备防治癌症的药物中的应用。
优选地,所述癌症包括胰腺癌、宫颈癌、结直肠癌、肺癌、肝细胞癌、肾癌、胃癌和胆管癌等。
进一步优选地,所述癌症包括肝细胞癌、宫颈癌。
本发明的有益效果为:
(1)本发明原料来源安全无毒,以电为能源,对环境友好反应条件绿色温和高效,对环境友好反应条件绿色温和高效,为有机化学研究者们提供了1,3-二芳巯基中氮茚衍生物的新途径。
(2)本发明通过电催化直接合成1,3-二芳巯基中氮茚衍生物,制备得到的1,3-二芳巯基中氮茚衍生物具有一定的抗癌细胞增殖活性,在克级反应中具有良好的产率。
(3)本发明技术方案制备的1,3-二芳巯基中氮茚衍生物的方法,操作简单,不含金属参与,反应底物适用范围广,区域选择性好,收率高,可绿色高效的合成一系列1,3-二芳巯基中氮茚衍生物,在农用化学品、药物制备和荧光材料方面具有广阔的应用前景。
附图说明
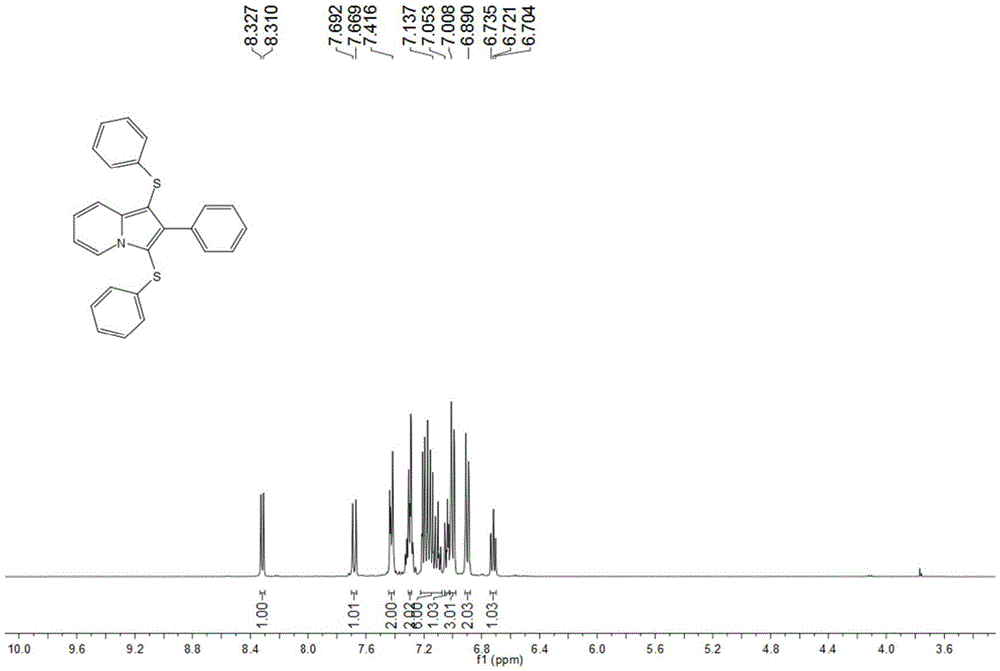
图1为本发明实施例1制备的化合物的氢谱图。
图2为本发明实施例1制备的化合物的碳谱图。
图3为本发明实施例2制备的化合物的氢谱图。
图4为本发明实施例2制备的化合物的碳谱图。
图5为本发明实施例3制备的化合物的氢谱图。
图6为本发明实施例3制备的化合物的碳谱图。
图7为本发明实施例4制备的化合物的氢谱图。
图8为本发明实施例4制备的化合物的碳谱图。
图9为本发明实施例5制备的化合物的氢谱图。
图10为本发明实施例5制备的化合物的碳谱图。
图11为本发明实施例6制备的化合物的氢谱图。
图12为本发明实施例6制备的化合物的碳谱图。
图13为本发明实施例7制备的化合物的氢谱图。
图14为本发明实施例7制备的化合物的碳谱图。
图15为本发明实施例8制备的化合物的氢谱图。
图16为本发明实施例8制备的化合物的碳谱图。
图17为本发明实施例9制备的化合物的氢谱图。
图18为本发明实施例9制备的化合物的碳谱图。
图19为本发明实施例10制备的化合物的氢谱图。
图20为本发明实施例10制备的化合物的碳谱图。
图21为本发明实施例11制备的化合物的氢谱图。
图22为本发明实施例11制备的化合物的碳谱图。
具体实施方式
下面结合具体实施例,对本发明进行说明,下述实施例不用于限制本发明,仅用于说明本发明,以使本发明技术方案更易于理解掌握。下属实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明均可从商业途径获得。
实施例1
将19.4mg(0.1mmol)吲哚嗪、96.5mg-167.3mg(0.4mmol-0.7mmol)四丁基碘化铵、13.22mg-66.06mg(0.12mmol-0.6mmol)苯硫醇置于25mL带有搅拌子的试管中,于室温加入5mL乙腈和水的混合溶液后,在通电条件下搅拌6小时,旋干后快速柱层析得到目标产物,90%产率。
1H NMR(400MHz,Chloroform-d)δ8.32(d,J=7.2Hz,1H),7.68(d,J=8.8Hz,1H),7.42(dd,J=7.6,2.0Hz,2H),7.30(d,J=6.0Hz,2H),7.22-7.07(m,6H),7.05-7.03(m,1H),7.00(dd,J=8.4,1.2Hz,3H),6.90(d,J=7.2Hz,2H),6.74-6.70(m,1H).
13C NMR(101MHz,CDCl3)δ141.6,140.1,139.4,136.6,133.1,130.4,129.3,128.7,127.8,127.6,125.6,125.2,125.2,124.6,124.5,122.2,117.8,112.8,107.6,97.4.
HR-ESI-MS m/z calcd.for C26H19NS2[M+H]+:409.0959,found:409.0955.
实施例2
将19.4mg(0.1mmol)吲哚嗪、96.5mg-167.3mg(0.4mmol-0.7mmol)四丁基碘化铵、15.22mg-75.11mg(0.12mmol-0.6mmol)邻甲基苯硫醇置于25mL带有搅拌子的试管中,于室温加入5mL乙腈和水的混合溶液后,在通电条件下搅拌6小时,旋干后快速柱层析得到目标产物,80%产率。
1H NMR(400MHz,CDCl3)δ8.40(t,J=6.1Hz,1H),7.83-7.78(m,1H),7.58(s,2H),7.47-7.42(m,3H),7.33-7.25(m,2H),7.15(dd,J=15.0,6.0Hz,5H),6.88-6.81(m,2H),2.59(s,6H).
13C NMR(101MHz,CDCl3)δ141.98,139.64,139.28,135.76,134.83,134.39,133.29,130.66,130.49,130.18,130.01,129.69,127.85,127.62,126.92,126.44,125.39,124.66,124.39,124.10,122.13,118.01,112.82,107.00,97.05,19.78,19.66.
HR-ESI-MS m/z calcd.for C28H23NS2[M+H]+:438.1345,found:438.1340.
实施例3
将19.4mg(0.1mmol)吲哚嗪、96.5mg-167.3mg(0.4mmol-0.7mmol)四丁基碘化铵、15.4mg-61.4mg(0.12mmol-0.6mmol)3-氟苯硫醇置于25mL带有搅拌子的试管中,于室温加入5mL乙腈和水的混合溶液后,在通电条件下搅拌6小时,旋干后快速柱层析得到目标产物,84%产率。
1H NMR(400MHz,CDCl3)δ8.37(d,J=7.0Hz,1H),7.73(d,J=8.9Hz,1H),7.45-7.41(m,2H),7.39-7.34(m,3H),7.11-7.06(m,1H),6.98(dd,J=8.3,5.0Hz,2H),6.95-6.93(m,1H),6.92-6.85(m,5H),6.80(td,J=6.8,1.3Hz,1H).
13C NMR(101MHz,Chloroform-d)δ162.26(d,J=50.9Hz),159.83(d,J=49.5Hz),141.52,139.22,134.85(d,J=3.0Hz),133.00,131.41(d,J=3.2Hz),130.47,127.83,127.73,127.37(d,J=8.0Hz),127.29(d,J=8.1Hz),124.38,122.38,117.80,116.43(d,J=22.1Hz),115.80(d,J=22.0Hz),112.89,108.20,98.28.
HR-ESI-MS m/z calcd.for C26H17F2NS2[M+H]+:446.0843,found:446.0840.
实施例4
将19.4mg(0.1mmol)吲哚嗪、96.5mg-167.3mg(0.4mmol-0.7mmol)四丁基碘化铵、21.36mg-106.8mg(0.06mmol-0.3mmol)3-三氟甲基苯硫醇置于25mL带有搅拌子的试管中,于室温加入5mL乙腈和水的混合溶液后,在通电条件下搅拌6小时,旋干后快速柱层析得到目标产物,82%产率。
1H NMR(400MHz,CDCl3)δ8.36(d,J=7.0Hz,1H),7.70(d,J=8.9Hz,1H),7.40-7.36(m,3H),7.35-7.32(m,3H),7.31-7.26(m,3H),7.23(s,2H),7.14-
7.08(m,2H),6.93(d,J=7.8Hz,1H),6.83(t,J=6.8Hz,1H).
13C NMR(101MHz,CDCl3)δ154.99,142.37,141.53,139.55,138.25,132.61,131.88,131.56,130.97,130.24,129.76,129.11,128.22,128.03,127.97,124.43,124.22,122.97,122.53,122.06,121.84,121.49,117.76,113.31,108.69,106.93,96.71.
HR-ESI-MS m/z calcd.for C28H17F6NS2[M+H]+:546.0779,found:546.0775.
实施例5
将19.4mg(0.1mmol)吲哚嗪、96.5mg-167.3mg(0.4mmol-0.7mmol)四丁基碘化铵、25.6mg-128.1mg(0.12mmol-0.6mmol)3-三氟甲基-4-氯苯硫醇置于25mL带有搅拌子的试管中,于室温加入5mL乙腈和水的混合溶液后,在通电条件下搅拌6小时,旋干后快速柱层析得到目标产物,81%产率。
1H NMR(400MHz,CDCl3)δ8.26(d,J=7.0Hz,1H),7.60(d,J=8.4Hz,1H),7.28(s,5H),7.21(d,J=8.3Hz,3H),7.17-7.14(m,1H),7.08-7.02(m,1H),6.90(d,J=8.4Hz,1H),6.79-6.72(m,2H).
13C NMR(101MHz,CDCl3)δ142.45,139.62,139.54,136.28,132.26,131.63,130.13,129.06,128.97,128.16,128.10,124.33,124.05(q,J=5.6Hz).,123.77,123.70(q,J=5.6Hz),123.62,123.38,120.96,117.65,113.59,106.62,96.41.
实施例6
将21.02mg(0.1mmol)8-甲基-2-苯基吲哚嗪、96.5mg-167.3mg(0.4mmol-0.7mmol)四丁基碘化铵、13.22mg-66.06mg(0.12mmol-0.6mmol)苯硫醇置于25mL带有搅拌子的试管中,于室温加入5mL乙腈和水的混合溶液后,在通电条件下搅拌6小时,旋干后快速柱层析得到目标产物,80%产率。
1H NMR(400MHz,Chloroform-d)δ8.32(d,J=6.8Hz,1H),7.36(d,J=19.6Hz,5H),7.23(t,J=6.9Hz,4H),7.15(t,J=7.2Hz,1H),7.08(t,J=7.6Hz,1H),7.03(d,J=8.0Hz,2H),6.94(d,J=8.0Hz,2H),6.79(d,J=6.8Hz,1H),6.66(t,J=6.8Hz,1H),2.78(s,3H).
13C NMR(101MHz,CDCl3)δ143.0,142.7,138.3,136.7,133.4,130.6,130.0,129.2,128.8,127.6,127.5,125.5,125.2,124.7,124.2,123.3,123.0,112.23,108.1,97.7,19.6.
实施例7
将21.02mg(0.1mmol)7-甲基-2-苯基吲哚嗪、96.5mg-167.3mg(0.4mmol-0.7mmol)四丁基碘化铵、13.22mg-66.06mg(0.12mmol-0.6mmol)苯硫醇置于25mL带有搅拌子的试管中,于室温加入5mL乙腈和水的混合溶液后,在通电条件下搅拌6小时,旋干后快速柱层析得到目标产物,74%产率。
1H NMR(400MHz,CDCl3)δ8.22(d,J=6.8Hz,1H),7.43(d,J=23.2Hz,3H),7.30(s,3H),7.23-7.16(m,4H),7.13(d,J=7.2Hz,1H),7.07(d,J=7.6Hz,1H),7.01(d,J=7.6Hz,2H),6.91(d,J=7.2Hz,2H),6.59(d,J=6.8Hz,1H),2.34(s,3H).
13C NMR(101MHz,CDCl3)δ140.5,139.9,139.8,139.8,137.1,137.0,133.3,133.2,130.4,129.3,128.8,127.7,127.5,125.5,125.1,125.1,124.5,124.0,116.2,115.5,29.7.
实施例8
将22.1mg(0.1mmol)6,8-二甲基-2-苯基吲哚嗪、96.5mg-167.3mg(0.4mmol-0.7mmol)四丁基碘化铵、13.22mg-66.06mg(0.12mmol-0.6mmol)苯硫醇置于25mL带有搅拌子的试管中,于室温加入5mL乙腈和水的混合溶液后,在通电条件下搅拌6小时,旋干后快速柱层析得到目标产物,82%产率。
1H NMR(400MHz,Chloroform-d)δ8.09(s,1H),7.33-7.28(m,2H),7.28-7.22(m,3H),7.21-7.13(m,4H),7.09(t,J=7.2Hz,1H),7.02(d,J=7.6Hz,3H),6.97(d,J=7.2Hz,2H),6.89(d,J=7.2Hz,2H),6.61(s,1H),2.68(s,3H),2.18(s,3H).
13C NMR(101MHz,CDCl3)δ142.9,142.7,137.1,133.6,130.5,129.3,129.2,128.8,127.5,127.4,126.6,125.4,125.1,124.7,124.2,121.8,120.7,107.6,97.3,19.4,18.2.
ESI-MS m/z(%)437(100)[M+H]+;Anal.Calcd for C28H23NS2 C,76.85;H,5.30;N,3.20;Found:C,76.86;H,5.31;N,3.22.
实施例9
将25.3mg(0.1mmol)2-(3,4-二甲氧基苯基)吲哚嗪、96.5mg-167.3mg(0.4mmol-0.7mmol)四丁基碘化铵、13.22mg-66.06mg(0.12mmol-0.6mmol)苯硫醇置于25mL带有搅拌子的试管中,于室温加入5mL乙腈和水的混合溶液后,在通电条件下搅拌6小时,旋干后快速柱层析得到目标产物,71%产率。
1H NMR(400MHz,CDCl3)δ8.40(d,J=7.2Hz,1H),7.74(d,J=8.8Hz,1H),7.25-7.18(m,4H),7.14(t,J=7.2Hz,1H),7.07(t,J=7.2Hz,4H),7.02(d,J=6.4Hz,1H),6.95(d,J=7.6Hz,2H),6.91(s,1H),6.84(d,J=8.4Hz,1H),6.78(t,J=6.4Hz,1H),3.85(s,3H),3.38(s,3H).
13C NMR(101MHz,CDCl3)δ148.4,148.0,141.4,140.6,139.9,137.2,129.3,128.8,125.6,125.5,125.0,125.0,124.5,124.4,122.5,122.3,117.7,113.7,112.8,110.5,107.4,96.8,55.6,55.0.
实施例10
将23.8mg(0.1mmol)2-苯基-1,3-氧乙基吲哚嗪、96.5mg-167.3mg(0.4mmol-0.7mmol)四丁基碘化铵、13.22mg-66.06mg(0.12mmol-0.6mmol)苯硫醇置于25mL带有搅拌子的试管中,于室温加入5mL乙腈和水的混合溶液后,在通电条件下搅拌6小时,旋干后快速柱层析得到目标产物,85%产率。
1H NMR(400MHz,CDCl3)δ8.33(d,J=7.2Hz,1H),7.69(d,J=8.8Hz,1H),7.21(dt,J=15.2,7.6Hz,4H),7.13(t,J=7.2Hz,1H),7.09-7.03(m,2H),7.01(d,J=7.6Hz,2H),6.92(d,J=2.8Hz,3H),6.90(s,0H),6.77(dd,J=14.4,7.6Hz,2H),5.93(s,2H).
13C NMR(101MHz,CDCl3)δ147.2,147.0,141.4,140.1,139.4,136.6,129.32,128.79,126.79,125.62,125.22,125.17,124.65,124.49,124.30,122.23,117.8,112.8,110.9,107.9,107.6,100.9,97.4.
实施例11
将21.02mg(0.1mmol)2-(3-甲基苯基)吲哚嗪、96.5mg-167.3mg(0.4mmol-0.7mmol)四丁基碘化铵、13.22mg-66.06mg(0.12mmol-0.6mmol)苯硫醇置于25mL带有搅拌子的试管中,于室温加入5mL乙腈和水的混合溶液后,在通电条件下搅拌6小时,旋干后快速柱层析得到目标产物,80%产率。
1H NMR(400MHz,CDCl3)δ8.38(d,J=9.6Hz,1H),7.74(d,J=7.6Hz,1H),7.27-7.19(m,7H),7.16(d,J=6.8Hz,2H),7.12-7.03(m,4H),6.96(d,J=1.2Hz,2H),6.77(t,J=6.8Hz,1H),2.29(s,3H).
13C NMR(101MHz,CDCl3)δ141.8,140.3,139.4,137.1,136.8,133.0,131.3,129.2,128.7,128.4,127.6,127.4,125.5,125.4,125.3,124.6,124.5,122.1,117.8,112.7,107.7,97.6,21.4.
以上数据表明,根据本申请提供的制备方法制备的1,3-二芳巯基中氮茚衍生物产率在71%-90%之间,具有较高的产率。当原料为吲哚嗪,硫源为苯硫醇时,制得的目标产物产率最高,产率高达90%。
实验例1MTT测试
对实施例4、实施例9以及实施例11制备得到的1,3-二芳巯基中氮茚衍生物进行MTT测试,以奥希替尼(AZD-9291)作为阳性对照药,细胞选取HepG2(人肝癌细胞)和HeLa(宫颈癌细胞)。
具体操作方法如下:
(1)细胞复苏、培养与传代
将冻存细胞从冰箱中取出后,迅速置于37.5℃恒温水浴锅中解冻,随后在超净台中将解冻细胞加入至含1mL DMEM的ep管中,放入离心机中,以1000rpm的速度离心3分钟,完成后弃去上清液,加入完全培养基(DMEM培养基+10%牛血清蛋白FBS),吹打均匀后,取至培养瓶中。当培养基颜色发生变化时,则进行培养基的更换。当细胞长至培养瓶的90%时,则进行传代,首先倒掉旧培养基,缓慢加入2mL PBS清洗培养瓶,随后加入0.5mL胰酶消化,观察到细胞开始脱落时,加入培养基并轻轻吹打,直至细胞完全脱落,接着收集细胞悬液以1000rpm的速度离心3分钟,弃去上清液,加入完全培养基,吹打均匀后,分装至两个培养瓶中。
(2)MTT法
取对数生长的细胞,将其消化并吹打使细胞均匀分布,随后进行细胞计数,接着用完全培养基将其稀释至6×10
分析化合物的IC
表1部分化合物的体外抗增殖活性
以上数据表明,根据实施例4、实施例9以及实施例11制备得到的1,3-二芳巯基中氮茚衍生物对HepG2细胞和HeLa细胞均有一定的抑制作用。
上述详细说明是针对本发明其中之一可行实施例的具体说明,该实施例并非用以限制本发明的专利范围。应当指出的是,凡未脱离本发明所为的等效实施或变更,均应包含于本发明技术方案的范围内。因此,本发明专利的保护范围应以所附要求为准。
- 一种取代1,2,3三氮唑类二芳基嘧啶衍生物及其制备方法与应用
- 一种1,3-二取代-2-氟中氮茚衍生物的制备方法
- 一种1,3-二取代-2-氟中氮茚衍生物的制备方法