一种紫杉烷类化合物的含硅杂质化合物及其制备方法、应用
文献发布时间:2024-04-18 19:59:31
技术领域
本发明属于药物化学领域,具体涉及一种紫杉烷类化合物的含硅杂质化合物及其制备方法和应用。
背景技术
恶性肿瘤已成为严重威胁我国居民身体健康的一大类疾病,根据中国国家癌症中心统计的数据,近十年来恶性肿瘤的发病死亡呈持续上升趋势,恶性肿瘤的发病率每年增幅保持约3.9%,死亡率每年增幅保持约2.5%,2019年恶性肿瘤死亡占居民全部死因的24%左右,因恶性肿瘤所致的医疗花费超过2200多亿。肺癌、肝癌、上消化系统肿瘤及结直肠癌、女性乳腺癌等是我国主要的恶性肿瘤,其中肺癌位于男性发病第1位,而乳腺癌为女性发病首位。我国恶性肿瘤患者的5年相对生存率约41%,尽管与10年前相比总体提高了约10%,但是与发达国家相比仍有很大差距。总体来讲,我国恶性肿瘤防控形势严峻,国家、居民经济负担很大。
目前,药物治疗已成为临床肿瘤治疗的重要手段之一,临床使用的抗肿瘤药物约有80多种,大致分为以下6类:细胞毒类药物、激素类药物、生物反应调节剂、单克隆抗体药物、辅助药物以及其他类药物。
紫杉烷类药物是一类从红豆杉的树皮或树叶中提取或半合成的高效光谱细胞毒类抗肿瘤药物,为一款微管蛋白抑制剂。研究表明,该类药物通过作用于微管/微管蛋白系统,促进微管双聚体的装配和抑制多聚化过程使得微管更趋稳定,阻滞细胞于G2和M期,从而抑制肿瘤细胞的有丝分裂和增殖。
目前临床使用的紫杉烷类药物共有三种,分别是紫杉醇、多烯紫杉醇(多西他赛)和卡巴他赛,具体结构如下所示:
紫杉烷类抗肿瘤药物在使用过程中会出现多药耐药性,造成抗肿瘤效果下降。因此,开发新型的紫杉烷类药物具有很大的临床价值。在多西他赛的结构基础上,申请人进行了结构优化,经初步药效筛选得到一种新型紫杉烷类化合物,代号为JJH201601,该化合物已在申请号为CN201810185012.4的专利中公开并获得授权,其具体结构如下所示:
根据初步药效研究结果显示,裸鼠模型药效显著提高,且毒副作用明显下降,能够消除肿瘤(抑瘤率达到99%以上),停药后观察期内未发现肿瘤复发,该结果在肺癌A549、肝癌HepG2和胰腺癌Panc-1模型上得以验证,目前该化合物已进入行政审批阶段,预计将广泛应用于相关疾病的临床质量中。
而杂质的研究在药品的研发、仿制、申报过程中是必需且重要的环节,人用药品注册技术要求国际协会(ICH)将杂质定义为:药物中存在的、化学结构与药物不一样的任何成分。杂质含量是药品质量的关键质量属性,药物所含的杂质多数具有潜在生物活性,以及可能的成分之间的相互作用,影响药物的安全性及效能,甚至产生毒副作用。为了保证用药安全,活性药物成分中的每一个杂质都必须进行安全性评估,即建立保证安全性的杂质限度。根据人用药品注册技术标准国际协调会(ICH)要求,原料药或其制剂组合物中的单个杂质量如果超过0.05%,应需要报告;单个杂质量如超过0.1%,就需要进行确证;单个杂质量如超过0.15%,则需要有安全性数据支持。在药物研发过程中,需弄清杂质的结构、来源以及去向,需对工艺杂质、降解杂质等做到全面了解和控制,为药品质量的控制提供有效的理论依据。申请人在JJH201601原料药的开发过程中,通过强制降解试验、稳定性试验发现,JJH201601分子会发生降解或构型转化,部分杂质随着存储时间增加有增加趋势,而杂质具有生物活性、毒性均不可预料的特性以及会对原料药品质产生极大影响。
杂质与药品安全性的关系是很复杂的,手性化合物的光学异构体对药物安全性的影响尤为复杂,有的对映体药理作用相同但有程度差异,有的作用具有互补性,但不少光学异构体药物的不同光学异构体的存在可影响药效,甚至可能引起严重的不良反应。例如治疗感染的氧氟沙星的外消旋体的作用仅为左旋体的一半,再例如扎考必利的R-对映体和S-对映体作用相反,以及沙利度胺的S-异构体具有很强的胚胎毒性和致畸作用、而其R-异构体安全有效。
发明内容
针对如上所述的紫杉烷类化合物(JJH201601)进行大量的研究后,申请人发现了紫杉烷类化合物产生毒性及影响药效的关键并完成了本发明。
本发明为实现上述目的采用的技术方案是:一种紫杉烷类化合物的含硅杂质化合物,其结构式如式(I)所示:
进一步地,所述紫杉烷类化合物具有如式(II)所示结构:
本发明还公开了如上所述的紫杉烷类化合物的含硅杂质化合物的制备方法,具体步骤为用极性非质子溶剂溶解式III化合物得到混合物,再将混合物在加热或不加热条件下加入到含有酸性试剂的极性质子溶剂中,加热或不加热条件下搅拌或不搅拌至反应结束,经后处理制得如式I所示的紫杉烷类化合物的含硅杂质化合物
进一步地,所述极性非质子溶剂为二氯甲烷、氯仿、乙酸乙酯、四氢呋喃、丙酮、N,N-二甲基甲酰胺中的任一种;
再进一步地,所述极性非质子溶剂为二氯甲烷;
进一步地,所述酸性试剂为盐酸、氢溴酸或硫酸中的任一种;
再进一步地,为盐酸。
进一步地,所述极性质子溶剂为醇类溶剂;
再进一步地,为甲醇、乙醇、正丙醇、异丙醇、正丁醇、仲丁醇、异丁醇、叔丁醇中的一种或任几种;
再进一步地,为甲醇。
进一步地,反应温度为0~40℃,所述酸性试剂与极性质子溶剂的体积比为0.01~0.2:1;
再进一步地,所述反应温度为5~15℃,所述酸性试剂与极性质子溶剂的体积比为0.02~0.05:1。
进一步地,所述后处理为缓慢加入饱和碳酸氢钠溶液调节pH=7-9后萃取及柱层析;
再进一步地,所述柱层析为硅胶柱层析分离;
再进一步地,所述柱层析分离使用100-400目硅胶。
进一步地,所述柱层析分离时,洗脱剂为乙酸乙酯/正己烷的混合物、二氯甲烷/甲醇的混合物或乙酸乙酯/石油醚的混合物;
再进一步地,所述洗脱剂为乙酸乙酯和正己烷的混合物;
再进一步地,所述乙酸乙酯与正己烷的体积比为1:1~6;
再进一步地,所述乙酸乙酯与正己烷的体积比为1:3。
本发明还公开了如上所述的紫杉烷类化合物的含硅杂质化合物在控制紫杉烷类化合物或含有紫杉烷类化合物的药物组合物质量中的用途。
进一步地,如式(I)所示的紫杉烷类化合物的含硅杂质化合物在紫杉烷类化合物中的含量低于0.5%;
再进一步地,所述控制紫杉烷类化合物或含有紫杉烷类化合物的药物组合物质量具体为控制其药理活性、毒性或代谢速率。
根据JJH201601合成路线(如下所示),在DTX2水解反应步骤,由于侧链三乙基硅基更易脱去,生成中间体(式I所示分子)。作为工艺杂质,式I所示分子在成品中会有一定残留,控制如式(I)所示的化合物在JJH201601原料药或其药物组合物中的含量对于改善JJH201601的药理活性、毒性等均具有十分重大的意义。
有益效果
由于采用了上述技术方案,本发明的有益效果是:
1)本发明技术方案的式I所示杂质为工艺杂质,该杂质在原料或制剂中的含量均不会进一步增加。通过强制降解、稳定性试验,式I所示杂质含量不会增加。根据中试生成结果,该杂质在每批成品中都会残留,限度控制很有必要。
2)本发明提供的化合物可用于紫杉烷类化合物的杂质的检测,能够为紫杉烷类化合物的质量控制提供对照品;控制本发明提供的化合物含量能够明显改善降低紫杉烷类化合物作为药物应用时的毒性或副作用,并对提高紫杉烷类化合物作为药物应用时的药理活性有明显益处。
附图说明
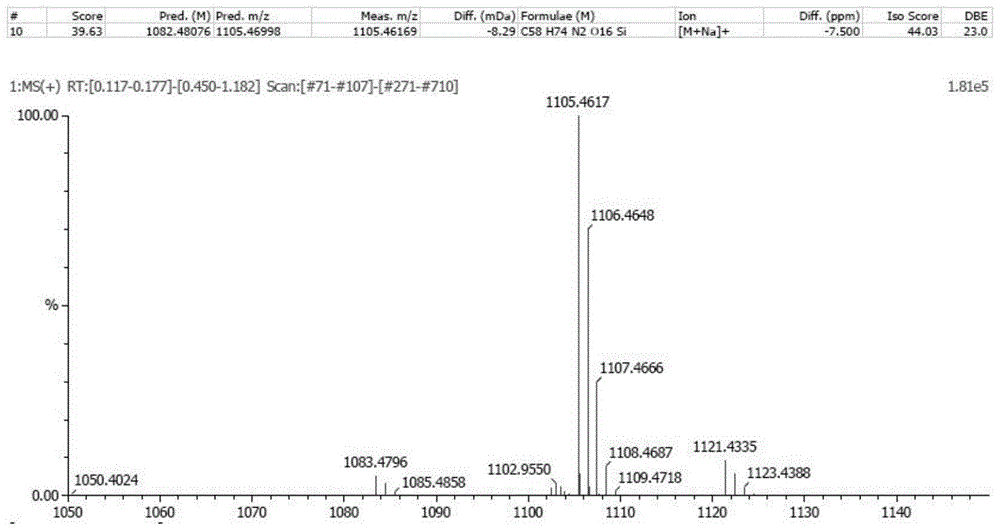
图1为式I所示结构的ESI(+)-TOFMS精密分子量测定谱图;
图2为式I所示结构在CDCl
图3为JJH201601及式I所示化合物的高效液相图谱。
具体实施例
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
式I所示化合物的制备
室温条件下,配制2%的盐酸甲醇溶液20mL加入到100mL三口瓶中,控温至5℃左右。称取500mg DTX2用1mL二氯甲烷溶解后滴加到盐酸甲醇溶液中,过程控制温度5~10℃。滴加结束保温搅拌至TLC监控反应结束。反应结束后,向反应中滴加饱和碳酸氢钠溶液至pH=7~8,加入20mL乙酸乙酯萃取两次。合并有机相,经适量无水硫酸钠干燥后过滤,滤液减压浓缩至干得粗品。粗品经柱层析分离(200~300目硅胶),洗脱剂乙酸乙酯/正己烷(1:4~1:3)。将所需洗脱液减压浓缩至干,得393mg白色粉末,摩尔收率87%。
实施例2
室温条件下,配制2%的盐酸甲醇溶液20mL加入到100mL三口瓶中,控温至0℃左右。称取500mg DTX2用1mL二氯甲烷溶解后滴加到盐酸甲醇溶液中,过程控制温度0~5℃。滴加结束保温搅拌至TLC监控反应结束。反应结束后,向反应中滴加饱和碳酸氢钠溶液至pH=7~8,加入20mL乙酸乙酯萃取两次。合并有机相,经适量无水硫酸钠干燥后过滤,滤液减压浓缩至干得粗品。粗品经柱层析分离(200~300目硅胶),洗脱剂乙酸乙酯/正己烷(1:4~1:3)。将所需洗脱液减压浓缩至干,得340mg白色粉末,摩尔收率75%。
实施例3
室温条件下,配制2%的盐酸甲醇溶液20mL加入到100mL三口瓶中,控温至10℃左右。称取500mg DTX2用1mL二氯甲烷溶解后滴加到盐酸甲醇溶液中,过程控制温度10~15℃。滴加结束保温搅拌至TLC监控反应结束。反应结束后,向反应中滴加饱和碳酸氢钠溶液至pH=7~8,加入20mL乙酸乙酯萃取两次。合并有机相,经适量无水硫酸钠干燥后过滤,滤液减压浓缩至干得粗品。粗品经柱层析分离(200~300目硅胶),洗脱剂乙酸乙酯/正己烷(1:4~1:3)。将所需洗脱液减压浓缩至干,得407mg白色粉末,摩尔收率90%。
实施例4
室温条件下,配制2%的盐酸甲醇溶液20mL加入到100mL三口瓶中,控温至25℃左右。称取500mg DTX2用1mL二氯甲烷溶解后滴加到盐酸甲醇溶液中,过程控制温度25~30℃。滴加结束保温搅拌至TLC监控反应结束。反应结束后,向反应中滴加饱和碳酸氢钠溶液至pH=7~8,加入20mL乙酸乙酯萃取两次。合并有机相,经适量无水硫酸钠干燥后过滤,滤液减压浓缩至干得粗品。粗品经柱层析分离(200~300目硅胶),洗脱剂乙酸乙酯/正己烷(1:4~1:3)。将所需洗脱液减压浓缩至干,得298mg白色粉末,摩尔收率66%。
实施例5
室温条件下,配制5%的盐酸甲醇溶液20mL加入到100mL三口瓶中,控温至5℃左右。称取500mg DTX2用1mL二氯甲烷溶解后滴加到盐酸甲醇溶液中,过程控制温度5~10℃。滴加结束保温搅拌至TLC监控反应结束。反应结束后,向反应中滴加饱和碳酸氢钠溶液至pH=7~8,加入20mL乙酸乙酯萃取两次。合并有机相,经适量无水硫酸钠干燥后过滤,滤液减压浓缩至干得粗品。粗品经柱层析分离(200~300目硅胶),洗脱剂乙酸乙酯/正己烷(1:4~1:3)。将所需洗脱液减压浓缩至干,得411mg白色粉末,摩尔收率91%。
实施例6
室温条件下,配制5%的盐酸甲醇溶液20mL加入到100mL三口瓶中,控温至5℃左右。称取500mg DTX2用1mL四氢呋喃溶解后滴加到盐酸甲醇溶液中,过程控制温度5~10℃。滴加结束保温搅拌至TLC监控反应结束。反应结束后,向反应中滴加饱和碳酸氢钠溶液至pH=7~8,加入20mL乙酸乙酯萃取两次。合并有机相,经适量无水硫酸钠干燥后过滤,滤液减压浓缩至干得粗品。粗品经柱层析分离(200~300目硅胶),洗脱剂乙酸乙酯/正己烷(1:4~1:3)。将所需洗脱液减压浓缩至干,得375mg白色粉末,摩尔收率83%。
实施例7
室温条件下,配制5%的盐酸甲醇溶液20mL加入到100mL三口瓶中,控温至5℃左右。称取500mg DTX2用2mL丙酮溶解后滴加到盐酸甲醇溶液中,过程控制温度5~10℃。滴加结束保温搅拌至TLC监控反应结束。反应结束后,向反应中滴加饱和碳酸氢钠溶液至pH=7~8,加入20mL乙酸乙酯萃取两次。合并有机相,经适量无水硫酸钠干燥后过滤,滤液减压浓缩至干得粗品。粗品经柱层析分离(200~300目硅胶),洗脱剂乙酸乙酯/正己烷(1:4~1:3)。将所需洗脱液减压浓缩至干,得375mg白色粉末,摩尔收率83%。
实施例8
室温条件下,配制5%的盐酸甲醇溶液30mL加入到100mL三口瓶中,控温至5℃左右。称取500mg DTX2用1mL二氯甲烷溶解后滴加到盐酸甲醇溶液中,过程控制温度5~10℃。滴加结束保温搅拌至TLC监控反应结束。反应结束后,向反应中滴加饱和碳酸氢钠溶液至pH=7~8,加入20mL乙酸乙酯萃取两次。合并有机相,经适量无水硫酸钠干燥后过滤,滤液减压浓缩至干得粗品。粗品经柱层析分离(200~300目硅胶),洗脱剂乙酸乙酯/正己烷(1:4~1:3)。将所需洗脱液减压浓缩至干,得389mg白色粉末,摩尔收率86%。
实施例9
室温条件下,配制5%的盐酸甲醇溶液20mL加入到100mL三口瓶中,控温至5℃左右。称取500mg DTX2用2mL的二氯甲烷溶解后滴加到盐酸甲醇溶液中,过程控制温度5~10℃。滴加结束保温搅拌至TLC监控反应结束。反应结束后,向反应中滴加饱和碳酸氢钠溶液至pH=7~8,加入20mL乙酸乙酯萃取两次。合并有机相,经适量无水硫酸钠干燥后过滤,滤液减压浓缩至干得粗品。粗品经柱层析分离(200~300目硅胶),洗脱剂乙酸乙酯/正己烷(1:4~1:3)。将所需洗脱液减压浓缩至干,得398mg白色粉末,摩尔收率88%。
实施例10
式(I)所示化合物的结构确认:
(1)高分辨质谱法(ESI(+)-TOFMS):
本品分子式为C
(2)核磁共振波谱法(NMR):
仪器型号:500M型核磁共振仪,溶剂:CDCl
式I所示结构原子编号如下所示:
核磁分析结果如表1所示:
表1式I所示结构的
如图2所示,在
综上所述,本品通过HRMS、
实施例11
JJH201601与式(I)所示的含硅杂质化合物检测:
色谱条件:以十八烷基硅烷键合硅胶为填充剂(AgilentEclipse Plus,4.6mm×150mm,3.5μm或效能相当的色谱柱);以甲酸铵缓冲液(5mM甲酸铵水溶液,用10%甲酸调节pH至4.0)-乙腈(30:70)为流动相A,以甲酸铵缓冲液(5mM甲酸铵水溶液,用10%甲酸调节pH至4.0)-乙腈(5:95)为流动相B;流速为1.2mL/min;柱温为40℃;检测波长为232nm。按下表进行梯度洗脱。
表2具体条件
如图3所示,JJH201601保留时间为2.571分钟,式I所示化合物保留时间为15.856分钟,具体参数如表3所示。
表3
<峰表>
检测器A 232nm
实施例12
静脉单次给予ICR小鼠不同含量式I所示杂质的JJH201601原料药的急性毒性研究:
取SPF级的ICR小鼠共60只,体重约20-25g,6-8周龄。共设置6组,每组10只,雌雄各半,分别设为:
受试组1:含0.1%式I所示杂质组:化合物JJH201601加适量吐温-80和乙醇溶解后,用5%葡萄糖注射液稀释至所需浓度;
受试组2:含0.25%式I所示杂质组:化合物JJH201601加适量吐温-80和乙醇溶解后,用5%葡萄糖注射液稀释至所需浓度;
受试组3:含0.50%式I所示杂质组:化合物JJH201601加适量吐温-80和乙醇溶解后,用5%葡萄糖注射液稀释至所需浓度;
受试组4:含0.75%式I所示杂质组:化合物JJH201601加适量吐温-80和乙醇溶解后,用5%葡萄糖注射液稀释至所需浓度;
受试组5:含1.0%式I所示杂质组:化合物JJH201601加适量吐温-80和乙醇溶解后,用5%葡萄糖注射液稀释至所需浓度;
溶媒对照组:5%葡萄糖注射用溶液;
受试组1-5分别按25mg/kg的给药剂量经ICR小鼠尾静脉注射药物,给药浓度为5mg/mL。给药后当天随时观察动物情况,并连续观察至14天,报告动物死亡情况。结果如下表4所示:
表4
由上述结果可知,化合物JJH201601中式I所示杂质含量超过0.75%时,单次静脉注射ICR小鼠可造成部分动物因急性毒性致死。因此,化合物JJH201601中式I所示杂质的啮齿类动物最大耐受含量为0.75%,结合制备工艺所能达到的质量情况,式I所示杂质的控制限度为≤0.5%。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进或润饰,这些改进和润饰也应视为本发明的保护范围。
- 一种衣物烘干装置
- 一种衣物烘干装置
- 一种家用干燥衣物装置
- 衣物整理装置、烘干系统及烘干方法
- 烘干系统、衣物处理装置及烘干系统控制方法
- 一种家用衣物安全烘干装置
- 一种具有衣物烘干功能的安全型家用电取暖器