丙二酰辅酶A代谢途径高可塑性的酵母底盘细胞的构建方法
文献发布时间:2024-04-18 20:02:18
技术领域
本发明属于生物技术领域,涉及一种丙二酰辅酶A代谢途径高可塑性的酵母底盘细胞的构建方法。
背景技术
中心碳代谢是所有生命体的关键代谢途径,合成胞内各种前体化合物、能量及辅因子,是细胞内代谢流量的调控中枢。工程化改造中心碳代谢,以调控细胞内代谢流量的分配,是构建高效微生物细胞工厂的关键需求之一。由于中心碳代谢涉及基因数量多,且调控具有鲁棒性和复杂性等特征,通常需要对其进行大量的基因修饰和反复测试,以获得高效的细胞工厂。遗憾的是,目前仍缺乏可实现对中心碳代谢途径多基因、多层次的快速高效的组合改造体系,大大限制了高效微生物细胞工厂的构建效率。因此,开发快速、高效的中心碳代谢途径高通量组合改造技术,是构建高效微生物细胞工厂亟待突破的瓶颈之一。
近年来,多种中心碳代谢途径工程化改造技术/策略相继被报道。在DNA和RNA水平上,基因调节元件(例如启动子、核糖体结合位点、终止子)工程化改造技术、RNAi和sRNA技术、动态调控策略及基于CRISPR系统的衍生技术(如(d)Cas9、CRISPRa、CRISPRi)已被成功应用于中心碳代谢途径的改造。在蛋白水平上,通过蛋白质标签工程能够实现蛋白酶浓度、空间分布的控制;而酶工程技术(如蛋白酶突变、异源替换、融合表达等)能够对蛋白酶活性进行调控。虽然通过以上单一水平的调控,能够在一定程度上提高目标产物产量,但仍难以满足构建高效微生物细胞工厂的需求。为了进一步提高中心碳代谢途径的改造效率,开发多基因、多层次的组合改造技术是该领域的研究热点之一。CRISPR系统是当前最新的多基因编辑技术,研究报道通过建立CRISPR-AID的三功能正交体系,能够同时实现基因敲除、转录激活和转录抑制。最近,该系统被进一步开发为多功能的全基因组进化技术(MAGIC)。虽然当前已建立的多基因组合改造技术进一步提高了细胞代谢流量的调控效率,但是获得高效的微生物细胞工厂,往往需要对中心碳代谢途径的大量基因进行系统性、多层次的组合改造与测试表征,而现有技术体系尚无法满足这一关键需求,因此亟需建立快速、高效的中心碳代谢途径高通量组合改造方法。
发明内容
基于以上技术问题,本发明提供一种丙二酰辅酶A代谢途径高可塑性的酵母底盘细胞的构建方法,可突破天然细胞的局限,为细胞中心碳代谢途径的高效组合改造提供新的方法体系,同时对进一步提升微生物细胞工厂的构建效率和强化工程化细胞的工作效能产生重要的促进作用。
本发明提供一种丙二酰辅酶A代谢途径高可塑性的酵母底盘细胞的构建方法,包括以下步骤:
将酵母中丙二酰辅酶A代谢途径的功能冗余基因删除,在质粒上重建主要功能基因的编码基因簇,然后将主要功能基因从基因组原位点删除,得到人工酵母细胞;
将酵母中丙二酰辅酶A代谢途径相关蛋白酶编码的主要功能基因分别构建至含有BsmBI位点的组装型质粒上,得到含有主要功能基因的不同大质粒;
将不同大质粒混合后连同靶向整合位点的gRNA质粒共转化至所述人工酵母细胞,转化产物经过孵育,得到改造后的酵母底盘细胞。
优选地,所述酵母为酿酒酵母。
优选的,所述质粒为PRS416质粒。
优选地,删除冗余基因及主要功能基因是利用CRISPR-Cas9基因编辑系统进行。
优选地,所述冗余基因为PDC5、PDC6、ACS1、HXK1、ALD6、TDH1、TDH2、PYK2、FBP1、GLK1及ENO1。
优选地,所述主要功能基因为PGI1、ACS2、GPM1、CDC19、HXK2、PFK2、PGK1、PDC1、TDH3、ENO2、FBA1、PFK1、TPI1及ACC1。
优选地,5个不同的大质粒分别包括5个不同的DNA大片段,5个不同的DNA大片段分别为ChrIX-HL-PGI1-ACS2-GPM1-P
其中,ChrIX-HL是酵母九号染色体上游同源臂,HR-ChrIX是酵母九号染色体下游同源臂,P
优选地,不同大质粒是按照等摩尔比进行混合。
优选地,所述孵育是先将转化产物涂布于SD-LEU培养基后,30℃孵育2~3d,获得转化子;再通过平板影印将转化子原位转移至SD-LEU+5FOA固体培养基上,30℃孵育2~4d。
本发明还提供一种根据上述方法构建得到的丙二酰辅酶A代谢途径高可塑性的人工酵母底盘细胞。
对比现有技术,本发明的有益效果为:
(1)本发明通过构建人工酵母底盘细胞,实现丙二酰辅酶A(Malonyl-CoA)代谢途径快速、高效及高通量的组合改造。
(2)本发明提供的方法,能够对Malonyl-CoA代谢途径进行快速的工程化组合改造,突破了目前多基因改造效率低下的限制,大大提高了细胞内代谢途径的改造效率,并突破了天然细胞的局限和现有技术的壁垒。
(3)本发明提供的方法,可应用于以Malonyl-CoA为前体物的酵母细胞工厂的高效构建。
(4)本发明提供的方法,可应用于以Malonyl-CoA代谢途径中间产物为前体物的酵母细胞工厂的高效构建。
(5)本发明提供的方法,不仅仅限制于Malonyl-CoA代谢途径,可以应用于细胞内其他代谢途径的高通量组合改造。
(6)本发明提供的方法,对必须或主要功能基因的致死突变和适宜工程化改造位点进行扫描,对基因序列和功能之间的映射关系进行研究。
附图说明
为了更清楚的说明本发明实施例或现有技术的实际方案,下面对实施例或现有技术描述所需要使用的附图作简单介绍。
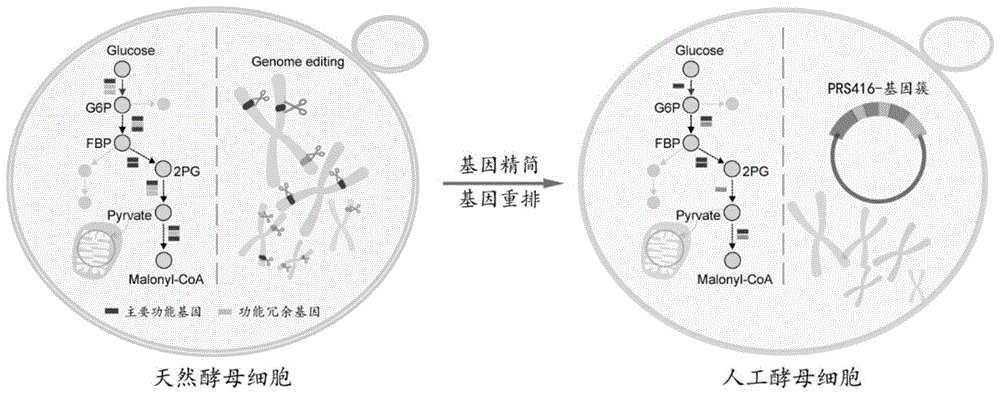
图1是人工酵母细胞的构建示意图;
图2是人工酵母菌株中25个基因(功能冗余基因和主要功能基因)敲除的PCR验证,M代表敲除后,W代表敲除前;
图3是组装型质粒的构建;
图4是大质粒的构建;
图5是将主要功能基因分别在PRS416质粒及染色体上拼接成基因簇;
图6是人工酵母基因簇的PCR验证;
图7是Malonyl-CoA代谢途径的高通量组合改造示意图;
图8是Malonyl-CoA代谢途径基因簇改造验证(A)和改造菌株生长表型分析(B),wt代表野生型酵母,MCP-0代表人工酵母,MCP-wt代表基因簇改造菌株;
图9是Malonyl-COA代谢途径高通量组合改造在细胞工厂高效构建的应用示例;A、Flaviolin高产菌株的筛选;B、Flaviolin高产菌株的产量分析;C、Flaviolin、β胡萝卜素和甜菜黄素的代谢途径示意图;D、胡萝卜素高产菌株的筛选;E、β胡萝卜素高产菌株的产量分析;F、甜菜黄素高产菌株的筛选;G、甜菜黄素高产菌株的产量分析。
具体实施方式
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合具体实施例及附图对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施的限制。
在酵母中,丙二酰辅酶A(Malonyl-CoA)代谢途径相关的25个基因分布在12条不同染色体上,且大部分基因之间具有同源性。研究报道,敲除转录水平较低的同源基因对细胞的生长和代谢流量没有显著影响。这表明,Malonyl-CoA途径相关基因具有数量多、分布散和功能冗余等特点,这些也是中心碳代谢途径相关基因的普遍特征,可能是其难以系统性组合改造的限制因素。
综合以上证据,本发明对位于不同染色体的基因进行系统性地编辑和重排,通过删除功能冗余基因,及成簇化汇聚主要功能基因,降低Malonyl-CoA代谢途径的复杂性和分散性,创建出含有精简型Malonyl-CoA代谢相关蛋白酶编码基因簇的人工酵母细胞。进一步,以此人工酵母作为底盘细胞,结合模块化的组合改造策略,可突破天然细胞的局限,进行Malonyl-CoA途径的高通量组合改造。
本发明所述酵母为酿酒酵母或者其他具有丙二酰辅酶A代谢途径的酵母,在此没有特殊限制。本发明下述实施例所用酿酒酵母为保藏编号为CGMCCNO.2.3868的酵母。
实施例1
人工酵母细胞构建
参照图1所示,包括以下步骤:
1、Malonyl-CoA代谢途径功能冗余基因的删除
利用CRISPR-Cas9基因编辑系统将Malonyl-CoA代谢途径的功能冗余基因PDC5(NCBI ID:850825)、PDC6(NCBI ID:852978)、ACS1(NCBI ID:851245)、HXK1(NCBI ID:850614)、ALD6(NCBI ID:856044)、TDH1(NCBI ID:853395)、TDH2(NCBI ID:853465)、PYK2(NCBI ID:854529)、FBP1(NCBI ID:851092)、GLK1(NCBI ID:850317)及ENO1(NCBI ID:853169)逐步删除,进行该途径的精简。具体操作如下:
(1)构建不同功能冗余基因的gRNA和donor单元质粒
出发质粒的gRNA表达盒由启动子(ptRNA
donor质粒包含上游同源臂(目标基因起始密码子上游大约500bp的DNA序列)、通用标签(通用标签序列为:GATGCGAGACGGAGTCACTTGCACGCGTCTCG TAGC,如SEQ ID NO:2所示)和下游同源臂(目标基因终止密码子下游大约300bp的DNA序列)。其中,上下游同源臂序列通过两对特异性的引物(如PDC6的上下游同源臂扩增引物为:PDC6-HL-F:5’-CTGATGGTCTCAGGCTGAATATGCAGATCGGCTGTGG-3’,如SEQ ID NO:3所示;PDC6-HL-R:5’-CTGATGGTCTCAAGTGACTCCGTCTCGCATCTTTGTTGGCAATATGTTTTT GT-3’,如SEQ ID NO:4所示;PDC6-HR-F:5’-CTGATGGTCTCACACTGCACGCGTCTCGTAGCGCCATTAGTAGTGTACTCA AACG-3’,如SEQ ID NO:5所示;PDC6-HR-R:5’-CTGATGGTCTCAGAGGGACGTGGCTGAACAACAGT-3’,如SEQ ID NO:6所示),以酵母基因组为模板进行PCR扩增获取,而通用标签则通过引物添加至同源敲除框中。所有基因的同源敲除框共享相同的载体骨架,经BsaI酶切处理,RFP被不同糖酵解基因的同源敲除框替换,通过菌落颜色快速筛选阳性克隆,质粒进行酶切验证。
(2)目标片段扩增
以上述新的gRNA质粒、donor质粒及载体质粒(含有半乳糖诱导自切割原件)为模板,按照常规PCR条件(即Tm值)利用通用引物分别扩增出末端含有~40bp同源序列的gRNA片段、donor片段和载体片段。
其中,通用引物为:
gRNA上游引物:5’
-CAGTGTGATCGTGGGCGTGATTTCGAAGTACGTGTTGCTAGCTGGAGCTcG TATATGTG-3’,如SEQ ID NO:7所示;
gRNA下游引物:5’
-GTTCGTCAGCGTGGTGGCTTATAGGCATAACGTACTGGTAGGATCCACAT ATTCCATAC-3’,如SEQ ID NO:8所示;
donor上游引物:5’
-TACCAGTACGTTATGCCTATAAGCCACCACGCTGACGAACGCactagtggcgtctc gTC-3’,如SEQ ID NO:9所示;
donor下游引物:5’
-CCAACTCGGTACCAAGAACACAACACCCAGAGAATTGACGCCTGCAGGcgt ctcAgaat-3’,如SEQ ID NO:10所示;
载体上游引物:5’
-CGTCAATTCTCTGGGTGTTGTGTTCTTGGTACCGAGTTGGTCACTGGCCGT CGTTTTAC-3’,如SEQ ID NO:11所示;
载体下游引物:5’
-TAGCAACACGTACTTCGAAATCACGCCCACGATCACACTGGTCATAGCTG TTTCCTG-3’,如SEQID NO:12所示。
(3)转化及鉴定
①将步骤(2)中得到的gRNA片段、donor片段和载体片段按照等摩尔共转化至酵母感受态细胞,涂布于相应的SD-URA平板上,30℃孵育2~3d;
②利用菌落PCR进行筛选,按照常规PCR条件(即Tm值)进行扩增,扩增产物为~900bp长度的菌落,鉴定为阳性菌株;
③将筛选的阳性菌株在YPG培养基上进行划线,然后将长出的单菌落分别在YPD和SD营养缺陷型平板上划短线,30℃孵育1~2d。在YPD上生长及在SD营养缺陷型平板不长的菌株,鉴定为质粒原件被消除的菌株(图2,即敲除功能冗余基因的人工酵母细胞),并作为下一轮基因敲除的出发菌株。
2、Malonyl-CoA途径的主要功能基因的基因组原位点删除及重建
将Malonyl-CoA途径的主要功能基因从基因组原位点删除,在PRS416质粒上集中重建Malonyl-CoA途径相关蛋白酶的编码基因簇。主要功能基因包括PGI1(NCBI ID:852495)、ACS2(NCBI ID:850846)、GPM1(NCBI ID:853705)、CDC19(NCBI ID:851193)、HXK2(NCBI ID:852639)、PFK2(NCBI ID:855245)、PGK1(NCBI ID:850370)、PDC1(NCBI ID:850733)、TDH3(NCBI ID:853106)、ENO2(NCBI ID:856579)、FBA1(NCBI ID:853805)、PFK1(NCBI ID:853155)、TPI1(NCBI ID:851620)及ACC1(NCBI ID:855750)14个基因。基因簇通过Golden-Gate与酵母介导的同源重组技术进行组装。具体操作如下:
(1)通过同义突变(突变NGG的任意一个G或紧邻NGG的gRNA序列中10个碱基的任意2~3个)进行主要功能基因的gRNA序列改造,得到突变后的基因PGI1’、ACS2’、GPM1’、CDC19’、HXK2’、PFK2’、PGK1’、PDC1’、TDH3’、ENO2’、FBA1’、PFK1’、TPI1’及ACC1’,然后启动子(~500bp)和终止子(~300bp)保留原始序列,经BsaI酶切,替换RFP序列,将突变后的基因分别构建在组装型质粒上(如图3所示,组装型质粒含有BsmBI位点,酶切后可产生互补粘末端),获得14个单元质粒,不同突变基因对应的单元质粒序列依次(gRNA序列及上下10bp)如下所示:
PGI1’:CATGGTCACTGAGGCTTTGAAGCAtTAtGCTGGTGTCTTG ACS2’:TCAACATTTTTACAACAGCCAACCtGGtAAGGGTTACGTT GPM1’:AACAGATCtTGGAGATTGAACGAAAGACATTACGGTGACTCDC19’:CAAAAGGCtAAGGCTATCATTGTCTTGTCCACTTCCGGTA HXK2’:TGAAGTTGTTGCTTTGATAAACGAtACcACCGGTACTTTG PFK2’:AATTTCTCCAtcaGACGTCCACAAAGTTCTAGTTGACAGA PGK1’:GTCACTGACAAGGAAGGTATTCCAGCcGGtTGGCAAGGGT PDC1’:TGTTGTTCtAGACAtGACGTCAAGGCTGAAACTAAGAAGT TDH3’:GACTACGCTGCTTACATGTTCAAGTACGACTCtACcCAtG ENO2’:AGAAGAATTGGGTGACAAGGCcGTtTAtGCCGGTGAAAAC FBA1’:GCTATTGCtGCTGCCCACTACATCAGATCCATTGCTCCAGPFK1’:CTGCCACtCGTGCCGCAACTCTATACTGTTTGTCTCACGG TPI1’:TAAGAAGCCACAAGTtACcGTtGGTGCTCAAAACGCCTAC ACC1’:TTGTTGAACTAGAATCTAAGGCTACtGCcAAaGTCGCTCT
上述单元质粒的序列依次如SEQ ID NO:13~26所示。
(2)将上述单元质粒经BsmBI酶切后,按照等摩尔配比并利用Golden-Gate组装技术将相关基因组装为5个大质粒(图4),5个大质粒分别包含5个不同的大片段:PGI1’-FBA1’-PFK2’-P
(3)将5个大质粒按相同的摩尔比混合(总质量约为4.5μg)后进行NotI酶切处理,然后连同线性化的PRS416质粒(末端分别为P
(4)通过主要功能基因上的特异性引物(即在基因启动子与终止子内部设计引物),按照常规PCR条件(即Tm值)扩增基因簇上的所有接口,通过片段的大小进行阳性菌株的筛选,所有片段符合理论值大小(~500kbp)的菌株被鉴定为阳性菌株(图6),特异性引物如下:
P1:5’-CAGGAAACAGCTATGACC-3’;
P2:5’-CCATCATAGTTGGTGAAAGTCTTGT-3’;
P3:5’-ACGATGCTTCTACCAACGGT-3’;
P4:5’-CACCGGTCTTTCTCTTTAAG-3’;
P5:5’-TGGAAACTTTCCGTACCACT-3’;
P6:5’-ATCAGGACGTAGAAGGATCTTCAA-3’;
P7:5’-TTAAGGGTTCACATGTCGT-3’;
P8:5’-CCGTGAGAGAAGTTCATACGG-3’;
P9:5’-AAGGCTAAGGAATTCGGTA-3’;
P10:5’-TTGGTGTATGGAGCATCCCAATGC-3’;
P11:5’-CGTCTCGTAGCCTTAAATGAGAAAAATTTCGTAATGAG-3’;
P12:5’-CGCTAAGAGAATGGACCTAT-3’;
P13:5’-GCGCTATTGCATTGTTCTTG-3’;
P14:5’-GCCAAGACAACGTATCTTGGGTG-3’;
P15:5’-CAGTCGGTCTCACGGTTGGCAAGGGTTGGACAATGG-3’;
P16:5’-GCAATCAAAGGTGTTTCGTG-3’;
P17:5’-CAGTCGGTCTCATACCACCGGTACTTTGGTTGCTTC-3’;
P18;5’-TCAAGGAATCTTGGGAAGTCCCT-3’;
P19:5’-CGTCTCGTAGCATGATTGCAATGAAAAGTTTAAGTT-3’;
P20:5’-GTCGCTGTCGTCATTTGCTG-3’;
P21:5’-GGTGCTCCAGCTAGATC-3’;
P22:5’-GGAGAAGACTCGAATAAGC-3’;
P23:5’-ATCAAGATGTTATCTACCGATGA-3’;
P24:5’-GGTTACTTGCATGCATTTCG-3’;
P25:5’-AATGGGACAAGTTGACCCA-3’;
P26:5’-AGCTCTGGACAACTTGGAAGTGT-3’;
P27:5’-TGGACCCAGAAGCTGCC-3’;
P28:5’-ACCACCGACAAAGAAAGTTC-3’;
P29:5’-TGAAGCCAGAATTTGTTGAT-3’;
P30:5’-TGTAAAACGACGGCCAGT-3’;
P31:5’-AAGAAGACGCATTGGCGT-3’;
P32:5’-AAACTTCTTGTGATTCTTTGCACT-3’;
P33:5’-CTTAAGTGCAAAGAATCACAAGAA-3’;
P34:5’-TTCCTCGTCAGATAATCACC-3’;
其中,P1~P34依次对应图6及图8A中的1~34,且序列依次如SEQ ID NO:27~60所示。
(4)以上述的阳性菌株为出发菌,基于迭代的CRISPR-Cas9介导的基因敲除来实现所有主要功能基因在基因组原位点的敲除(操作步骤与上述功能冗余基因的敲除流程一致),最终构建出Malonyl-CoA途径具有高可塑性的人工酵母细胞。
综上,最终获得的人工酵母细胞中Malonyl-CoA代谢途径的相关基因全部从基因组原位点敲除,主要功能基因以基因簇的形式存在于PRS416质粒上,基因顺序为PGI1-FBA1-PFK2-CDC19-ACS2-TDH3-PGK1-HXK2-PFK1-ENO2-ACC1-PDC1-GPM1-TPI1。
实施例2
Malonyl-CoA代谢途径的高通量组合改造
以实施例1中最终得到的人工酵母作为底盘细胞,采用模块化的组合改造策略,进行Malonyl-CoA代谢途径的高通量组合改造。参照图7,具体操作如下:
①将Malonyl-CoA代谢途径相关蛋白酶编码的主要功能基因分别构建至组装型质粒中,得到单元质粒(与实施例1中单元质粒构建方式一致,参照图3);
在构建单元质粒的过程中,可以对基因的启动子、编码序列和终止子序列进行设计和改造,包括替换、突变、融合和修饰等,因此可以形成不同设计的基因改造库,该实施例中以构建基因顺序不同的改造型基因簇进行示范(图5);
②如图4所示,单元质粒通过BsmBI酶切后,产生能够相互连接的粘端,进而利用Golden-Gate组装技术将相关基因组装为5个不同的大质粒,5个不同的大质粒分别包含5个不同的DNA大片段:ChrIX-HL(酿酒酵母九号染色体上游同源臂)-PGI1-ACS2-GPM1-P
需要说明的是,该组装方法的单元质粒不具有特异性,可以将相应基因任何形式的单元质粒改造库进行随机组合的组装;既可以进行特定形式的组合改造设计,生成特定的改造型基因簇;也可以进行组合库的高通量改造,生成基因簇的组合改造库。
③将上述大质粒按照相同的摩尔比进行混合后(总质量约为4.5μg),进行NotI酶切处理,连同800μg靶向整合位点的gRNA质粒共转化至实施例1构建的人工酵母细胞,转化产物涂布于SD-LEU培养基后,30℃孵育2d,通过平板影印将转化子原位转移至SD-LEU+5-FOA固体平板上(SD-LEU+5-FOA固体平板中5-FOA的终质量浓度为0.1%),30℃孵育3d后,获得的转化子将PRS416质粒(包含野生型基因簇)丢失,仅含有工程化改造的Malonyl-CoA代谢途径基因簇,并通过PCR扩增进行改造型基因簇的组装验证,同时进行生长表型的分析,完成Malonyl-CoA代谢途径高通量的组合改造(图8)。
实施例3
由于Flaviolin、β胡萝卜素和甜菜黄素都具有颜色,因此可以通过细胞的颜色变化进行高产菌株的筛选。相对于对照菌株(wt),Flaviolin、β胡萝卜素和甜菜黄素具有不同颜色变深的菌株被筛选(图9)。通过进一步的定量分析,确定了高产菌株的表型。以上结果表明上述方法可应用于高产细胞工厂的快速构建。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 一株高产丙二酰辅酶A的基因工程菌及其构建方法和应用
- 一株高产丙二酰辅酶A的基因工程菌及其构建方法和应用