多序列保守区间的探测方法、简并引物的设计方法、相关装置和电子设备
文献发布时间:2024-04-18 19:58:21
技术领域
本申请涉及生物技术领域,具体涉及多序列保守区间的探测方法、简并引物的设计方法、相关装置和电子设备。
背景技术
如果一个基因存在于多个物种,那么不同物种间该基因的差异可以用来进行物种鉴定、环境多样性评估和系统发育生物学等相关研究。相同或不同物种的同一基因存在序列上的差异,所以需要通过多序列比对将所有序列有序地排列在一起(对齐为相同长度),从而进行多序列之间保守区间的探测,用来进行简并引物的设计。
通过简并引物(多重引物)进行特定基因序列的检测已经成为一种越来越流行的生物学研究方法,广泛应用于物种鉴定、临床诊断、环境多样性评估和系统发育生物学等研究领域中。然而,由于不同基因和样本的复杂性,进行简并引物设计需要准确度很高的方法,尽可能地探测所有可能的保守区间。
目前一般是通过一致性序列方法来探测保守区间,从而完成引物设计。一致性序列(consensus sequence)是2018年全国科学技术名词审定委员会公布的生物物理学名词,是指在一组多序列比对结果中每个位置出现频率最高的核苷酸或氨基酸序列。
然而,一致性序列方法存在着保守区间探测不完全、保守区间评估不准确等问题,需要开发一种更全面、更准确的多序列保守区间的探测方法,用以提高简并引物的覆盖度。
发明内容
有鉴于此,本发明提供了多序列保守区间的探测方法、简并引物的设计方法、相关装置和电子设备。该探测方法实现了对多序列比对结果保守区间的更全面和准确的探测,从而提高了设计的简并引物的覆盖度。
为了实现上述发明目的,本发明提供以下技术方案:
第一方面,本发明提供了一种多序列保守区间的探测方法,包括:
获取多条核苷酸序列的多序列比对文件;
计算多序列比对文件中各个位点的香农熵F(X),其中,X表示多序列比对文件中某一列碱基的相对位置;计算保守位点的香农熵最大阈值N;判断各个位点的F(X)是否小于等于N,根据所得的判断结果判定待判定位点是否为保守位点,得到保守位点信息;
根据保守位点信息,得到若干个保守区间。
核苷酸共有4种碱基,分别为A、T、C、G,多序列比对中空缺或间隔用短横线“-”表示,因此多序列比对结果中每个位置最多有上述5种符号。发明人发现,一致性序列方法一般只考虑每个位置上最大频率或者频率较高的核苷酸,但是这也带来了以下几个问题:
(1)使用一致性序列方法探测保守区间,导致过高评估探测区间的保守性,获得保守性较低的保守区间。如一个位点有四种碱基,A占比70%,C占比10%,T占比10%,G占比10%,通常一致性序列会保留频率大于等于10%的碱基,会将包含四种碱基的上述位点作为保守区间的一部分。因此,该方法由于对位点的保守性评估不准确,导致获得保守性较低的保守区间;
(2)使用一致性序列方法探测保守区间,会漏掉占比较小的核苷酸种类,从而降低了最终多重引物的覆盖率。比如一个位点有两种碱基,A占比98%,G占比2%,通常一致性序列方法会只考虑A,而忽略了碱基G。因此,该方法由于保守性评估多采用最高碱基频率,丧失了碱基组成复杂度信息。
基于上述原因,一致性序列方法存在着保守区间评估不准确、保守区间碱基复杂度丢失等问题。
本发明方法使用了香农熵来描述一个位点的系统复杂度,能够全面地考虑每个位点的所有符号(A、T、C、G、空缺),很好地评估每个位点的符号种类多样性,准确地计算每个位点的保守性,提高保守区间鉴定的准确性,进而提高引物设计的覆盖度、分辨力和特异性。因此,本发明方法可以更准确地评估每个位点的多样性和复杂性,实现了对多序列比对结果保守区间的更全面和准确的探测,从而提高了设计的简并引物的覆盖度。
在本发明实施方式中,若干个是指1个或1个以上。例如为1、2、5、10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、1000中的任一数值或上述任意两两数值组成的范围值内的任一数值。
在本发明实施方式中,计算多序列比对文件中各个位点的香农熵F(X)中的位点是指多序列比对文件中某一列碱基所在的位点。如第一列碱基所在的位点为位点1,第二列碱基所在的位点为位点2,以此类推。
在本发明实施方式中,香农熵F(X)的计算公式如下:
上述公式为归一化香农熵公式。其中,x表示某一列碱基中的碱基种类,包括A、T、C、G或“-”中的至少一种,“-”表示多序列比对中的空缺;P(x)表示某一列碱基中某个碱基的频率。
在本发明实施方式中,保守位点的香农熵最大阈值N的获取方法如下:设置一种碱基的最大频率阈值为n∈[0.5,1.0],基于n的最小多样性频率数组为{n,1.0-n,0,0,0},根据公式I计算{n,1.0-n,0,0,0}的香农熵,记为N。
在本发明实施方式中,一种碱基的最大频率阈值为n∈[0.5,1.0],例如为0.5、0.55、0.6、0.65、0.7、0.75、0.8、0.85、0.9、0.95、1.0中的任一数值或上述任意两两数值组成的范围值中的任一数值。最大频率阈值越大,表明对保守区间中位点的保守性要求越高。
在本发明实施方式中,判断各个位点的F(X)是否小于等于N,根据所得的判断结果判定待判定位点是否为保守位点包括:
若待判定位点的F(X)小于等于N,则判定该待判定位点为保守位点;
若待判定位点的F(X)大于N,则判定该待判定位点为非保守位点。
在本发明实施方式中,保守区间的个数为2个或2个以上,则在得到若干个保守区间之后还包括:
计算相邻的两个保守区间之间所有非保守位点的香农熵之和,记为F
根据判定结果执行合并和/或不合并的操作,得到若干个保守区间。
本发明根据不同保守区间及区间之内各个位点的香农熵之和来评估整个区域的保守性,指导合并不同的保守区间,能够更好地容忍多样性很高的少数位点导致的保守区间中断,更全面地探测所有可能的保守区间,增加引物设计的目的区间。
在本发明实施方式中,非保守位点的香农熵之和的最大阈值为m∈[0,3]。例如为0、0.001、0.005、0.01、0.05、0.1、0.2、0.4、0.6、0.8、1、1.2、1.4、1.6、1.8、2、2.2、2.4、2.6、2.8、3中的任一数值或上述任意两两数值组成的范围值中的任一数值。最大阈值越大,表明对合并条件的保守性要求越低。
在本发明实施方式中,判断F
若F
若F
在本发明实施方式中,采用上述第一方面提供的探测方法得到的保守区间,其碱基个数至少为1,例如1、2、3、4、5、6、7、8、9、10、15、20、25、30、35、40、45、50中的任一数值或上述任意两两数值组成的范围值内的任一数值。
第二方面,本发明提供了一种多序列保守区间的探测装置,包括:
获取单元,用于获取多条核苷酸序列的多序列比对文件;
第一判定单元,用于计算多序列比对文件中各个位点的香农熵F(X),其中,X表示多序列比对文件中某一列碱基的相对位置;计算保守位点的香农熵最大阈值N;判断各个位点的F(X)是否小于等于N,根据所得的判断结果判定待判定位点是否为保守位点,得到保守位点信息;
探测单元,用于根据保守位点信息,得到若干个保守区间。
在本发明实施方式中,探测装置还包括:
第二判定单元,用于计算相邻的两个保守区间之间所有非保守位点的香农熵之和,记为F
合并单元,用于根据判定结果,执行合并和/或不合并的操作,得到若干个保守区间。
第三方面,本发明提供了一种简并引物的设计方法,包括:
采用上述多序列保守区间的探测方法,获得若干个保守区间;
根据若干个保守区间,设计得到简并引物。
作为优选,在“获得若干个保守区间”与“根据若干个保守区间,设计得到简并引物”之间还包括“筛选符合引物设计原则的保守区间”的操作步骤。
在本发明实施方式中,筛选符合引物设计原则的保守区间具体为:筛选碱基个数为15个或15个以上的保守区间。例如筛选碱基个数为15个、16个、17个、18个、19个、20个、22个、24个、26个、28个、30个、32个、34个、36个、38个、40个、50个、60个、70个、80个、90个、100个、200个、300个、400个或500个的保守区间,以满足引物设计原则。
第四方面,本发明提供了一种简并引物的设计装置,包括:
上述探测装置,用于获得若干个保守区间;
引物设计单元,用于根据若干个保守区间,设计得到简并引物。
第五方面,本发明提供了一种电子设备,包括:
存储器,用于存储至少一组指令;
处理器,用于执行至少一组指令,以执行上述多序列保守区间的探测方法或简并引物的设计方法。
第六方面,本发明提供了一种计算机可读存储介质,计算机可读存储介质存储有指令,指令用于执行上述多序列保守区间的探测方法或简并引物的设计方法。
与现有技术相比,本发明具有的有益效果为:
(1)使用本发明方法探测保守区间,能够全面地考虑每个位点的所有符号,很好地评估每个位点的符号种类多样性,准确地计算每个位点的保守性,提高保守区间鉴定的准确性,进而提高引物设计的覆盖度、分辨力和特异性;
(2)使用本发明方法探测保守区间,不会漏掉占比较小的核苷酸种类,从而提高了最终多重引物的覆盖率;
(3)进一步地,本发明更全面地探测所有可能的保守区间,并通过保守区间的合并,能够更好地容忍多样性很高的少数位点导致的保守区间中断,增加引物设计的目的区间。
附图说明
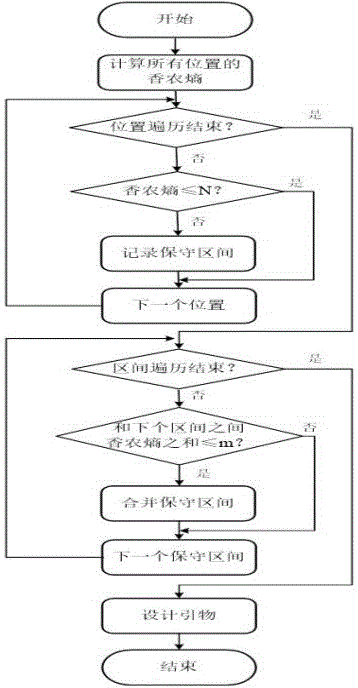
图1为本发明实施例3多序列保守区间探测和引物设计流程。
图2为试验例1中10条核苷酸序列的多序列比对文件。
图3为试验例2中15条核苷酸序列的多序列比对文件。
其中,每一行为一条核苷酸序列,每一列为一个位置上的所有碱基,“*”表示同一位点(同一列)的碱基完全相同。
具体实施方式
本发明公开了多序列保守区间的探测方法、简并引物的设计方法、相关装置和电子设备,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
术语解释:
多序列比对,是指将两个或多个序列排列在一起,标明其相似之处。序列比对中可以通过插入间隔(通常用短横线“-”表示)实现多个相似序列有序地排列在一起。每列(每个位点)是多条序列在该位置对应的相同或最相似的符号(在核酸序列中是A, T, C, G);每行是一条序列。
多序列比对中的保守区间,指的是多序列比对结果中一段连续的高相似区域,其中每个位点包含的符号种类尽可能的少。通过保守区间设计的简并引物可以尽可能多地覆盖所有序列。
保守位点,指的是在不同物种或个体DNA序列中都高度保守、相似的位点。
非保守位点,指的是在不同物种或个体DNA序列中不保守、不同的位点。
香农熵,也被称为信息熵,是由克劳德·艾尔伍德·香农提出,表示了某一系统的复杂度,香农熵越大,代表系统越复杂。香农熵解决了对信息的量化度量问题。对于随机变量而言,其取值是不确定的。在做随机试验之前,只了解各取值的概率分布,而做完随机试验后,就确切地知道了取值,不确定性完全消失。这样,通过随机试验获得了信息,且该信息的数量恰好等于随机变量的熵。在这个意义上,可以把熵作为信息的量度。
碱基频率,是指不同物种间的同一位点处碱基的频率。例如,发挥相近功能的基因的某一位点,在10个物种中检测,有6个是A,2个是T,1个是C,1个是G,那么对于这10个物种,这一位点的碱基频率分别为:A为60%;T为20%,C为10%;G为10%。
简并引物,是指代表编码区域内不同碱基序列所有可能性组合的引物混合物。
覆盖度,是指设计出的简并引物存在于样本中的序列占全样本的比例。
多序列对比中碱基的相对位置:把多序列比对看作一张二维表,表中每一行代表一个序列,每一列代表一个位置上的所有碱基。将序列依照下列规则填入表中:(a)一个序列所有碱基的相对位置保持不变;(b)将不同序列间相同或相似的碱基放入同一列,即尽可能将序列间相同或相似碱基上下对齐。称比对前序列中碱基的位置为绝对位置。相应地,称比对后序列中碱基的位置为相对位置。显然,同一列中所有碱基的相对位置相同,而每个碱基的绝对位置可能不同,因为它们来自不同的序列。需要说明的是,绝对位置是序列本身固有的属性,或者说是比对前的位置,而相对位置则是经过比对后的位置,也就比对过程赋予它的属性。
本发明中所用仪器、软件等均可通过商业渠道获得。
下面结合实施例,进一步阐述本发明:
实施例1多序列保守区间的探测方法(包括合并保守区间的操作)
步骤(1):采用序列比对软件,获取多条核苷酸序列的多序列比对文件;
步骤(2):根据公式I计算多序列比对文件中各个位点的香农熵F(X),香农熵F(X)的计算公式如下:
其中,X表示多序列比对文件中某一列碱基的相对位置;x表示某一列碱基中的碱基种类,包括A、T、C、G或“-”中的至少一种,“-”表示多序列比对中的空缺或间隔;P(x)表示某一列碱基中某个碱基的频率。
计算保守位点的香农熵最大阈值N。设置一种碱基的最大频率阈值为n∈[0.5,1.0],基于n的最小多样性频率数组为{n,1.0-n,0,0,0},根据公式I计算{n,1.0-n,0,0,0}的香农熵,记为N。
判断各个位点的F(X)是否小于等于N:
若待判定位点的F(X)小于等于N,则判定该待判定位点为保守位点;
若待判定位点的F(X)大于N,则判定该待判定位点为非保守位点。
步骤(3):根据上述得到的保守位点信息,得到若干个保守区间,这里的“若干个”表示2个或2个以上。
步骤(4):根据公式I计算相邻的两个保守区间之间所有非保守位点的香农熵之和,记为F
设置非保守位点的香农熵之和的最大阈值为m,m∈[0,3];
判断F
若F
若F
步骤(5):重复上述操作步骤(4),直至合并操作完成,得到若干个合并后的新保守区间。
实施例2多序列保守区间的探测方法(不包括合并保守区间的操作)
步骤(1):采用序列比对软件,获取多条核苷酸序列的多序列比对文件;
步骤(2):根据公式I计算多序列比对文件中各个位点的香农熵F(X),香农熵F(X)的计算公式如下:
其中,X表示多序列比对文件中某一列碱基的相对位置;x表示某一列碱基中的碱基种类,包括A、T、C、G或“-”中的至少一种,“-”表示多序列比对中的空缺或间隔;P(x)表示某一列碱基中某个碱基的频率。
计算保守位点的香农熵最大阈值N。设置一种碱基的最大频率阈值为n∈[0.5,1.0],基于n的最小多样性频率数组为{n,1.0-n,0,0,0},根据公式I计算{n,1.0-n,0,0,0}的香农熵,记为N。
判断各个位点的F(X)是否小于等于N:
若待判定位点的F(X)小于等于N,则判定该待判定位点为保守位点;
若待判定位点的F(X)大于N,则判定该待判定位点为非保守位点。
步骤(3):根据上述得到的保守位点信息,得到若干个保守区间,这里的“若干个”表示1个或1个以上。
对比例1一致性序列方法
步骤(1):采用序列比对软件,获取多条核苷酸序列的多序列比对文件;
步骤(2):统计一组多序列比对结果中每个位置出现频率最高的碱基的频率;
使用最大碱基频率阈值(例如0.5-1.0),鉴定出符合阈值的最大一致性区间为保守区间;
步骤(3):基于上述保守区间探测结果,在保守区间设计得到简并引物。
需要说明的是,在实际的多序列比对中,所比对的序列很长,不利于展示。因此,以下试验例从多序列比对文件中截取了部分序列作为效果验证的例子。
试验例1技术效果验证
10条核苷酸序列分别为:
序列1:ATGGCTAAAGAAAAATTTCA
序列2:TTGGCTAATAAAAAATTTGT
序列3:TTGGCTAATAAAAAATTTCA
序列4:TTGGCTAAACAAAAATTTGA
序列5:ATGGCTAACGAAAAATTTGA
序列6:ATGGCTAACGAAAAATTTCT
序列7:ATGGCTAAGGAAAAATTTGA
序列8:TTGGCTAAGCAAAAATTTGA
序列9:ATGCCTAAATAAAAATTTCT
序列10:TTGGCTAAATAAAAATTTGA
每条核苷酸的长度为20bp,针对10条核苷酸序列探测保守区间。
一、实验方法
实验方法分为实验组方法和对照组方法。
(一)实验组方法
实验组方法采用实施例1的方法,具体如下:
步骤(1):采用序列比对软件,获取多条核苷酸序列的多序列比对文件,如图2所示;
步骤(2):根据公式I计算多序列比对文件中各个位点的香农熵F(X),香农熵F(X)的计算公式如下:
其中,X表示多序列比对文件中某一列碱基的相对位置;x表示某一列碱基中的碱基种类,包括A、T、C、G或“-”中的至少一种,“-”表示多序列比对中的空缺或间隔;P(x)表示某一列碱基中某个碱基的频率。
计算保守位点的香农熵最大阈值N。设置一种碱基的最大频率阈值为n=0.9,基于n的最小多样性频率数组为{0.9,0.1,0,0,0},根据公式I计算{0.9,0.1,0,0,0}的香农熵,记为N。
从第一个位点(首位置)开始,判断各个位点的F(X)是否小于等于N:
若第一个位点的F(X)小于等于N,则判定该位点为保守区间的起始位置;并继续判断下一个位点的F(X)是否小于等于N,直到出现一个位点的F(X)大于N,则这个位点的上一个位点为保守区间的终止位置;
若第一个位点的F(X)大于N,则判定该位点为非保守位点,继续判断下一个位点的F(X)是否小于等于N,直到出现一个位点的F(X) 小于等于N,则这个位点为保守区间的起始位置,重复上述操作。
继续上述判断规则,直到遍历结束。
步骤(3):根据上述得到的保守位点信息,得到若干个保守区间。
步骤(4):根据公式I计算第一个保守区间和第二个保守区间之间的所有非保守位点的香农熵之和,记为F
设置非保守位点的香农熵之和的最大阈值为m,m=2;
判断F
若F
若F
步骤(5):重复上述操作步骤(4),直至遍历剩余所有保守区间,合并操作完成,得到若干个合并后的新保守区间。
步骤(6):基于上述保守区间探测结果,在保守区间设计得到简并引物。
(二)对照组方法
对照组方法采用对比例1的方法,具体如下:
步骤(1):采用序列比对软件,获取多条核苷酸序列的多序列比对文件;
步骤(2):统计一组多序列比对结果中每个位置出现频率最高的碱基的频率;
使用最大碱基频率阈值(此例为>=0.9),鉴定出符合阈值的最大一致性区间为保守区间;
步骤(3):基于上述保守区间探测结果,在保守区间设计得到简并引物。
二、实验结果
(一)实验组结果
采用本发明方法,该多序列比对结果各个位点的香农熵F(X)依次为:[0.43067655807339306, 0.0, 0.0, 0.2019854079986195, 0.0, 0.0, 0.0, 0.0,0.8277293767706428, 0.8277293767706428, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0,0.0, 0.4181656600790516, 0.37955133114208027]。
本方法使用最大碱基频率阈值参数n=0.9,计算获得香农熵最大阈值参数N=0.2019854079986195。
根据F(X)小于等于N的判断规则,鉴定出两个保守区间,分别为:
保守区间1:位置2-8;
保守区间2:位置11-18。
位置9和位置10为非保守位点,其香农熵之和F
使用保守区间合并香农熵阈值m=2,根据F
最后,基于保守区间,进行简并引物设计。
(二)对照组结果
采用一致性序列方法,该多序列比对结果各个位点的最大碱基频率值依次为:[0.5, 1.0, 1.0, 0.9, 1.0, 1.0, 1.0, 1.0, 0.4, 0.4, 1.0, 1.0, 1.0, 1.0, 1.0,1.0, 1.0, 1.0, 0.6, 0.7]。
使用最大碱基频率阈值(此例为>=0.9),鉴定出符合阈值的最大一致性区间为8bp(位置11-18),该保守区间不满足引物设计最小长度(15bp),无法设计引物。
三、结果分析
通过上述结果可知,相较于一致性序列方法,本发明更全面地探测所有可能的保守区间,并通过保守区间的合并,能够更好地容忍多样性很高的少数位点导致的保守区间中断,增加引物设计的目的区间。
试验例2技术效果验证
15条核苷酸序列分别为:
序列11:ATAGCTAAAGAAAAATTTCACCGG
序列12:ACACCGAAAGAAAAATTTCACCGG
序列13:CCTGCGATAGAAAAATTTCACCGG
序列14:CGAGCTATAGAAAAATTTCACCGG
序列15:AGAGCATCAGAAAAATTTCACCGG
序列16:ATAGCATCAGAAAAATTTCACCGG
序列17:ATAGCTAGAGAATAATTTCACCGG
序列18:TTTGCTCGGGAAAAATTTCACCGG
序列19:TTAGGTCAGGAAAAATTTCACCGG
序列20:ATACGTAACGAAAAATTTCACCGG
序列21:ATCGATAACGAAAAATTTCACCGG
序列22:ATAAATAATGAAAAATTTCACCGG
序列23:ATCACTAATGAAAAATTTCACCGG
序列24:ATAGCTAAAGAAAAATTTCACCGG
序列25:ATAGCTAAAGCAAAATTTCACCGG
每条核苷酸的长度为24bp,针对15条核苷酸序列探测保守区间。
一、实验方法
实验方法分为实验组方法和对照组方法。
(一)实验组方法
实验组方法采用实施例2的方法,具体如下:
步骤(1):采用序列比对软件,获取多条核苷酸序列的多序列比对文件,如图3所示;
步骤(2):根据公式I计算多序列比对文件中各个位点的香农熵F(X),香农熵F(X)的计算公式如下:
其中,X表示多序列比对文件中某一列碱基的相对位置;x表示某一列碱基中的碱基种类,包括A、T、C、G或“-”中的至少一种,“-”表示多序列比对中的空缺或间隔;P(x)表示某一列碱基中某个碱基的频率。
计算保守位点的香农熵最大阈值N。设置一种碱基的最大频率阈值为n=0.7,基于n的最小多样性频率数组为{0.7,0.3,0,0,0},根据公式I计算{0.7,0.3,0,0,0}的香农熵,记为N。
从第一个位点(首位置)开始,判断各个位点的F(X)是否小于等于N:
若第一个位点的F(X)小于等于N,则判定该位点为保守区间的起始位置;并继续判断下一个位点的F(X)是否小于等于N,直到出现一个位点的F(X)大于N,则这个位点的上一个位点为保守区间的终止位置;
若第一个位点的F(X)大于N,则判定该位点为非保守位点,继续判断下一个位点的F(X)是否小于等于N,直到出现一个位点的F(X) 小于等于N,则这个位点为保守区间的起始位置,重复上述操作。
继续上述判断规则,直到遍历结束。
步骤(3):根据上述得到的保守位点信息,得到若干个保守区间。
步骤(4):基于上述保守区间探测结果,在保守区间设计得到简并引物。
(二)对照组方法
对照组方法采用对比例1的方法,具体如下:
步骤(1):采用序列比对软件,获取多条核苷酸序列的多序列比对文件;
步骤(2):统计一组多序列比对结果中每个位置出现频率最高的碱基的频率;
使用最大碱基频率阈值(此例为>=0.7),鉴定出符合阈值的最大一致性区间为保守区间;
步骤(3):基于上述保守区间探测结果,在保守区间设计得到简并引物。
二、实验结果
(一)实验组结果
采用本发明方法,
该多序列比对结果的各个位点的香农熵F(X)依次为:[0.4751686372360966,0.4751686372360966, 0.4751686372360966, 0.4751686372360966,0.4751686372360966, 0.4751686372360966, 0.4751686372360966,0.6912081378734457, 0.6912081378734457, 0.0, 0.15218358217012873, 0.0,0.15218358217012873, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0]。
本方法使用最大碱基频率阈值参数n=0.7,计算获得香农熵最大阈值参数N=0.3795513311420802。
根据F(X)小于等于N的判断规则,鉴定出1个保守区间:
保守区间1:位置10-24。
(二)对照组结果
采用一致性序列方法,该多序列比对结果各个位点的最大碱基频率值依次为:[0.73, 0.73, 0.73, 0.73, 0.73, 0.73, 0.73, 0.6, 0.6, 1.0, 0.93, 1.0, 0.93,1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0, 1.0]。
使用最大碱基频率阈值(此例为>=0.7),鉴定一致性区间,符合阈值的一致性区间为:
保守区间1:7bp(位置1-7),
保守区间2:15bp(位置10-24)。
三、结果分析
首先,采用一致性序列方法得到的保守区间1中,位置1的A、T、C、G各碱基频率为0.73、0.13、0.13、0;位置2的A、T、C、G各碱基频率为0、0.73、0.13、0.13;位置3的A、T、C、G各碱基频率为0.73、0.13、0.13、0;位置4的A、T、C、G各碱基频率为0.13、0、0.13、0.73;位置5的A、T、C、G各碱基频率为0.13、0、0.73、0.13;位置6的A、T、C、G各碱基频率为0.13、0.73、0、0.13;位置7的A、T、C、G各碱基频率为0.73、0.13、0.13、0。可见,采用一致性序列方法得到的保守区间1的保守性并不高。
采用本申请方法,排除掉了一致性序列方法得到的保守性不高的保守区间1,鉴定出的保守区间长度为15bp(位置10-24),与一致性序列方法得到的保守区间2完全一致(此后称为共有保守区间,即位置10-24),然后使用共有保守区间中的所有序列用以设计简并引物,增强了简并引物的可用性。
其次,共有保守区间中位置11的A、T、C、G各碱基频率为0.93、0、0.07、0,使用最小碱基频率阈值(此例为>=0.10)生成共有保守区间的一致性序列,仅包含位置11中的A,不包含位置11中的C,导致不能覆盖第15条序列(序列25),覆盖度降低。
共有保守区间中位置13的A、T、C、G各碱基频率为0.93、0.07、0、0,使用最小碱基频率阈值(此例为>=0.10)生成共有保守区间的一致性序列,仅包含位置13中的A,不包含位置13中的T,导致不能覆盖第7条序列(序列17),覆盖度降低。
通过上述结果可知,相较于一致性序列方法,使用本发明方法探测保守区间,能够全面地考虑每个位点的所有符号,很好地评估每个位点的符号种类多样性,准确地计算每个位点的保守性,提高保守区间鉴定的准确性,进而提高引物设计的覆盖度、分辨力和特异性。
实施例3简并引物的设计方法
步骤(1):采用序列比对软件,获取多条核苷酸序列的多序列比对文件;
步骤(2):根据公式I计算多序列比对文件中各个位点的香农熵F(X),香农熵F(X)的计算公式如下:
其中,X表示多序列比对文件中某一列碱基的相对位置;x表示某一列碱基中的碱基种类,包括A、T、C、G或“-”中的至少一种,“-”表示多序列比对中的空缺或间隔;P(x)表示某一列碱基中某个碱基的频率。
计算保守位点的香农熵最大阈值N。设置一种碱基的最大频率阈值为n∈[0.5,1.0],基于n的最小多样性频率数组为{n,1.0-n,0,0,0},根据公式I计算{n,1.0-n,0,0,0}的香农熵,记为N。
判断各个位点的F(X)是否小于等于N:
若待判定位点的F(X)小于等于N,则判定该待判定位点为保守位点;
若待判定位点的F(X)大于N,则判定该待判定位点为非保守位点。
步骤(3):根据上述得到的保守位点信息,得到若干个保守区间,这里的“若干个”表示2个或2个以上。
步骤(4):基于上述保守区间探测结果,筛选碱基个数为15个或15个以上的保守区间作为引物设计的保守区间,在保守区间设计得到简并引物。
实施例4简并引物的设计方法
步骤(1):采用序列比对软件,获取多条核苷酸序列的多序列比对文件;
步骤(2):根据公式I计算多序列比对文件中各个位点的香农熵F(X),香农熵F(X)的计算公式如下:
其中,X表示多序列比对文件中某一列碱基的相对位置;x表示某一列碱基中的碱基种类,包括A、T、C、G或“-”中的至少一种,“-”表示多序列比对中的空缺或间隔;P(x)表示某一列碱基中某个碱基的频率。
计算保守位点的香农熵最大阈值N。设置一种碱基的最大频率阈值为n∈[0.5,1.0],基于n的最小多样性频率数组为{n,1.0-n,0,0,0},根据公式I计算{n,1.0-n,0,0,0}的香农熵,记为N。
判断各个位点的F(X)是否小于等于N:
若待判定位点的F(X)小于等于N,则判定该待判定位点为保守位点;
若待判定位点的F(X)大于N,则判定该待判定位点为非保守位点。
步骤(3):根据上述得到的保守位点信息,得到若干个保守区间,这里的“若干个”表示2个或2个以上。
步骤(4):根据公式I计算相邻的两个保守区间之间所有非保守位点的香农熵之和,记为F
设置非保守位点的香农熵之和的最大阈值为m,m∈[0,3];
判断F
若F
若F
步骤(5):重复上述操作步骤(4),直至合并操作完成,得到若干个合并后的新保守区间。
步骤(6):基于上述保守区间探测结果,筛选碱基个数为15个或15个以上的保守区间作为引物设计的保守区间,在保守区间设计得到简并引物。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 一种多鱗鯔鰕虎魚线粒体基因组全序列引物及设计、系统进化分析全序列扩增方法
- 监控序列相关数据的可变抽样区间CUSUM控制图的设计方法
- 为识别SARS相关冠状病毒设计的靶序列RT-PCR引物、短柄圆环探针、试剂盒及检测方法和应用