用于乳腺癌新辅助化疗获益和/或预后评估的标志物组合及其应用
文献发布时间:2024-04-18 19:58:26
技术领域
本发明属于生物医药技术领域,具体涉及用于乳腺癌预后和/或治疗获益评估的标志物组合及其应用。
背景技术
新辅助化疗是乳腺癌的一种重要治疗策略,但存在耐药性发展的缺点。化疗具有副作用,与耐药性相结合可能会导致总体生存率降低。本研究通过乳腺癌新辅助化疗Sensitive和Resistant样本进行差异分析,筛选影响新辅助化疗疗效关键的基因,并基于关键基因构建疗效预测模型。
发明内容
本发明第一方面的目的,在于提供一种标志物组合。
本发明第二方面的目的,在于提供上述标志物组合或检测上述标志物组合的试剂的应用。
本发明第三方面的目的,在于提供一种产品。
本发明第四方面的目的,在于提供一种乳腺癌患者风险评分预后和/或治疗获益预测的系统。
本发明所采取的技术方案是:
本发明的第一方面,提供一种标志物组合,所述标志物组合包括RLN2、MSLN、SAPCD2、LY6D、CACNG4、TUBA3E、LAMP3、GNMT、KLHDC7B。
本发明的第二方面,提供本发明第一方面所述标志物组合或定量检测本发明第一方面所述标志物组合的试剂在制备乳腺癌预后和/或治疗获益预测产品中的应用。
优选地,所述治疗包括新辅助化疗、免疫治疗、传统化疗、新辅助靶向治疗中的至少一种。
优选地,所述治疗为新辅助化疗。
本发明的第三方面,提供一种产品,所述产品包含定量检测本发明第一方面所述标志物组合的试剂。
优选地,所述定量检测标志物组合的试剂包含在基因水平上检测标志物组合的试剂。
优选地,所述试剂包括通过测序技术、核酸杂交技术、核酸扩增技术定量检测标志物组合的试剂。
优选地,所述检测标志物组合的试剂选自下组中的至少一种:对标志物组合中的标志物具有特异性的物质,标志物组合中的标志物特异性的探针、引物等。
优选地,所述产品包含试剂、试剂盒、试纸或芯片。
优选地,所述产品的受试样品选自血液、组织、细胞样品、尿液、粪便中至少一种;进一步为组织。优选地,所述组织包含癌组织。
本发明的第四个方面,提供一种乳腺癌患者治疗获益和/或预后风险预测的系统,包括以下模块:
a)数据收集模块:收集患者的样本,测定本发明第一方面的标志物组合中标志物的表达量,并将标志物的表达量数据输出至模型计算模块;
b)模型计算模块:计算患者的风险分数;所述风险分数计算公式如下:
风险分数=-0.145*RLN2表达量+0.066*MSLN表达量+0.254*SAPCD2表达量+0.079*LY6D表达量-0.08*CACNG4表达量-0.156*TUBA3E表达量-0.243*LAMP3表达量-0.178*GNMT表达量-0.234*KLHDC7B表达量;
c)输出预测模块,根据计算得到的患者的风险分数来预测患者的预后情况。
其中患者的风险分数越高,则预后和/或治疗获益越好;将风险分数与阈值比较,若高于阈值,则预测其预后和/或治疗获益越好,对治疗方式更为敏感;若低于阈值,则预测其预后和/或治疗获益不佳,对治疗方式耐药。优选地,所述阈值为0。
优选地,所述治疗包括新辅助化疗、免疫治疗、传统化疗、新辅助靶向治疗中的至少一种。
优选地,所述新辅助化疗药物包括蒽环类化疗药、紫杉类化疗药、铂类药物、HER2靶向药中的至少一种。
优选地,所述样本为血液、组织、细胞样品、尿液、粪便中至少一种;进一步为组织。优选地,所述组织包含癌组织。
本发明的有益效果是:
本发明公开了用于乳腺癌预后和/或治疗获益评估的标志物组合,包含以下标志物:RLN2、MSLN、SAPCD2、LY6D、CACNG4、TUBA3E、LAMP3、GNMT、KLHDC7B,通过该标志物组合构建了风险评分模型,通过风险评分模型的ROC曲线可知该模型可用于乳腺癌预后和/或治疗获益评估;该模型具有较强的鲁棒性,并且独立于临床病理学特征,在独立的数据集中均发挥稳定的预测效能;可以应用于临床试验,为乳腺癌的医疗决策提供科学依据。
附图说明
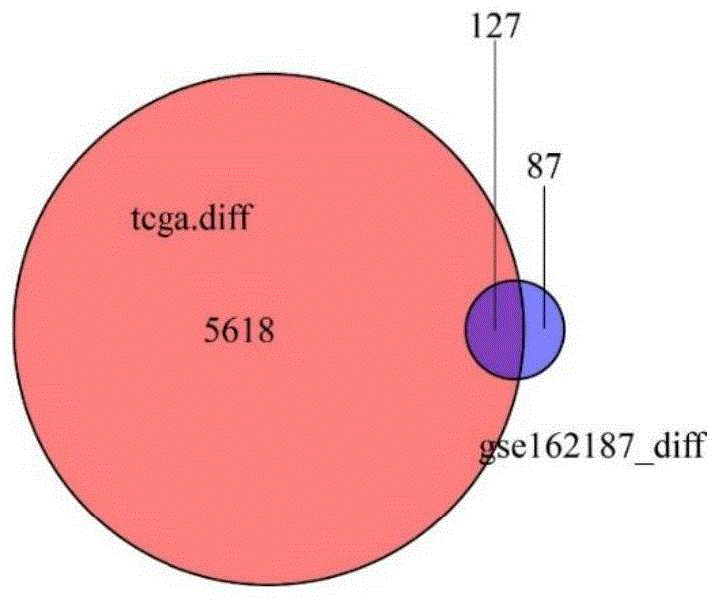
图1-图7为12个基因在癌组织和癌旁组织的表达;图1:TCGA差异基因与GSE162187差异基因的venn图;图2:预后相关关键基因的森林图;图3:原发肿瘤样本中存在12个关键基因突变和野生组之间的生存分析;图4:色氨酸代谢相关基因在原发肿瘤样本中的突变图谱;图5:原发肿瘤样本中的12个关键基因的拷贝数变异汇总;图6:原发肿瘤样本中不同的拷贝数变异类型之间的基因表达水平差异分析结果;图7:12个关键基因在原发肿瘤样本和癌旁正常组织样本中的转录表达水平差异分析结果。
图8-图12为三个分子亚型的预后特征;图8:TCGA队列样本CDF曲线;图9:TCGA队列样本CDF Delta area曲线,一致性聚类的德尔塔面积曲线,表明与k–1相比,每个类别数k在累积分布函数(CDF)曲线下的面积相对变化。横轴表示类别编号k,纵轴表示CDF曲线下面积的相对变化;图10:k=3时样本聚类热图;图11:TCGA三个亚型预后的关系KM曲线;图12:在GSE20685队列中三个亚型的预后的KM曲线。
图13为TCGA队列中分子亚型的临床信息分布。
图14-图15为TCGA队列分子亚型的基因组改变。图14:在TCGA队列中不同分子亚型的体细胞突变分析(Fisher's exact test);图15:比较TCGA队列不同分子亚型中的同源重组缺陷、部分改变、片段数目以及肿瘤突变负荷的差异。
图16为TCGA数据集不同亚型GSVA富集分析热图。
图17-图20为不同亚型之间在免疫治疗的差异;图17:TCGA队列22种免疫细胞评分在不同分子亚型之间的差异;图18:TCGA队列ESTIMATE免疫浸润在不同分子亚型之间的差异;图19:TCGA队列中不同分组之间差异表达的免疫检查点;图20:TCGA队列中不同分组之间的TIDE分析结果差异。
图21-图27为新辅助治疗表现关键基因的鉴定;图21:TCGA队列clust1 vs no_clust1差异分析火山图;图22:TCGA队列clust2 vs no_clust2差异分析火山图;图23:TCGA队列clust3 vs no_clust3差异分析火山图;图24:A total of 774promising candidateswere identified among the DEGs;图25:每个自变量随着lambda变化的轨迹;图26:lambda下的置信区间;图27:多因素cox分析,预后相关基因的系数。
图28-图29为风险模型的建立以及预后效果验证;图28:TCGA数据集9个基因构建风险模型的ROC曲线和KM曲线;图29:GSE20685数据集9个基因构建风险模型的ROC曲线和KM曲线。
图30-图31为风险评分在不同的临床病理特征的表现;图30:TCGA队列中不同表型的RiskScore之间的差异(wilcox.test);图31:TCGA队列RiskScore分组之间的临床表型的比较。
图32-图33为TCGA队列分子亚型的基因组改变。图32:在TCGA队列中高低风险组的体细胞突变分析(Fisher's exact test);图33:比较TCGA队列高低风险组中的同源重组缺陷、部分改变、片段数目以及肿瘤突变负荷的差异。
图34-图39为风险模型的免疫特征;图34:TCGA队列中不同风险组在CIBERSORT预测细胞评分的差异;图35:TCGA队列中不同风险组在免疫评分和基质评分中的差异;图36:TCGA队列中不同分组之间差异表达的免疫检查点;图37:TCGA队列中细胞评分与风险得分的相关性分析;
图38:TCGA 队列中ImmuneScore、StromalScore和ESTIMATEScore和风险得分的相关性分析;
图39:TCGA-BRCA数据集中药物IC50值的箱式图。(wilcox.test统计方法)。
具体实施方式
以下将结合实施例对本发明的构思及产生的技术效果进行清楚、完整地描述,以充分地理解本发明的目的、特征和效果。显然,所描述的实施例只是本发明的一部分实施例,而不是全部实施例,基于本发明的实施例,本领域的技术人员在不付出创造性劳动的前提下所获得的其他实施例,均属于本发明保护的范围。
实施例1差异基因的筛选
1、数据收集
通过TCGA GDC API下载TCGA-BRCA的突变数据和拷贝数变异数据通过TCGA GDCAPI下载TCGA-BRCA的RNA-Seq数据,经过筛选最后共包含988例原发肿瘤样本和113个癌旁样本。
从NCBI的GEO官网下载GSE162187数据集的表达谱数据,其中包含了13个Resistant样本和9个Sensitive样本。
从NCBI的GEO官网下载GSE20685数据集的表达谱数据和生存数据,最终获取了327个肿瘤组织和23520个基因。
2、数据预处理
对TCGA的RNA-seq数据做以下几步预处理:
1)去掉没有临床随访信息的样本;
2)去掉没有进展时间的样本;
3)去掉没有进展状态的样本;
4)将Ensembl转换为Gene symbol;
5)具有多个Gene Symbol的表达情况取其均值;
6)去除进展时间大于10年的样本,且保留进展时间大于30天以上的样本;
对GEO数据做以下几步预处理
1)去掉正常组织样本;
2)将探针通过平台注释文件转化为基因symbol,并去除一个探针对应多个基因名及对多个探针对应一个基因名的取均值;
3)去掉没有临床随访信息的样本;
4)去掉没有生存时间数据的样本;
5)去掉没有生存状态的样本;
6)去除生存时间大于10年的样本,且保留生存时间大于30天以上的样本;
3、新辅助治疗相关基因的筛选
新辅助治疗药物涉及蒽环类化疗药、紫杉类化疗药、铂类药物、HER2靶向药,所用数据以herceptin为主。
通过limma包对数据进行差异分析,并通过log2(Fold Change)>log2(1.5)且p<0.05筛选新辅助治疗数据集的差异基因,最终筛选到59个上调基因和155个下调基因=;并通过对TCGA数据集进行差异分析使用log2(Fold Change)>log2(1.5)且p<0.05筛选肿瘤发生的关键基因,最终筛选到2739个表达上调的基因,3006个表达下调的基因。
通过重叠分析最终筛选到127个肿瘤发生关键基因且与新辅助治疗相关的基因(图1),接着通过survival包基于TCGA数据集针对127个关键基因进行单因素cox分析筛选预后相关的基因共12个;这12个基因单因素cox分析的森林图如图2。
为了确定这12个基因改变在乳腺癌中的作用,评估了12个基因中体细胞突变的基因突变率。在985个TCGA-BRCA的原发肿瘤样本中,34个(3.45%)样本发生了基因突变(图4)。然而具有这些基因突变的乳腺癌患者的总生存期与没有突变的患者相比无显著差异(图3);对数秩检验,p=0.574)。
然后检查了原发性肿癌中12个基因的体细胞拷贝数变化,仅有部分样本发生了CNV突变(图5)。同时比较了CNV扩增、缺失和未发生CNV突变患者关于12个基因表达的情况(图6,anova)。图7为12个基因在癌组织和癌旁组织的表达(t.test)。
实施例2分子亚型的构建
1、相关基因的分子亚型的构建
通过一致性聚类(ConsensusClusterPlus)构建一致性矩阵,对样本进行聚类分型[PMID:20427518]。利用前面筛选到12个相关基因的表达数据,得到样本的分子亚型。利用“pam”算法和“spearman”作为度量距离,并进行了500次bootstraps,每个bootstraps过程包括80%的训练集患者。设定聚类数为2到10,通过计算一致性矩阵和一致性累积分布函数来确定最佳分类,得到样本的分子亚型。
根据累计分布函数(CDF)确定最佳聚类个数,观察CDF Delta area曲线从中可以看出Cluster选择为3时具有较为稳定的聚类结果(图8-图9),最终选择k=3得到三种分子亚型(图10)。进一步的分析这三个分子亚型的预后特征,观察到他们存在显著的预后差异如图11,总的来说,clust1预后最好,clust2次之,clust3预后最差。此外,使用同样的方法对GSE20685数据进行分子分型,可以观察到这三类分子分型预后也存在显著的差异如图12,这与TCGA数据集一致。
进一步分析了TCGA队列中不同分子亚型之间的临床病理特征差异,比较了不同临床特征在三种分子亚型中的分布,查看临床特征在不同的亚型中分布差异,结果发现,在T分期、Stage和患者的存活状态,ER、Her2、PR中,三个亚型存在显著性差异(图13)。
实施例3风险模型的构建
1)通过前面鉴定的分子亚型识别出亚型之间差异的相关基因;
2)选取预后显著的差异表达基因(|logfc|>1&FDR<0.05);
3)进一步,通过lasso回归的方法进一步降低基因数目,得到表型相关的预后显著基因;
4)风险模型的建立。
具体如下:
1、申请人研究了TCGA队列中不同分子亚型之间基因组改变的差异;下载了TCGA的mutect2软件处理的突变数据集,筛选出在突变频次大于3的基因共包含5598个基因如tcga.subtype.mut.gene.csv,使用fisher检验筛选在各个亚型中显著高频突变的基因,选择阈值为p<0.05,最终得到了471个基因,前20个基因在各个亚型中的突变特征如图14。此外,比较了亚型之间的同源重组缺陷、片段改变、片段数目变化和肿瘤突变负荷的分布情况,同源重组缺陷、片段改变、片段数目变化和肿瘤突变负荷在亚型中同样具有差异(图15)。
2、分子亚型的通路分析
为了研究不同分组中不同生物过程的通路,基于h.all.v7.5.1.symbols.gmt基因集进行GSVA分析,并通过kruskal.test检验筛选在不同亚型中具有统计学意义的通路共有50个(P<0.05),图16为在三个亚型中具有统计学差异的通路富集情况。
3、分子亚型的免疫特征
为了进一步阐明不同分子亚型之间患者的免疫微环境的差异,通过使用免疫细胞中基因的表达水平来评估TCGA队列中患者的免疫细胞浸润程度,首先采用了CIBERSORT计算了22种免疫细胞的相对丰度的差异如图17(kruskal.test),可以观察到部分免疫细胞类型在亚型之间存在显著差异。
同时也采用ESTIMATE评估免疫细胞浸润情况如图18(kruskal.test),可以看到clust1亚型的“ImmuneScore”显著低于其他的亚型,具有较低的免疫细胞浸润。
进一步分析了TCGA队列中不同分子亚型之间对免疫治疗的是否存在差异。首先比较了亚型之间的免疫检查点的表达是否存在差异,结果如图19,可以看到大部分的免疫检查点基因在亚型之间差异表达。
还分析不同亚型之间在免疫治疗的差异。采用TIDE(http://tide.dfci.harvard.edu/)软件评估免疫治疗在自定义的分子亚型中潜在的临床效应。TIDE预测评分越高,表示免疫逃逸可能性越高,提示患者从免疫治疗中获益的可能性越小。如图20所示(wilcox.test),可以发现在TCGA队列中clust1和clust2亚型中的TIDE评分高于clust3,提示clust1和clust2亚型免疫逃逸的可能性较高,从免疫治疗当中获益的可能性较小。
4、新辅助治疗表型关键基因的鉴定
在前文分析中,通过新辅助治疗预后相关基因构建分子亚型鉴定了三个不同的分子亚型,接下来使用limma包计算clust1和no_clust1亚型、clust2和no_clust2亚型、clust3和no_clust3亚型之间存在差异表达的基因(FDR<0.05and|log2FC|>1),最终clust1vs no_clust1中筛选到142个表达上调的基因,20个表达下调的基因,clust2 vs no_clust2中筛选到34个表达上调的基因,6个表达下调的基因,clust3 vs no_clust中筛选到351个表达上调的基因,635个表达下调的基因。最终共筛选到991个差异基因进行下一步分析。图21-图23为差异分析的火山图。
对991个差异基因通过survival包的coxph函数进行单因素cox分析,共识别出了15个对预后影响较大的基因(P<0.05),其中包含258个“Risk”和1个“Protective”基因,图24为991个基因单因素cox分析的结果。图25为15个预后相关基因单因素cox分析的森林图。
使用lasso回归对TCGA数据集中的这259个基因进行进一步压缩,以减少风险模型的基因数量。Lasso(Least absolute shrinkage and selection operator,Tibshirani(1996))方法是一种压缩估计。它通过构造一个罚函数得到一个较为精炼的模型,使得它压缩一些系数,同时设定一些系数为零。因此保留了子集收缩的优点,是一种处理具有复共线性数据的有偏估计,可以在参数估计的同时实现变量的选择,较好的解决回归分析中的多重共线性问题,本实施例使用R软件包glmnet进行了lasso cox回归。首先分析每个自变量的变化轨迹如图25所示,从中可以看出随着lambda的逐渐增大,自变量系数趋于0的个数也逐渐增多,使用10-fold交叉验证进行模型构建,分析每个lambda下的置信区间如图26所示,从图中可以看出当lambda=0.0121时模型达到最优,为此选择lambda=0.0121时的23个基因作为下一步的目标基因。
进一步基于lasso分析结果中的23个基因,利用逐步多因素回归分析,逐步回归利用AIC赤池信息准则,它考虑了模型的统计拟合度以及用来拟合的参数数目,MASS包中的stepAIC方法从最复杂的模型开始,并依次删除一个变量以降低AIC,该值越小,模型越优,它说明模型用较少的参数获得了足够的拟合度。最终,确定了9个基因作为影响预后的相关基因,如图27。这9个基因为RLN2、MSLN、SAPCD2、LY6D、CACNG4、TUBA3E、LAMP3、GNMT、KLHDC7B。
采用以下公式计算每个患者的风险评分:RiskScore=Σβi×Expi),i指的是表型预后相关基因特征的基因表达水平,β是相应基因Cox回归系数。
最终9-gene signature公式如下:RiskScore=-0.145*RLN2+0.066*MSLN+0.254*SAPCD2+0.079*LY6D-0.08*CACNG4-0.156*TUBA3E-0.243*LAMP3-0.178*GNMT-0.234*KLHDC7B。
实施例4风险模型的预后分析
1、临床预后模型的建立以及验证
以TCGA数据作为训练数据集,通过9个基因表达水平分别计算每个样本的风险得分。然后使用R软件包timeROC对RiskScore进行预后分类的ROC分析,分别分析了1,3,5年的预后预测分类效率,其中1,3,5年的AUC都达到了0.7,同时对Riskscore进行zscore,根据阈值“0”,将zscore化后Riskscore大于零的样本划分为高风险组,小于零的样本低风险组,并绘制KM曲线,结果发现存在极显著的差异p<0.0001(图28)。
为了更好地验证模型的鲁棒性,使用GSE20685数据集使用同样的方法进行验证,结果得到了类似的结果(图29)。
2、RiskScore在不同的临床病理特征的表现
为了检验RiskScore评分与肿瘤临床特征之间的关系,在TCGA数据集中分析了不同临床表型之间RiskScore评分的差异。结果显示:风险得分随着临床等级的加深而增加(图30)。同时也比较了TCGA队列中RiskScore分组之间的临床病理特征差异,发现类似的结果(图31)。
4、风险模型的突变特征
进一步探讨了TCGA队列中高低风险组之间基因组改变的差异。下载了TCGA的mutect2软件处理的突变数据集,筛选出在突变频次大于3的基因共包含5598个基因如tcga.risk.gene.csv,使用fisher检验筛选在各个亚型中显著高频突变的基因,选择阈值为p<0.05,最终得到了888个基因,前20个基因在各个亚型中的突变特征如图32。此外,比较了亚型之间的同源重组缺陷、片段改变、片段数目变化和肿瘤突变负荷的分布情况同源重组缺陷、片段改变、片段数目变化和肿瘤突变负荷在风险组中同样具有差异(图33)。
4、风险模型的免疫特征
为了阐明RiskScore分组中患者的免疫微环境的差异,比较了RiskScore高低分组中CIBERSORT预测的细胞的相对丰度的差异如图34(wilcox.test),可以观察到部分的免疫细胞在RiskScore高低分组中存在显著差异。此外也采用ESTIMATE评估免疫细胞浸润情况如图35(wilcox.test),可以看到”High”组中的“ImmuneScore”低于“Low”组,高风险组具有较低的免疫细胞浸润。
接着分析了TCGA队列中高低风险组之间对免疫治疗的是否存在差异。
首先比较了亚型之间的免疫检查点的表达是否存在差异,如图36,可以看到大部分的免疫检查点基因在高低风险之间差异表达,且高风险组表达更低(wilcox.test)。同时通过Hmisc包rcorr函数通过pearson的方法分别计算风险得分与细胞的评分的相关性和显著性,结果发现,风险得分与部分细胞存在显著相关(图37),接着比较了风险得分与ImmuneScore、StromalScore和ESTIMATEScore的相关性,结果发现其与风险得分存在显著的负相关(图38)。此外,也分析了高低风险对传统的化疗药物的响应程度,发现高风险对于这些传统的药物更为敏感如图39。
上述具体实施方式对本发明作了详细说明,但是本发明不限于上述实施例,在所属技术领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下作出各种变化。此外,在不冲突的情况下,本发明的实施例及实施例中的特征可以相互组合。
- 评估三阴性乳腺癌对新辅助化疗药物敏感性和/或预后的标志物及其应用
- 评估三阴性乳腺癌对新辅助化疗药物敏感性和/或预后的标志物及其应用