基因编辑蛋白和组合物向细胞和组织的无载体递送
文献发布时间:2024-01-17 01:28:27
本申请是2016年12月22日递交的申请号为201680082683.7,发明名称为“基因编辑蛋白和组合物向细胞和组织的无载体递送”的分案申请。
相关申请
本申请要求于2015年12月30日提交的美国临时申请No.62/273,284的优先权,其全部内容通过引用并入本文。
技术领域
本文描述的主题涉及基因编辑化合物和复合物向细胞和组织的无载体递送。
序列表通过引用并入
2016年12月22日创建的大小为64.2KB的文本文件“48831_506001WO_ST25.txt”的内容通过引用全部并入本文。
发明背景
成簇的规律间隔的短回文重复序列(CRISPR)-Cas基因组工程系统(以下简称“CRISPR-Cas系统”)使研究人员能够对细胞内的基因组DNA进行修饰。这个系统需要三个组分:Cas9,可能来源于化脓性链球菌(Streptococcus pyogenes),是一种内切核酸酶,其与CRISPR RNA(crRNA)和转录激活crRNA(tracrRNA)复合以形成RNA引导的内切核酸酶(RGEN)。这些RGEN切割细胞中的染色体DNA,产生位点特异性双链断裂(DSB)。这些DSB的修复可以通过内源性高保真同源重组(HR)或易错非同源末端连接(NHEJ)进行,导致靶向诱变和染色体重排。RGEN的特异性由crRNA和靶DNA之间的沃森-克里克(Watson-Crick)碱基配对和NGG-三核苷酸前间区序列相邻基序(PAM)的Cas9识别决定。crRNA和tracrRNA可以融合形成单引导RNA(gRNA或sgRNA)。与其他基因组编辑核酸酶相比,CRISPR-Cas9的一个主要优势是通过替换crRNA而容易地重新编程Cas9的特异性的能力,与之不同,锌指核酸酶(ZFN)和类转录激活因子效应物核酸酶(TALEN),他们的DNA靶向特异性必须通过蛋白质工程策略改变。
CRISPR-Cas系统最常见的是以质粒DNA的形式递送到细胞中,质粒DNA编码三种组分,或者作为单独的质粒,或者作为组合质粒。但是质粒与脱靶编辑相关。质粒也可以随机整合到基因组或Cas9产生的位点。虽然后一个事件至少可以预测和监控,但前一个事件可能难以检测,因此可能更成问题。不管整合的位点如何,这些外源序列都会引起宿主免疫反应,这给基因编辑干细胞或原代细胞在临床应用中的使用带来了挑战。此外,用于临床应用的质粒转染的细胞被监管当局认为是遗传修饰的,因此受到冗长且昂贵的监管程序。
发明内容
下面描述的装置、方法和系统通过提供用于递送基因编辑系统的构建块(例如蛋白质或基因编辑复合体)的可靠和有效的系统,从而避免递送质粒DNA或编码这种蛋白质的其它载体,解决了与先前基因编辑方法相关的许多问题和缺点。本系统还提供优于其他方法(诸如电穿孔、磁转染、脂质体介导的转染、慢病毒介导的转染或细胞内注射方法)的优点。
尽管一些基因编辑系统使用Cas9和引导RNA的DNA质粒形式,但是本文所述的本发明递送RNP[例如(基因编辑蛋白和引导RNA)],以实现更短暂的基因编辑效果,从而导致更少的脱靶效应。尽管电穿孔可用于将蛋白质(例如基因编辑蛋白)递送至细胞,但本文所述的方法、系统和组合物与电穿孔相比提供了若干优点。例如,与电穿孔相比,本发明的乙醇介导的喷雾递送系统以更高的效率递送基因编辑蛋白和复合物,例如10%、20%、50%、75%、2倍或更多,并且与电穿孔相比,递送后细胞的存活力相当或更好,基因编辑效率相当或更好。此外,与电穿孔相比,使用乙醇喷雾方法更好地保持细胞功能,例如干细胞分化或免疫细胞活化(例如10%、20%、50%、75%、2倍或更多)。此外,与本文所述的系统不同,其他方法,诸如脂质体介导的转染或电穿孔,后者的方法要求粘附的细胞从它们的基质上分离,以便使用例如电穿孔仪转染它们,并且这种额外的操作步骤损害细胞的无菌性和生物学/功能。此外,使用其他方法(例如电穿孔)不可能多次定量给予递送货物(基因递送蛋白质和/或基因递送复合物),这是由于与此类程序相关的高水平细胞损伤。这里描述的组合物和方法允许2、3、4、5或更多剂量(递送喷雾,随后是停止溶液),这一过程增加了货物向细胞的有效递送和那些基因编辑组合物进入靶细胞的量。靶细胞包括原代细胞(例如直接从其源器官组织培养的或从血液获得的那些细胞)以及永生化细胞,例如细胞系和粘附细胞以及悬浮(例如自由漂浮)细胞。原代人细胞直接从其来源器官组织或血细胞中培养。
电穿孔是一种广泛使用的无载体(vector)/载体(carrier)方法,但是尽管它可以有效地将核酸和/或蛋白质递送到某些细胞类型,但是毒性可能很高,特别是在原代细胞中。因此需要替代的膜破坏方法。如上所述,本文所述的装置、方法和系统优于这种其它技术,尤其优于电穿孔。
本主题的方面提供了跨细胞的质膜递送基因编辑组合物的方法。在优选的实施方式中,该方法不包含载体,例如慢病毒载体、脂质体或电穿孔。在一些实施方式中,该方法包括接触细胞或将编码基因编辑蛋白的质粒DNA递送至细胞;在其他实施方式中,该方法不包括接触细胞或将编码基因编辑蛋白的质粒DNA递送至细胞。在一些实施方式中,该方法包括接触细胞或将编码基因编辑蛋白的RNA递送至细胞;在其他实施方式中,该方法不包括接触细胞或将编码基因编辑蛋白的RNA递送至细胞。这里描述的方法包括提供细胞群,并将细胞群与一定体积的水溶液接触,例如,该水溶液含有基因编辑蛋白和/或复合物。水溶液可以包括基因编辑组合物和浓度为至少约2%的醇。在各种实施方式中,该体积随以下各项变化:(I)细胞群的暴露表面积;或(ii)细胞群中的细胞数目。
优选地,溶液以喷雾或雾的形式递送到细胞中,例如对于每个细胞,多个1、2、3、4、5、6、7、8、9、10或更多的含水颗粒。例如,细胞涂有喷雾,但没有浸泡或浸没在含有递送化合物的溶液中。喷雾或雾包括以微滴形式通过空气吹入或驱动的液体。
实现涂覆细胞群的涂覆的条件的实例包括递送细颗粒喷雾,例如,该条件排除在细胞上滴下或吸移团体积的溶液使得大量细胞被一定体积的流体浸泡或浸没。因此,雾或喷雾包括流体体积与细胞体积的比率。可替代地,该条件包括雾或喷雾的体积与暴露的细胞面积的比率,暴露的细胞面积例如当细胞作为汇合层或基本汇合层存在于基本平坦的表面上时暴露的细胞膜面积,该基本平坦的表面诸如组织培养容器的底部,例如组织培养板的孔,例如微量滴定组织培养板。
因此,需要提供一种无载体(例如,无病毒载体)的方法,用于跨质膜递送生物相关载荷物(例如,化合物或组合物)并进入细胞。“货物”或“载荷物”是用于描述通过水溶液被跨细胞质膜递送并进入细胞内部的化合物或组合物的术语。
在一些实例中,水溶液包括浓度在约2%至约50%之间的醇;至少约2%且小于约20、19、18、17、16或15%;或至少约2、2.5、3、3.5、4、4.5、5、5.5、6、6.5、7、7.5、8、8.5、9、9.5、10、11、12、13、14或15%。在一些方面,醇的浓度小于约50、49、48、47、46、45、44、43、42、41、40、39、38、37、36、35、34、33、32、31、30、29、28、27、26、25、24、23、22、21、20、19、18、17、16、15、14、13、12、11、10、9、8、7、6、5、4、3或2%。在一些方面,醇,例如乙醇,浓度不超过50%。在一些方面,醇,例如乙醇,浓度不超过70%。在一些实例中,醇浓度约为25%。醇浓度可以根据细胞类型而变化,例如醇浓度可以是25%、27%或33%。例如,某些靶细胞,例如T细胞诸如原代T细胞,其特征在于对乙醇具有更高的耐受性,并在使用大于25%乙醇(例如27%、30%、33%、35%或更多)的递送缓冲液处理后保持存活力和功能性。NK细胞,例如原代NK细胞,对于货物递送缓冲液中乙醇的存在/浓度也可以具有这样的耐受水平。
以下一个或多个特征可以包括在任何可行的组合中。待递送到细胞中的溶液的体积是多个单位,例如喷雾,例如多个水性颗粒微滴。相对于单个细胞或相对于汇合或亚汇合(例如至少75%、至少80%汇合,例如85%、90%、95%、97%、98%、100%)的细胞群的暴露表面积描述体积。例如,体积可以在每个细胞6.0×10
细胞汇合是指细胞在表面上相互接触。例如,它可以表示为估计的(或计数的)百分比,例如10%的汇合意味着10%的表面,例如组织培养容器,被细胞覆盖,100%意味着它被完全覆盖。例如,粘附细胞在组织培养孔、板或烧瓶的表面上二维生长。非粘附细胞可以被离心下来,通过真空或组织培养基细胞群的顶部抽吸下来,或者通过抽吸或真空移除从容器底部移除来移除。
使细胞群与一定体积的水溶液接触可以通过气体推动水溶液形成喷雾来进行。气体可以包括氮气、环境空气或惰性气体。喷雾可以包括尺寸从10nm到100μm,,例如直径30-100μm的体积的离散单位。喷雾包括直径为约30-50μm的体积离散单位。20μl水溶液的总体积可以喷雾形式递送到约1.82cm
24孔=约2.0cm
48孔=约0.8cm
96孔=约0.3cm
在本实例中,测量位于每个孔尺寸中的溶液体积。通常使用“体积与表面积”或“体积与细胞数目”的比率。
另一方面,培养板可包括选自1、6、9、12、24、48、96、384和1536孔的样品孔,特别是培养板具有96个样品孔的实例。样品孔的直径可以在0.1mm至100mm的范围内;例如,24孔培养板可具有约15.6mm的直径,48孔培养板可具有约11mm的直径,96孔培养板可具有约6.4mm的直径。下面示出近似孔尺寸和喷雾递送体积。
单孔近似直径单孔近似生长每单孔递送喷雾
*方形孔;**2.5μl,由位于每口井尺寸中的溶液体积确定(见上文)
典型地,水溶液包括待跨细胞膜递送并进入细胞的载荷物,第二体积是不包含载荷物的缓冲液或培养基。可替代地,第二体积(缓冲区或培养基)也可以包含载荷物。在一些实施方式中,水溶液包括载荷物和醇,并且第二体积不含醇(并且可选地不含载荷物)。在加入第二体积的缓冲液或培养基以浸没或悬浮所述细胞群之前,细胞群可以与所述水溶液接触0.01-10分钟,例如0.1-10分钟。缓冲液或培养基可以是磷酸盐缓冲盐水(PBS)。在加入第二体积的缓冲液或培养基以浸没或悬浮细胞群之前,细胞群可以与水溶液接触2秒至5分钟。在加入第二体积的缓冲液或培养基(例如不含载荷物)以浸没或悬浮细胞群之前,细胞群可以与水溶液(例如包含载荷物)接触30秒至2分钟。在加入第二体积的缓冲液或培养基以浸没或悬浮细胞群之前,细胞群可以与喷雾溶液接触约1-2分钟。在一些方面,水溶液在递送之前小于约30、29、28、27、26、25、24、23、22、21、20、19、18、17、16、15、14、13、12、11、10、9、8、7、6、5、4、3、2或1分钟制备。
在一些实施方式中,向水溶液中加入醇可以在培养5、10、15、20、25、30、45分钟(例如20-30分钟)后进行。
在喷雾细胞和加入缓冲液或培养基之间的时间内,细胞通过来自喷雾体积的湿气层保持水合。
基因编辑组合物可以包括编辑基因组DNA的化合物。例如,基因编辑组合物可以包括切割、缺刻、拼接、重排、转位、重组或以其他方式改变基因组DNA的化合物或复合物。替代地或另外,基因编辑组合物可以包括:(i)可以包括在切割、缺刻、拼接、重排、转位、重组或以其他方式改变基因组DNA的基因编辑复合物的化合物;或者(ii)可以被处理或改变为包括在切割、缺刻、拼接、重排、转位、重组或以其他方式改变基因组DNA的基因编辑复合物中的化合物的化合物。在各种实施方式中,基因编辑组合物包括以下一种或多种:(a)基因编辑蛋白;(b)RNA分子;和/或(c)核糖核蛋白(RNP)。
在一些实施方式中,基因编辑组合物包括基因编辑蛋白,基因编辑蛋白是锌指核酸酶(ZFN)、类转录激活因子效应物核酸酶(TALEN)、Cas蛋白、Cre重组酶、Hin重组酶或Flp重组酶。在另外的实施方式中,基因编辑蛋白可以是结合归巢核酸内切酶和组件式DNA结合结构域的融合蛋白(megaTAL)。例如,megaTAL可以作为蛋白质递送,或可替代地,megaTAL蛋白编码mRNA被递送到细胞。
在各种实施方式中,基因编辑组合物包括RNA分子,RNA分子包括sgRNA、crRNA和/或tracrRNA。
在某些实施方式中,基因编辑组合物包括RNP,RNP包括Cas蛋白和sgRNA或crRNA和tracrRNA。
本主题的方面对于控制特定基因编辑化合物在细胞中何时存在以及存在多长时间特别有用。
在本主题的各种实现方式中,基因编辑组合物在细胞群或其后代中在(a)细胞群与水溶液接触后约0.5、1、2、3、4、5、6、7、8、9、10、12、24、48、60、72、0.5-2、0.5-6、6-12或0.5-72小时,或(b)细胞群与水溶液接触后小于约0.5、1、2、3、4、5、6、7、8、9、10、12、24、48、60、72、0.5-2、0.5-6、6-12或0.5-72小时可检测到。
在一些实施方式中,细胞群中的细胞基因组或其后代包括Cre重组酶、Hin重组酶或Flp重组酶的至少一个位点特异性重组位点。
本发明的方面涉及包括一种基因编辑化合物的细胞,并向该细胞中插入另一种基因编辑化合物。例如,RNP的一个组分可以被引入到表达或者已经包含RNP的另一个组分的细胞中。例如,细胞群中的细胞或其后代可以包括sgRNA、crRNA和/或tracrRNA。在一些实施方式中,细胞群或其后代表达sgRNA、crRNA和/或tracrRNA。可替代地或另外,细胞群中的细胞或其后代表达Cas蛋白。
本文主题的各种实现方式包括Cas蛋白。在一些实施方式中,Cas蛋白是Cas9蛋白或其突变体。本文描述了示例性Cas蛋白(包括Cas9和Cas9突变体的非限制性实例)。
在各个方面,Cas9蛋白的浓度可以在约0.1至约25μg的范围内。例如,Cas9的浓度可以是约1μg、约5μg、约10μg、约15μg或约20μg。可替代地,Cas9的浓度可以在约10ng/μL至约300ng/μL的范围内;例如约10ng/μL至约200ng/μL;或约10ng/μL至约100ng/μL,或约10ng/μL至约50ng/μL。
在某些实施方式中,基因编辑组合物包括(a)第一sgRNA分子和第二sgRNA分子,其中第一sgRNA分子的核酸序列不同于第二sgRNA分子的核酸序列;(b)包括第一sgRNA的第一RNP和包括第二sgRNA的第二RNP,其中第一sgRNA分子的核酸序列不同于第二sgRNA分子的核酸序列;(c)第一crRNA分子和第二crRNA分子,其中第一crRNA分子的核酸序列不同于第二crRNA分子的核酸序列;(d)第一crRNA分子和第二crRNA分子,其中第一crRNA分子的核酸序列不同于第二crRNA分子的核酸序列,并且还包括tracrRNA分子;或者(e)包括第一crRNA和tracrRNA的第一RNP和包含第二crRNA和tracrRNA的第二RNP,其中第一crRNA分子的核酸序列不同于第二crRNA分子的核酸序列。
在方面中,Cas9蛋白与引导RNA的比率可以是1:1、1:2、1:3、1:4、1:5、1:6、1:7、1:8、1:9或1:10。
在实施例中,增加细胞通过递送过程的次数(替代地,增加剂量的数量),可以增加百分比编辑;其中,在一些实施方式中,剂量的数量可以包括1、2、3、4、5、6、7、8、9或10个剂量。
在各种实施方式中,第一和第二sgRNA或第一和第二crRNA分子一起包括与基因侧翼的靶序列互补的核酸序列、外显子、内含子、染色体外序列或基因组核酸序列,其中基因、外显子、内含子、染色体外序列或基因组核酸序列的长度为约1、2、3、4、5、6、10、20、30、40、50、60、70、80、90、100、1-100千碱基或长度为至少约1、2、3、4、5、6、10、20、30、40、50、60、70、80、90、100、1-100千碱基。在一些实施方式中,使用包含第一和第二sgRNA或第一和第二crRNA分子的成对RNP可用于产生包括基因、外显子、内含子、染色体外序列或基因组核酸序列的多核苷酸分子。
在某些实施方式中,sgRNA或crRNA的靶序列为约12至约25,或约12、13、14、15、16、17、18、19、20、21、22、23、24、25、17-23或18-22个核苷酸长。在一些实施方式中,靶序列为20个核苷酸长或约20个核苷酸长。
在各种实施方式中,第一和第二sgRNA或第一和第二crRNA分子与表达载体内染色体外序列侧翼的序列互补。
本主题的方面涉及基因编辑复合体的多个组分的递送,其中多个组分不复合在一起。在一些实施方式中,基因编辑组合物包括至少一种基因编辑蛋白和至少一种核酸,其中基因编辑蛋白和核酸不彼此结合或复合。
本主题允许高基因编辑效率,同时保持高细胞存活力。在一些实施方式中,至少约10、20、30、40、50、60、70、80、90、95、99%、1-99%,或更多的细胞群或其后代在与水溶液接触后变成遗传修饰的。在各种实施方式中,至少约10、20、30、40、50、60、70、80、90、95、99%、1-99%,或更多的细胞群或其后代在与水溶液接触后是活的。
在某些实施方式中,基因编辑组合物诱导细胞内DNA中的单链或双链断裂。在一些实施方式中,基因编辑组合物还包括修复模板多核苷酸。在各种实施方式中,修复模板包括(a)包括与在单链或双链断裂的一侧的约40至约90个碱基对互补的序列中的核苷酸的第一侧翼区和包括与在单链或双链断裂的另一侧约40至约90个碱基对互补的序列中的核苷酸的第二侧翼区;或(b)包括与在单链或双链断裂的一侧的至少约20、25、30、35、40、45、50、60、70、80或90个碱基对互补的序列中的核苷酸的第一侧翼区,以及包括与在单链或双链断裂的另一侧的至少约20、25、30、35、40、45、50、60、70、80或90个碱基对互补的序列中的核苷酸的第二侧翼区。Ran等人(2013)Nat Protoc.2013Nov;8(11):2281–2308中讨论了使用CRISPR-Cas系统进行基因编辑(包括修复模板)的非限制性描述,其全部内容通过引用并入本文。涉及修复模板的实施方式不限于包括CRISPR-Cas系统的那些。
在本主题的各种实现方式中,水溶液的体积以喷雾的形式递送到细胞群。体积在每个细胞6.0×10
在一些实施方式中,RNP具有大约
例如,使细胞群与一定体积的水溶液接触可以通过气体推动水溶液形成喷雾来进行。在某些实施方式中,在加入第二体积的缓冲液或培养基以浸没或悬浮所述细胞群之前,细胞群与所述水溶液接触0.01-10分钟(例如0.1-10分钟)。
在一些实施方式中,缓冲液或培养基包括磷酸盐缓冲盐水(PBS)。在各种实施方式中,水溶液包括约5%至约50%的乙醇浓度。在非限制性实例中,所述水溶液包括以下一种或多种:(a)约75%至约98%H
在各种实施方式中,细胞群包括原代或永生化细胞中的至少一种。例如,细胞群可以包括间充质干细胞、肺细胞、神经元细胞、成纤维细胞、人脐静脉(HUVEC)细胞和人胚肾(HEK)细胞、原代或永生化造血干细胞(HSC)、T细胞、自然杀伤(NK)细胞、细胞因子诱导杀伤(CIK)细胞、人脐带血CD34+细胞、B细胞。T细胞的非限制性实例可包括CD8+或CD4+T细胞。在一些方面,使用CD3+T细胞的CD8+亚群。CD8+T细胞可以通过使用抗CD8珠的阳性分离从PBMC群体中纯化。在一些方面,原代NK细胞从PBMC中分离,GFP mRNA可以通过平台递送技术递送(即,24小时时3%的表达和96%的存活力)。在另外的方面,可以使用NK细胞系,例如NK92。
细胞类型还包括先前被修饰的细胞,例如T细胞、NK细胞和MSC,以增强它们的治疗效果。例如:表达嵌合抗原受体的T细胞或NK细胞(分别为CAR T细胞、CAR NK细胞);表达修饰T细胞受体(TCR)的T细胞;经病毒或非病毒修饰以过量表达补充其固有特性的治疗蛋白的MSC(例如,使用慢病毒载体递送Epo或使用AAV-6递送BMP-2)(在Park等人Methods,2015Aug;84-16中综述。);用分别用于增强效力和外部调节的靶向的非肽类药物或磁性纳米颗粒引发的MSC(Park等人,2015);用靶向部分功能化以利用酶修饰(例如岩藻糖基转移酶)、化学缀合(例如通过使用N-羟基琥珀酰亚胺(NHS)化学修饰MSC上的SLe
本主题的方面还提供一种用于跨细胞的质膜递送载荷物的组合物,该组合物包括含有载荷物和浓度为至少2%的醇的水溶液。在一些实施方式中,组合物还包括镁盐。在某些实施方式中,组合物包括约0.1至约10mM的镁盐或约0.1至约10mM的氯化镁。镁盐包括镁离子。
本主题的方面还提供了一种用于跨细胞的质膜递送载荷物的组合物,该组合物包括含有载荷物、浓度至少为2%的醇、小于约46mM的缺少镁离子的盐、小于约121mM的糖、小于19mM的缓冲剂和约0.1mM至约10mM的镁盐的水溶液。
在某些实施方式中,缓冲剂是弱酸或弱碱,其有效地将水溶液的pH调节或保持在约6.5、6.75、7、7.25、7.5、7.75、8、8.25或8.5的pH。在各种实施方式中,醇是甲醇、乙醇、异丙醇、丁醇或苯甲醇。在一些实施方式中,缺少镁离子的盐包括Na或K离子。在一些实施方式中,盐是NaCl、KCl、Na
本发明的方面提供了跨细胞的质膜递送基因编辑组合物的设备。在各种实施方式中,该设备包括:在压力下产生气体的气动发生器;可操作地联接到气动发生器的雾化器;被配置为包含一定体积的水溶液的贮存器,该水溶液包括基因编辑组合物和浓度大于2%的醇;以及气动发生器和雾化器之间的阀。在一些实施方式中,阀可在用于防止压力下的气体激活雾化器的关闭位置和用于允许压力下的气体激活雾化器以从水溶液产生胶体微滴的打开位置之间切换。在各种实施方式中,阀具有小于约250毫秒的切换速度。在某些实施方式中,雾化器定向成使细胞群与水溶液接触,并且其中该体积随以下变化:(i)细胞群的暴露表面积;或(ii)细胞群中的细胞数目。
还提供了本文描述或说明的附加的设备、系统、方法、组合物和制品。
一方面,跨细胞的质膜递送载荷物包括提供细胞群和使细胞群与一定体积的水溶液接触。水溶液包括载荷物和浓度大于百分之5的醇含量。水溶液的体积可以随细胞群暴露表面积而变化,或者随细胞群中细胞的数目而变化。
另一方面,一种用于跨细胞的质膜递送载荷物的组合物包括水溶液,该水溶液包括载荷物、浓度大于5%的醇、小于46mM的盐、小于121mM的糖和小于19mM的缓冲剂。例如,醇,例如乙醇,浓度不超过50%。
以下一个或多个特征可以包括在任何可行的组合中。待递送到细胞中的溶液的体积是多个单位,例如喷雾,例如多个微滴或水性颗粒。相对于单个细胞或相对于汇合或亚汇合(例如至少75%、至少80%汇合,例如85%、90%、95%、97%、98%、100%)的细胞群的暴露表面积描述体积。例如,体积可以在每个细胞6.0×10
细胞汇合是指细胞在表面上相互接触。例如,它可以表示为估计的(或计数的)百分比,例如10%的汇合意味着10%的表面,例如组织培养容器,被细胞覆盖,100%意味着它被完全覆盖。例如,粘附细胞在组织培养孔、板或烧瓶的表面上二维生长。非粘附细胞可以被离心下来,通过真空或组织培养基细胞群的顶部抽吸下来,或者通过抽吸或真空移除从容器底部移除来移除。
使细胞群与一定体积的水溶液接触可以通过气体推动水溶液形成喷雾来进行。气体可以包括氮气、环境空气或惰性气体。喷雾可以包括尺寸从10nm到100μm,,例如直径30-100μm的体积的离散单位。喷雾包括直径为约30-50μm的体积离散单位。例如,20μl水溶液的总体积可以喷雾形式递送到约1.9cm
水溶液可以包括5至30%的乙醇浓度。水溶液可以包括75至98%的H2O、2至45%的乙醇、6至91mM的蔗糖、2至35mM的KCl、2至35mM乙酸铵和1至14mM(4-(2-羟乙基)-1-哌嗪乙磺酸)(HEPES)中的一种或多种。
细胞群可以包括粘附细胞或非粘附细胞。粘附细胞可以包括原代间充质干细胞、成纤维细胞、单核细胞、巨噬细胞、肺细胞、神经元细胞、成纤维细胞、人脐静脉(HUVEC)细胞、中国仓鼠卵巢(CHO)细胞和人胚肾(HEK)细胞或永生细胞(诸如细胞系)中的至少一种。细胞群可以包括非粘附细胞。非粘附细胞可以包括原代造血干细胞(HSC)、T细胞、自然杀伤(NK)细胞、细胞因子诱导杀伤(CIK)细胞、人脐带血CD34+细胞、B细胞或细胞系诸如JurkatT细胞系中的至少一种。
细胞群可以基本上汇合,诸如大于百分之75的汇合。细胞汇合是指细胞在表面上相互接触。例如,它可以表示为估计的(或计数的)百分比,例如,10%的汇合意味着10%的表面,例如组织培养容器,被细胞覆盖,100%意味着它被完全覆盖。例如,粘附细胞在组织培养孔、板或烧瓶的表面上二维生长。非粘附细胞可以被离心下来,通过真空或组织培养基细胞群的顶部抽吸下来,或者通过抽吸或真空移除从容器底部移除来移除。细胞群可以形成单层细胞。
在一些实施方式中,本主题的组合物或溶液包括醇;盐;糖;缓冲剂;和/或乙酸铵。醇可选自例如甲醇、乙醇、异丙醇、丁醇和苯甲醇。盐可选自例如NaCl、KCl、Na
本主题涉及一种跨质膜递送基因编辑化合物和复合物的方法。本主题的方法包括将化合物或复合物引入水性组合物中以形成基质;将基质雾化成喷雾;以及使基质与质膜接触。
这里描述的示例性方法包括载荷物,其中载荷物包括醇。术语“醇”是指包括附连到至少一个碳原子上的羟基(-OH)官能团的多原子有机化合物。醇可以是一元醇,并且可以包括至少一个碳原子,例如甲醇。醇可以包括至少两个碳原子(例如乙醇)。在其他方面,醇包括至少三个碳(例如异丙醇)。醇可以包括至少四个碳原子(例如丁醇),或者至少七个碳原子(例如苯甲醇)。示例性载荷物可以包括不超过50%(v/v)的醇,更优选地,载荷物包括2-45%(v/v)的醇、5-40%的醇和10-40%的醇。载荷物可以包括20-30%(v/v)的醇。
最优选地,载荷物包括25%(v/v)的醇。可替代地,载荷物可以包括2-8%(v/v)的醇或2%的醇。醇可以包括乙醇,且载荷物包括5、10、20、25、30、40或50%(v/v)的乙醇。示例性方法可以包括甲醇作为醇,且载荷物可以包括5、10、20、25、30、40或50%(v/v)的甲醇。载荷物可以包括2-45%(v/v)的甲醇、20-30%(v/v)或25%(v/v)甲醇。优选地,载荷物包括20-30%(v/v)的甲醇。另外,可替代地,醇是丁醇,且载荷物包括2、4或8%(v/v)的丁醇。
在本主题的一些方面,载荷物在低渗溶液或缓冲液中。载荷物溶液可以具有171mOsm/L的渗透浓度。根据示例性方法,载荷物溶液在室温下具有171mOsm/L的渗透浓度。
根据本主题,载荷物可以包括至少一种盐。盐可选自NaCl、KCl、Na
根据本主题的示例性方法,载荷物可以包括糖(例如蔗糖或二糖)。根据示例性方法,载荷物包括小于121mM的糖、6-91mM或26-39mM的糖。还另外,载荷物包括32mM的糖(例如蔗糖)。任选地,糖是蔗糖,且载荷物包括6.4、12.8、19.2、25.6、32、64、76.8或89.6mM的蔗糖。
根据本主题的示例性方法,载荷物可以包括缓冲剂(例如弱酸或弱碱)。缓冲剂可以包括两性离子。根据示例性方法,缓冲剂是4-(2-羟乙基)-1-哌嗪乙磺酸。载荷物可以包括小于19mM的缓冲剂(例如,1-15mM,或4-6mM或5mM的缓冲剂)。根据示例性方法,缓冲剂是4-(2-羟乙基)-1-哌嗪乙磺酸,且载荷物包括1、2、3、4、5、10、12、14mM的4-(2-羟乙基)-1-哌嗪乙磺酸。还优选地,载荷物包括5mM的4-(2-羟乙基)-1-哌嗪乙磺酸。
根据本主题的示例性方法,载荷物包括乙酸铵。载荷物可以包括小于46mM的乙酸铵(例如,在2-35mM之间、10-15mM之间或12mM的乙酸铵)。载荷物可包括2.4、4.8、7.2、9.6、12、24、28.8或33.6mM的乙酸铵。
本文描述的方法包括本主题的第二方面,其中提供了包括68mM NaCl、1.4mM KCl、5mM Na
在各种实施方式中,由推动水溶液的气体执行的水溶液的体积可以包括例如压缩空气(例如环境空气)。这些和其他实现方式可以包括惰性气体,例如氦、氖和氩。
在本主题的某些方面,细胞群可包括粘附细胞(例如肺、肾、免疫细胞诸如巨噬细胞)或非粘附细胞(例如悬浮细胞)。
在本主题的某些方面,细胞群可以基本上汇合,并且基本上可以包括大于75%的汇合。在优选的实现方式中,细胞群可以形成单个单层。
在2015年10月23日提交的PCT国际专利申请No.PCT/US2015/057247中描述了用于跨细胞质膜递送化合物的方法、系统、设备和装置的附加的非限制性描述,其全部内容通过引用并入本文。
本主题还涉及用于跨质膜递送载荷物的设备、系统、技术和制品。本主题还涉及一种用于跨质膜递送载荷物诸如蛋白质或蛋白质复合物的设备。本主题可在细胞内递送领域中找到用途,并可应用于例如将基因编辑组合物递送至靶位点,诸如细胞、组织或器官。
在一些实现方式中,用于跨质膜递送载荷物的设备可以包括雾化器,该雾化器具有至少一个雾化发射器和相对于雾化器定向的支撑件。该方法还可以包括在使质膜与载荷物接触之前雾化载荷物的步骤。
在各种实施方式中,雾化器可以选自例如机械雾化器、超声雾化器、电喷雾、喷雾器和文丘里管。雾化器可以是市售雾化器。雾化器可以是鼻内粘膜雾化装置。雾化器可以是可从美国NC的LMA Teleflex商购的鼻内粘膜雾化装置。雾化器可以是可从美国NC的LMATeleflex商购的鼻内粘膜雾化装置,产品编号为MAD300。
在某些实施方式中,雾化器可适于在质膜与载荷物接触之前提供直径为30-100μm的颗粒的胶体悬浮液。在各种实施方式中,雾化器可适于提供直径为30-80μm的颗粒的胶体悬浮液。例如,雾化器可适于提供直径为50-80μm的颗粒的胶体悬浮液。
在一些实施方式中,雾化器包括气体贮存器。例如,雾化器可以包括具有保持在压力下的气体的气体贮存器。在某些实施例方式,气体选自空气、二氧化碳和氦。在各种实施方式中,气体贮存器包括固定压头发生器。在一些实施方式中,气体贮存器可以与雾化发射器流体连通。在本主题的各种实现方式中,气体贮存器包括可与雾化发射器流体连通的气体引导件。在一些实施方式中,气体引导件适于允许气体从其通过。气体引导件可以例如包括中空体。例如,气体引导件可以是具有开口端的中空体。在一些实施方式中,气体引导件包括具有第一和第二开口端的中空体。在各种实施方式中,气体引导件是具有第一和第二相对的开口端的中空体。在某些实施方式中,第一开口端的直径不同于第二开口端的直径。在各种实施方式中,第一开口端的直径可以小于或大于第二开口端的直径。在各种实现方式中,第一开口端与气体贮存器流体连通,或者第二开口端与雾化发射器流体连通。
在某些实施方式中,所述设备包括样品贮存器。在非限制性示例中,样品贮存器与雾化器流体连通。在各种实施方式中,样品贮存器与雾化发射器流体连通。例如,气体贮存器和样品贮存器都可以与雾化发射器流体连通。
在一些实施方式中,该设备包括位于样品贮存器和气体贮存器之间的样品阀。在这些和其他实施方式中,该设备可以包括位于样品贮存器和气体引导件之间的样品阀。在某些实施方式中,样本阀可适于调节来自样品贮存器的样品流动。在一些实例中,样品阀可适于允许连续或半连续样品流动。在各种实现方式中,样品阀可适于允许限定量的半连续样品流动。例如,样品阀可适于允许0.5-100μL或10μL的半连续样品流动。在一个非限制性实例中,样品阀适于允许1L的半连续样品流动至0.065-0.085cm
在各种实施方式中,雾化器和支撑件间隔开。支撑件可以包括固体支撑件。在一些实施方式中,支撑件可以包括具有样品孔的板。例如,支撑件可以包括板,该板包括从1、6、9、12、24、48、384和1536个孔中选择的样品孔。在某些实施方式中,固体支撑件由惰性材料形成。例如,固体支撑件可以由塑料材料、金属或金属合金或其组合形成。在一些实施方式中,支撑件包括加热元件和/或电阻元件。在某些实施方式中,支撑件可往复地安装到设备上。在各种实现方式中,支撑件可相对于设备往复移动。例如,支撑件可以相对于雾化器往复移动。在各种实施方式中,支撑件可以相对于雾化发射器往复移动。在一些实施方式中,支撑件包括支撑致动器以使支撑件相对于雾化器往复移动。在各种实施方式中,支撑件包括支撑致动器以使支撑件相对于雾化发射器往复移动。在非限制性实例中,支撑件可以包括支撑致动器,以使支撑件相对于雾化发射器的纵向轴线往复移动。在一些实施方式中,支撑件可以包括支撑致动器,以使支撑件横向于雾化发射器的纵向轴线往复移动。
在一些实施方式中,喷雾区的纵向轴线与支撑件的纵向轴线或中心点和/或支撑件的圆形孔同轴,载荷物将被递送到圆形孔。在某些实施方式中,雾化发射器的纵向轴线与支撑件的纵向轴线或中心点和/或支撑件的圆形孔同轴。在各种实施方式中,雾化发射器的纵向轴线、支撑件的纵向轴线和喷雾区的纵向轴线均同轴。在某些实施方式中,喷雾区(例如,载荷物将被递送到的细胞的区域)的纵向长度大于喷雾区的圆形基部的直径(可以大于两倍)。
在各种实施方式中,该设备包括位于气体贮存器和雾化器之间的阀。在一些实施方式中,阀是电磁操作的阀。在某些实施方式中,阀是电磁阀或气动阀。在一些实施方式中,阀位于气体引导件处。例如,阀可适于调节气体引导件内的气体流动。在各种实施方式中,阀适于允许连续或半连续的气体流动。在非限制性实例中,阀适于允许限定时间间隔(诸如一秒时间间隔)的半连续气体流动。在某些实施方式中,该设备包括至少一个过滤器。在一些实施方式中,过滤器包括小于10μm的孔径或10μm的孔径。在各种实施方式中,过滤器位于气体引导件处。在一些实施方式中,过滤器与气体引导件流体连通。
在某些实施方式中,该设备包括至少一个调节器。调节器可以是例如电调节器或机械调节器。在一些实施方式中,调节器位于气体引导件处。在一些实施方式中,调节器与气体引导件流体连通。在各种实施方式中,调节器是调节阀。在一些实施方式中,气体引导件的压力为约1.0-2.0巴或约1.5巴。在非限制性实例中,气体引导件内的压力为1.0-2.0巴,雾化器和支撑件之间的距离小于或等于31mm。例如,气体引导件内的压力可以是1.5巴,雾化器和支撑件之间的距离可以是31mm。在各种实现方式中,气体引导件内的压力对于每毫米雾化器和支撑件之间的距离为约0.05巴。在一些实施方式中,调节阀可适于将气体引导件内的压力调节至约1.0-2.0巴或约1.5巴。在某些实施方式中,每个调节阀可适于将气体引导件内的压力保持在约1.0-2.0巴或约1.5巴。
在一些实施方式中,该设备包括两个调节器。例如,该设备可以包括第一和第二调节器。在各种实现方式中,第一和第二调节器可以位于气体引导件处。在一些实现方式中,第一和第二调节器可以与气体引导件流体连通。在某些实施方式中,第一调节器位于贮存器和过滤器之间。在各种实施方式中,第一调节器可适于将气体引导件内的来自气体贮存器的压力调节到约2.0巴,或者将气体引导件内的压力保持在约2.0巴。在一些实施方式中,第二调节器可以位于过滤器和阀之间。
在各种实施方式中,雾化发射器可适于提供锥形喷雾区(例如,大致圆形的锥形喷雾区)。例如,雾化发射器可适于提供30°锥形喷雾区。在一些实施方式中,该设备还可以包括微处理器以控制该设备的任何或所有部分。在各种实施方式中,微处理器可以布置成控制样品阀、支撑件致动器、阀和调节器中的任何一个或全部。在一些实现方式中,该设备可以包括具有至少一个雾化发射器的雾化器;和相对于雾化器定向的支撑件;雾化器可以选自机械雾化器、超声雾化器、电喷雾、喷雾器和文丘里管。在某些实施方式中,雾化器可适于提供直径为30-100μm的颗粒的胶体悬浮液。在非限制性实力中,设备可以包括样品贮存器和气体引导件,以及位于样品贮存器和气体引导件之间的样品阀。在一些实施方式中,样品阀可适于允许10-100μL的半连续样品流动。在各种实现方式中,雾化器和支撑件可以间隔开并在其间限定大致锥形喷射区;雾化器与支撑件之间的距离可以是分子将被递送到的细胞的区域圆形基部直径的大约两倍;雾化器和支撑件之间的距离可以是31mm,分子将被递送到的细胞的区域的圆形基部的直径可以是15.5mm。在某些实施方式中,该设备可以包括气体引导件,并且气体引导件内的压力为1.0-2.0巴。在一些实施方式中,该设备可以包括至少一个孔径小于10μm的过滤器。
变型的非限制性实例包括:保持在移动板定位器上方的静态框架上的一个或多个雾化器;保持在可调节的x-y-z-r轴线上的一个或多个雾化器(其中r是从0-360度的旋转偏移);保持在移动板定位器上方的静态支撑件上的一个或多个雾化器;保持在可调节x-y轴线上的一个或多个雾化器;保持在移动板定位器上方的静态框架上的一个或多个雾化器;和保持在可调节x轴线上的一个或多个雾化器。
在一些实现方式中,一个或多个雾化器定位在导管或棒的远端,目的是离体或体内对准组织或组织的部分。例如,本主题的方面涉及将基因编辑载荷物递送到暴露的组织,例如动物的真皮或眼组织,或者内部组织,例如哺乳动物,例如人或人以外的哺乳动物的包括腔的组织,诸如肺组织(气道)、胃肠组织(食道、肠)或心血管组织(动脉或静脉)。
在各种实施方式中,一个或多个雾化器定位在导管或棒的远端,目的是对准孔板表面的部分或子部分(例如,板的一部分或多孔板的特定孔)。在某些实施方式中,一个或多个雾化器定位在静态支撑件上,该静态支撑件上定位在连续移动的衬底上方。衬底可以与细胞培养相容,或者包括培养细胞的板或孔。
在一些实施方式中,本主题的装置或设备包括与雾化器或多个雾化器共入射的可见低功率激光器和透镜装置,以指示喷雾正在或将要递送到何处。在各种实施方式中,激光器和透镜可适于采用与雾化器相同的分散模式。在一些实施方式中,分散模式是锥形分散模式。
本主题的各种实施方式包括液体分配器和移液管,其能够在微控制、可视化、操纵和/或调整微滴/颗粒尺寸的情况下产生微升、纳升或皮升的滴并将滴单独定位到细胞上。例如,具有纳升或皮升范围体积的微滴/含水颗粒(例如,包含一种或多种RNP)在显微镜引导下被施用至单个细胞(单独或作为邻接细胞单层或垫的成员)。使用这种方法,质膜不像显微注射那样被针刺穿。而是将微滴/颗粒滴或施加到细胞上,并实现进入细胞质,而无需对质膜进行物理操作,例如显微注射。这样,微滴/颗粒在尺寸(例如,直径500纳米至100微米)或体积(例如,在1.0×10
本发明的方面涉及将基因编辑化合物和复合物无表达载体地递送到细胞和组织,诸如递送用于原代人T细胞和造血干细胞(HSC)中基因组编辑的Cas-gRNA核糖核蛋白。
CRISPR-Cas系统的各个方面在本领域中是已知的。该系统的非限制性方面描述在例如2015年5月5日发布的美国专利No.9,023,649;2015年7月7日发布的美国专利No.9,074,199;2014年4月15日发布的美国专利No.8,697,359;2015年1月13日发布的美国专利No.8,932,814;2015年8月27日出版的PCT国际专利申请公开No.WO2015/071474;Cho等人(2013)Nature Biotechnology Vol 31No 3pp 230-232(含补充资料);和Jinek等人(2012)cience Vol 337No 6096pp 816-821中,其每个的全部内容通过引用并入本文。
Cas蛋白的非限制性实例包括Cas1、Cas1B、Cas2、Cas3、Cas4、Cas5、Cas6、Cas7、Cas8、Cas9(也称为Csn1和Csx12)、Cas10、Csy1、Csy2、Csy3、Cse1、Cse2、Csc1、Csc2、Csa5、Csn2、Csm2、Csm3、Csm4、Csm5、Csm6、Cmr1、Cmr3、Cmr4、Cmr5、Cmr6、Csb1、Csb2、Csb3、Csx17、Csx14、Csx10、Csx16、CsaX、Csx3、Csx1、Csx15、Csf1、Csf2、Csf3、Csf4、其同源物或修饰形式。这些酶是已知的;例如,化脓性链球菌(S.pyogenes)Cas9蛋白的氨基酸序列可以在SwissProt数据库中以登录号Q99ZW2(SEQ ID NO:1)找到,在NCBI数据库中以登录号Q99ZW2.1找到。UniProt数据库登录号A0A0G4DEU5和CDJ55032提供了Cas9蛋白氨基酸序列(SEQ ID NO:18)的另一个实例。另一个非限制性实例是嗜热链球菌(Streptococcusthermophilus)Cas9蛋白,其氨基酸序列可以在UniProt数据库中以登录号Q03JI6.1(SEQID NO:19)找到。在一些实施方式中,未修饰的CRISPR酶具有DNA切割活性,诸如Cas9。在某些实施方式中,CRISPR酶是Cas9,并且可以是来自化脓性链球菌(S.pyogenes)或肺炎链球菌(S.pneumoniae)的Cas9。在各种实施方式中,CRISPR酶在靶序列的位置指导一条或两条链的切割,例如在靶序列内和/或在靶序列的互补序列内。在一些实施方式中,CRISPR酶指导从靶序列的第一个或最后一个核苷酸切割约1、2、3、4、5、6、7、8、9、10、15、20、25、50、100、200、500或更多碱基对内的一条或两条链。在一些实施方式中,载体编码相对于相应的野生型酶突变的CRISPR酶,使得突变的CRISPR酶缺乏切割包含靶序列的靶多核苷酸的一条或两条链的能力。例如,在来自化脓性链球菌(S.pyogenes)的Cas9的RuvC I催化结构域中,天冬氨酸至丙氨酸的取代(D10A,其中氨基酸编号如SEQ ID NO:1所示)将Cas9从切割两条链的核酸酶转化为切口酶(切割单条链)。导致Cas9切口酶的突变的其它实例包括但不限于H840A、N854A和N863A(其中氨基酸编号如SEQ ID NO:1所示)。在本发明的方面,可以通过同源重组将切口酶用于基因组编辑。
在某些实施方式中,Cas9切口酶可与分别靶向DNA靶的有义链和反义链的引导序列(例如两个引导序列)组合使用。该结合使得两条链都被切开并用于诱导NHEJ。
作为另一个例子,Cas9的两个或更多催化结构域(RuvC I、RuvC II和RuvC III)可被突变以产生基本上缺乏所有DNA切割活性的突变Cas9。D10A突变可与H840A、N854A或N863A突变中的一种或多种结合,以产生基本上缺乏所有DNA切割活性的Cas9酶。在某些实施方式中,当突变酶的DNA切割活性小于约25%、10%、5%、1%、0.1%、0.01%或更低(相对于其非突变形式)时,CRISPR酶被认为基本上缺乏所有DNA切割活性。其它突变可能是有用的;当Cas9或其它CRISPR酶来自化脓性链球菌以外的物种时,相应氨基酸的突变可以达到类似的效果。
在某些实施方式中,被递送的蛋白质(例如Cas蛋白或其变体)可以包括亚细胞定位信号。例如,RNP内的Cas蛋白可以包括亚细胞定位信号。根据上下文,包含例如Cas9和核定位信号的融合蛋白在本文中可称为“Cas9”,而没有指定包括核定位信号。在一些实施方式中,载荷物(诸如RNP)包括含有定位信号的融合蛋白。例如,融合蛋白可包含核定位信号、核仁定位信号或线粒体靶向信号。这样的信号在本领域中是已知的,并且在Kalderon等人(1984)Cell 39(3Pt 2):499–509;Makkerh et al.,(1996)Curr Biol.6(8):1025–7;Dingwall et al.,(1991)Trends in Biochemical Sciences 16(12):478–81;Scott etal.,(2011)BMC Bioinformatics 12:317(7pages);Omura T(1998)J Biochem.123(6):1010-6;Rapaport D(2003)EMBO Rep.4(10):948-52;and Brocard&Hartig(2006)Biochimica et Biophysica Acta(BBA)-Molecular Cell Research 1763(12):1565–1573中描述了非限制性实例,其每个内容通过引用并入本文。在各种实施方式中,Cas蛋白可包括多于一个的定位信号,诸如2、3、4、5或更多个核定位信号。在一些实施方式中,定位信号位于Cas蛋白的N末端,而在其他实施方式中,定位信号位于Cas蛋白的C末端。
核定位信号的非限制性实例包括GGSGPPKKKRKV(SEQ ID NO:2)、PKKKRKV(SEQ IDNO:5)、KR[PAATKKAGQA]KKKK(SEQ ID NO:6)、KR[XXXXXXXXXX]KKKK(SEQ ID NO:7)、KKXK(SEQ ID NO:8)、KRXK(SEQ ID NO:9)、KKXR(SEQ ID NO:10)、KRXR(SEQ ID NO:11)、AVKRPAATKKAGQAKKKKLD(SEQ ID NO:12)、MSRRRKA NPTKLSENAKKLAKEVEN(SEQ ID NO:13)、PAAKRVKLD(SEQ ID NO:14)、PPKKKRKV(SEQ ID NO:15)和KLK IKRPVK(SEQ ID NO:16)。线粒体定位信号的非限制性实例包括MLSLRQSIRFFKPATRTLCSSRYLL(SEQ ID NO:17)。
在一些实施方式中,编码CRISPR酶的酶编码序列被密码子优化用于在特定细胞诸如真核细胞中表达。真核细胞可以是或来源于特定生物体诸如哺乳动物的真核细胞,包括但不限于人、小鼠、大鼠、兔、狗或非人灵长类动物。一般而言,密码子优化是指通过用在宿主细胞基因中使用频率较高或最高的密码子替换天然序列的至少一个密码子(例如,约1、2、3、4、5、10、15、20、25、50或更多密码子),同时保持天然氨基酸序列,来修饰核酸序列以增强在目的宿主细胞中的表达的过程。各种物种对特定氨基酸的某些密码子表现出特别的偏好。密码子偏好(生物体之间密码子使用的差异)通常与信使RNA(mRNA)翻译效率相关,这反过来被认为尤其取决于被翻译密码子的特性和特定转运RNA(tRNA)分子的可用性。细胞中选定tRNA的优势通常反映肽合成中最常用的密码子。
因此,可以基于密码子优化来定制基因,以在给定生物体中获得最佳的基因表达。密码子使用表很容易获得,例如在“密码子使用数据库”中,这些表可以通过多种方式改编。参见Nakamura,Y.等人“Codon usage tabulated from the international DNA sequencedatabases:status for the year 2000”Nucl.Acids Res.28:292(2000)。用于密码子优化特定序列以在特定宿主细胞中表达的计算机算法也是可用的,诸如Gene Forge(Aptagen;Jacobus,Pa.)也是可用的。在一些实施方式中,编码CRISPR酶的序列中的一个或多个密码子(例如,1、2、3、4、5、10、15、20、25、50或更多,或所有密码子)对应于特定氨基酸的最常用密码子。
一般而言,引导序列是具有与靶多核苷酸序列足够互补性以与靶序列杂交并且指导CRISPR复合物序列特异性结合靶序列的任何多核苷酸序列。在一些实施方式中,当使用合适的比对算法进行最佳比对时,引导序列与其对应的靶序列之间的互补性程度为约50%、60%、75%、80%、85%、90%、95%、97.5%、99%或更多。在一些实施方式中,互补性程度为100%。最佳比对可以通过使用用于比对序列的任何合适的算法来确定,其非限制性实例包括Smith-Waterman算法、Needleman-Wunsch算法、基于Burrows-Wheeler变换的算法(例如Burrows Wheeler比对器)、ClustalW、Clustal X、BLAT、Novoalign(NovocraftTechnologies、ELAND(Illumina,San Diego,Calif.)、SOAP(在soap.genomics.org.cn可获取)和Maq(在available at maq.sourceforge.net可获取)。在一些实施方式中,引导序列的长度为约5、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、35、40、45、50、75或更多核苷酸。在某些实施方式中,引导序列长度小于约75、50、45、40、35、30、25、20、15、12或更少的核苷酸。引导序列指导CRISPR复合物序列特异性结合靶序列的能力可以通过任何合适的分析来评估。例如,可以将足以形成CRISPR复合物的CRISPR系统的组分,包括待测试的引导序列,提供给具有相应靶序列的宿主细胞,诸如通过用编码CRISPR序列组分的载体转染,然后评估靶序列内的优先切割,诸如通过本文所述的Surveyor分析。类似地,可以通过提供靶序列、CRISPR复合物的组分(包括待测试的引导序列和不同于测试引导序列的对照引导序列),以及比较测试和对照引导序列反应之间靶序列的结合或切割速率,在试管中评估靶多核苷酸序列的切割。
可以选择引导序列来靶向任何靶序列。在一些实施方式中,靶序列是细胞基因组内的序列。示例性靶序列包括在靶基因组中独特的那些。例如,对于化脓性链球菌Cas9,基因组中的独特靶序列可以包括形式为MMMMMMMMNNNNNNNNNNNNXGG(SEQ ID NO:20)的Cas9靶位点,其中NNNNNNNNNNNNX GG(N是A、G、T或C;且X可以是任何碱基)在基因组中只有一次出现。基因组中唯一的靶序列可以包括形式为MMMMM MMMMNNNNNNNNNNNXGG的化脓性链球菌Cas9靶位点,其中NNNNNNNNNNNXGG(N是A、G、T或C;且X可以是任何碱基)在基因组中只有一次出现。对于嗜热链球菌CRISPR1Cas9,基因组中的独特靶序列可以包括形式MMMMMMMMNNNNNNNNNNNNXXAGAAW(SEQ ID NO:21)的Cas9靶位点,NNNNNNNNNNNNXXAGAAW(N是A、G、T或C;X可以是任何碱基;且W是A或T)在基因组中只有一次出现。基因组中唯一的靶序列可以包括形式为MMMMMMMMMNNNNNNNN NNNXXAGAAW的嗜热链球菌CRISPR1 Cas9靶位点,其中NNNNNNNNNNNXXAGAAW(N是A、G、T或C;X可以是任何碱基;且W是A或T)在基因组中只有一次出现。对于化脓性链球菌Cas9,基因组中的独特靶序列可以包括形式为MMMMMMMMNNNNNNNNNNNNXGGXG(SEQ ID NO:22)的Cas9靶位点,其中NNNNNNNNNNNNXGGXG(N是A、G、T或C;且X可以是任何碱基)在基因组中只有一次出现。基因组中唯一的靶序列可以包括形式为MMMMMMMMMNNNNNNNNNNNX GGXG的化脓性链球菌Cas9靶位点,其中NNNNNNNNNNNXG GXG(N是A、G、T或C;且X可以是任何碱基)在基因组中只有一次出现。在这些序列的每一个中,“M”可以是A、G、T或C,并且在将序列确定为唯一序列时不需要考虑。在一些实施方式中,选择引导序列以降低引导序列内的二级结构的程度。二级结构可以通过任何合适的多核苷酸折叠算法来确定。一些程序是基于计算最小吉布斯自由能。一种这样的算法的实例是mFold,如Zuker和Stiegler(Nucleic Acids Res.9(1981),133-148)所描述的另一个折叠算法的实例是维也纳大学理论化学研究所(Th eoretical Chemistry at the University ofVienna)开发的在线网络服务器RNAfold,它使用质心结构预测算法(参见,例如A.R.Gruber等人,2008,Cell 106(1):23-24;and PA Carr and GM Church,2009,NatureBiotechnology 27(12):1151-62)。
促进了广泛的多种细胞类型的基因组工程的CRISPR-Cas技术正在迅速发展。最近已经显示,与递送编码Cas9和gRNA的质粒相比,递送核糖核蛋白(RNP)形式的Cas9-gRNA编辑工具产生若干益处。好处包括编辑更快、效率更高、脱靶的效果更少以及毒性更小。RNP已经通过脂质体转染和电穿孔来递送,但是这些递送方法仍然存在局限性,特别是对于某些临床相关的细胞类型,该局限性包括毒性和低效率。因此,需要提供一种无载体(例如,无病毒载体)的方法,用于跨质膜递送生物相关载荷物(例如,RNP)并进入细胞。“货物”或“载荷物”是用于描述通过水溶液被跨细胞质膜递送并进入细胞内部的化合物或组合物的术语。
当前的主题涉及便于向细胞递送宽范围载荷物的低毒性递送技术。基因组编辑可以通过使用当前主题的一些方面将RNP递送到细胞来实现。Cas9和两个Alexa488标记的gRNA可单独递送至细胞,诸如人肺细胞系A549的细胞。Cas9可在递送后1小时通过蛋白质印迹在细胞中检测。此后水平下降,直到Cas9不能再被检测到。递送技术本身不会有害地影响Jurkat和原代T细胞的存活力或功能。当前的主题使得能够通过Cas9 RNP在临床相关细胞类型中以最小毒性编辑基因。
与表达载体介导的递送相比,CRISPR/Cas组分诸如Cas和/或gRNA的瞬时和直接递送具有优势。例如,与使用表达载体相比,可以在更精确的时间和有限的时间内添加一定量的Cas、gRNA或RNP。从载体表达的组分可以以各种的量和各种的时间量产生,使得在没有脱靶编辑的情况下难以实现一致的基因编辑。另外,预先形成的Cas和gRNA(RNP)复合物不能与表达载体一起递送。
在示例性实施方式中,水溶液可进一步包括添加剂。附加的非限制性添加剂,包括次优的那些,诸如甘油,可以包括甘油(例如,水溶液中的浓度为0.01至25%v/v)、NDSB(例如,水溶液中的浓度为约0.01M至约1M)、山梨醇(例如,水溶液中的浓度为0.01至10%w/v)诸如D-山梨醇、甘露醇(例如,水溶液中的浓度为0.01至10%w/v)诸如D-山梨醇或约1至400M,或约1至300M,或约1至200M,或α-晶体蛋白(例如水溶液中的浓度为1至500μM;或约1至400μM,或约1至300μM,或约1至200μM,或约1至100μM,或约1至50μM,或约1至20μM,或约1至10μM)诸如从牛眼晶状体纯化的α-晶体蛋白,明胶(例如,水溶液中的浓度为0.01至10%w/v)诸如明胶A或明胶B,甘氨酸(例如水溶液中浓度为约0.01M至约1M),脯氨酸(例如,在水溶液中浓度为约1mM至约200mM),吐温20 (例如,在水溶液中浓度为约0.01%至约5%v/v),L-组氨酸(例如,水溶液中的浓度为约1mM至约200mM),肌醇(例如,水溶液中浓度为0.01至10%w/v)和海藻糖(例如,水溶液中浓度为约1mM至约200mM)。
在优选的实施方式中,添加剂可以是α-晶体蛋白。与α-晶体蛋白氨基酸序列相关的登录号和人α-晶体蛋白的实例包括:CAA32891.1,ACP18852.1,AAB22011.1,AAB22010.1,AMM63587.1,EAX09498.1,EAW67163.1,AAI13599.1,AAP35416.1,AMM63591.1,andAAB22009.1
在一些实例中,α-晶体蛋白的氨基酸序列包括登录号CAA32891.1(EMBLL X14789mRNA.翻译:CAA32891.1),其中氨基酸序列如(SEQ ID NO:23)所示:
1mdvtiqhpwfkrtlgpfypsrlfdqffgeglfeydllpflsstispyyrqslfrtvldsg
61isevrsdrdkfvifldvkhfspedltvkvqddfveihgkhnerq
示例性区域或片段包括但不限于氨基酸1-50和60至104。
在一些实例中,α-晶体蛋白的氨基酸序列包括登录号CAA32891.1(EMBLFJ876064mRNA.翻译:ACP18852.1.),其中氨基酸序列如(SEQ ID NO:24)所示:
1mdiaihhpwihrpffpfhspsrlfdqffgehllesdlfptstslspfylrppsflrapsw61fdtglsemrlekdrfsvnldvkhfspeelkvkvlgdvievhgkheerqdehgfisrefhr121kyripadvdpltitsslssdgvltvngprkqvsgpertipitreekpavtaapkk
示例性区域或片段包括但不限于氨基酸1至51、14至161或67-50。
在一些实施方式中,市售α-晶体蛋白可以具有两种异构体,cry-aA或cry-aB。在一些实施方式中,cry-aA与cry-aB的比率可以是1:1、1:2、1:3、1:4、1:5或2:1、3:1、4:1或5:1。在晶状体中,α-晶体蛋白是由3:1比例的αA与αB亚单位组成的多分散分子。在一些实施方式中,α-晶体蛋白可以从牛晶状体中纯化,例如牛α晶(Sigma C4163)。
α-晶体蛋白A链CRYAA(牛)的氨基酸序列如(SEQ ID NO:25)所示:
MDIAIQHPWFKRTLGPFYPSRLFDQFFGEGLFEYDLLPFLSSTISPYYRQSLFRTVLDS
GISEVRSDRDKFVIFLDVKHFSPEDLTVKVQEDFVEIHGKHNERQDDHGYISREFHR
RYRLPSNVDQSALSCSLSADGMLTFSGPKIPSGVDAGHSERAIPVSREEKPSSAPSS
α-晶体蛋白B链CRYAB(牛)的氨基酸序列如(SEQ ID NO:26)所示:
MDIAIHHPWIRRPFFPFHSPSRLFDQFFGEHLLESDLFPASTSLSPFYLRPPSFLRAPSW
IDTGLSEMRLEKDRFSVNLDVKHFSPEELKVKVLGDVIEVHGKHEERQDEHGFISRE
FHRKYRIPADVDPLAITSSLSSDGVLTVNGPRKQASGPERTIPITREEKPAVTAAPKK
在一些实施方式中,人αA或B晶体蛋白可以从任何合适的供应商购买,包括细胞科学(Cell Sciences)。在一些实例中,α晶体蛋白A(编号CRC167)可以具有氨基酸序列(SEQID NO:27)
MDVTIQHPWF KRTLGPFYPS RLFDQFFGEG LFEYDLLPFL SSTISPYYRQSLFRTVLDSGISEVRSDRDK FVIFLDVKHF SPEDLTVKVQ DDFVEIHGKHNERQDDHGYI SREFHRRYRL PSNVDQSALSCSLSADGMLT FCGPKIQTGLDATHAERAIP VSREEKPTSA PSS
在其他实例中,α晶体蛋白B(编号CRC168)可以具有氨基酸序列(SEQ ID NO:28)
MDIAIHHPWI RRPFFPFHSP SRLFDQFFGE HLLESDLFPT STSLSPFYLRPPSFLRAPSWFDTGLSEMRL EKDRFSVNLD VKHFSPEELK VKVLGDVIEVHGKHEERQDE HGFISREFHR KYRIPADVDPLTITSSLSSD GVLTVNGPRKQVSGPERTIP ITREEKPAVT AAPKK
在一些实施方式中,人αA或B晶体蛋白可以从任何合适的供应商购买,也包括Abcam,其中αA(ab48778)具有氨基酸序列(SEQ ID NO:29)
MDVTIQHPWF KRTLGPFYPS RLFDQFFGEG LFEYDLLPFL SSTISPYYRQSLFRTVLDSGISEVRSDRDK FVIFLDVKHF SPEDLTVKVQ DDFVEIHGKHNERQDDHGYI SREFHRRYRL PSNVDQSALSCSLSADGMLT FCGPKIQTGLDATHAERAIP VSREEKPTSA PSS
在其他方面中,来自Abcam(ab48779)的α晶体蛋白B具有氨基酸序列(SEQ ID NO:30)
MDIAIHHPWIRRPFFPFHSP SRLFDQFFGE HLLESDLFPT STSLSPFYLRPPSFLRAPSWFDTGLSEMRL EKDRFSVNLD VKHFSPEELK VKVLGDVIEVHGKHEERQDE HGFISREFHR KYRIPADVDPLTITSSLSSD GVLTVNGPRKQVSGPERTIP ITREEKPAVT AAPKK
在一些实施方式中,MgCl
本文描述的组合物和方法的另一个优点包括基因编辑蛋白货物(例如Cas9)沉淀的最小化或减少。在某些条件下,已经观察到细菌蛋白质Cas9从溶液中沉淀出来,这种沉淀可能降低其功能或活性。为了减少、最小化或消除Cas9沉淀,递送缓冲组合物任选地包括保护蛋白质免于沉淀的赋形剂或添加剂。这种保护剂包括热休克蛋白,例如α-晶体蛋白。其它热休克蛋白保护剂包括Hsp60、Hsp72和Hsp73(Whitley等人,1999,J.of Vascular Surgery29(4):748-751)。
其它赋形剂或添加剂包括糖醇,例如山梨醇。α-晶体蛋白和/或山梨醇的加入提高了Cas-9:gRNA在暴露于醇基例如乙醇基递送溶液后的核酸酶活性,从而克服了Cas 9在某些缓冲液中沉淀/活性降低的问题和缺点。
因此,本发明包括用于向细胞递送基因编辑蛋白或基因编辑复合物的组合物(例如缓冲液),其包含至少2%(但小于70%)的醇,例如乙醇,并且任选地包含热休克蛋白,诸如α-晶体蛋白和/或糖醇,诸如山梨醇。一种在细胞中编辑靶基因的方法,包括将细胞(其中需要基因编辑)与包括基因编辑蛋白或复合物的微滴的雾的喷雾接触,将细胞温育一段时间,例如约0-10分钟、0.1-10分钟、1-5分钟,例如约2分钟,以使细胞的质膜不稳定,从而允许货物穿过质膜进入细胞,将相同的细胞与终止溶液接触一段时间,例如0-5分钟、0.1-5分钟、0.2-4分钟、0.3-3分钟、0.4-2分钟、0.5-1分钟,例如30秒,以反转细胞膜透化/去稳定化过程,并将细胞在细胞培养基(例如标准细胞培养基)中温育,在此期间发生基因编辑,例如,细胞可在培养期间保留在该溶液中。如上所述,所述方法的一个显著优点是,这一系列步骤可任选地重复几次(2、3、4、5、6、8、10或更多次),以增加递送到细胞中的货物量和/或基因编辑效率。任选地,在货物递送喷雾递送事件之间,剂量之间的恢复周期(例如,下一次递送喷雾)在1-8小时,例如2-6小时,例如,约4小时。
本文所述的方法和组合物优于其它递送方法,因为从细胞处理到适于临床使用的活的基因编辑细胞的持续时间较短。使用本文描述的乙醇喷雾递送系统递送货物的速度与电穿孔相当;然而,细胞恢复比电穿孔快。对于T细胞,本发明的方法不延迟增殖,而电穿孔延迟2天。对于MSC,用本发明的技术的细胞分化与未处理的对照相当,而电穿孔延迟细胞分化。就存活力和功能性而言,细胞在使用本文所述喷雾技术处理后4小时即可使用。
因此,处理待随后使用(例如递送给人或动物受试者进行临床治疗)的细胞的速度,与需要用于使处理的细胞恢复正常健康/功能的长时间段的程序相比,具有优势。例如,电穿孔递送损害了一些细胞诸如MSC和T细胞的功能。电穿孔的这一缺点可能是由于大的电梯度(特别是在小细胞中)和局部加热,这触发DNA损伤和内源性修复机制(Meaking等人,1995,Biochimica et Biophysics Acta 1264:357-362,和Branzei等人,2008,NatureReviews Molecular Cell Biology 9:297-308)。例如,在电穿孔后,一些细胞的功能完整性受损,例如分化降低、活性降低,例如电穿孔后在长达数周内功能完整性降低。相反,使用该方法和组合物处理的细胞的存活力和功能受到最小程度损害或没有受到损害,并且细胞在数小时(处理后1、2、6、12、18、24小时)或数天(处理后1、2、3、4、5、6、7天)内即可使用。因此,本文所述的基因编辑技术的总体关键优点是避免了由电梯度引起的膜破坏。此外,电穿孔参数可以根据细胞类型和实验条件而宽范围变化。相比之下,本发明的方法和系统在宽范围的细胞类型上工作良好,该方法可以在几分钟内完成,并且在细胞友好的条件下进行。使用本文所述的喷雾递送方法和组合物处理的细胞的存活力和功能受到最小程度损害或没有受到损害,并且细胞在数小时(处理后1、2、6、12、18、24小时)或数天(处理后1、2、3、4、5、6、7天)内即可使用。
本文公开的每个实施方式被认为可应用于其他公开的实施方式中的每一个。在一些方面,水溶液不包括氯化镁和/或甘油。因此,本文描述的各种元素的所有组合都在本发明的范围内。
本文描述的主题的一个或多个变型的细节在附图和下面的描述中阐述。从说明书和附图以及从权利要求书中,本文描述的主题的其它特征和优点将是显而易见的。本文引用的所有参考文献,包括蛋白质或核酸序列的登录号引用的内容,都通过引用并入本文。
附图说明
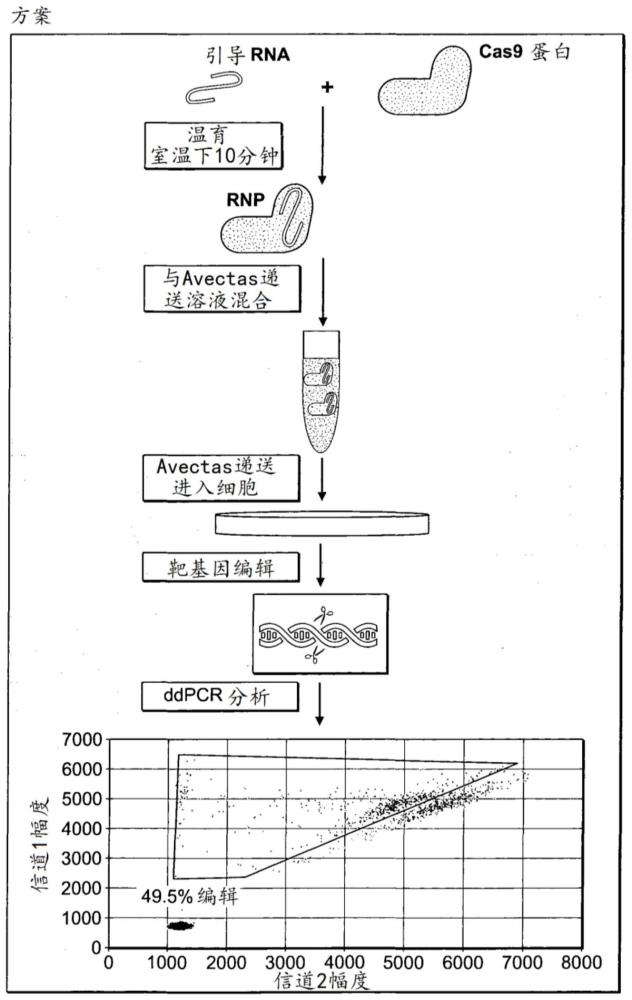
图1是说明编辑A549细胞中靶基因的方案的草图。引导RNA以sgRNA的形式递送。双RNA是化学合成的。sgRNA1在体外转录。引导RNA与Cas9一起温育。使用递送平台将Cas9:gRNA复合物递送到A549细胞中。在用Cas9切割和非同源末端连接(NHEJ)之后,从细胞中收获DNA,并使用微滴式数字PCR(ddPCR)和脱落探针分析(drop-off probe assay)检测编辑。
图2是显示Cas9蛋白在体外编辑分析中功能的电泳凝胶的图像。从Toolgen购买的重组Cas9的功能在体外编辑分析中得到证实。对于阳性对照,安捷伦试剂盒中提供了DNA、gRNA和Cas9,并使用这些试剂产生了预测的DNA片段1和2。来源于Toolgen(韩国首尔Toolgen有限公司)的Cas9也使用对照DNA和gRNA成功地产生了DNA片段1和2。
图3是显示递送溶液中Cas9蛋白在体外编辑分析中功能的电泳凝胶的图像。安捷伦体外分析是在带有安捷伦试剂盒的缓冲液中进行的,或在递送溶液w/o乙醇(S缓冲液)中进行。预测的DNA片段1和2是使用安捷伦缓冲液产生的,而不是使用S缓冲液产生的。
图4是显示在递送溶液与试剂盒缓冲液的各种比率的情况下Cas9蛋白在体外编辑分析中功能的电泳凝胶的图像。使用安捷伦体外分析测试了“递送溶液w/o乙醇(S缓冲液)”:“安捷伦试剂盒缓冲液”的各种比率。比率见表2。预测的DNA片段1和2是在所有比率组合存在下产生的,除非安捷伦试剂盒缓冲液不存在(泳道6)。
图5是电泳凝胶的图像,显示了在体外编辑分析中递送溶液中的MgCl
图6是本文所述方法的图示,其中描述了递送机构。
图7是说明使用本文所述的方法和递送平台在NK细胞中GFP mRNA表达百分比的柱状图。
图8是验证含有15% EtOH和2.5mMMgCl
图9是一组蛋白质印迹,说明与脂质体转染的质粒DNA相比,Cas9蛋白的递送导致快速和瞬时的蛋白表达。Cas9蛋白被递送到A549细胞,并通过细胞裂解物的蛋白质印迹检测。当使用递送平台时,在递送后1小时检测到Cas9,此后水平降低。使用GAPDH作为加载对照。相反,当编码Cas9的质粒通过脂质体转染递送到A549细胞时,表达在6小时内不明显。
图10A和B是说明Cas9蛋白-剂量递送响应的蛋白质印迹。当递送增加剂量的Cas9蛋白时,在(图10A)A549细胞和(图10B)Jurkat细胞中检测到Cas9水平的增加。
图11A-E是说明Cas9蛋白在递送到A549细胞后的亚细胞定位的图像。(图11A)将Cas9-Cy3蛋白(红色)递送至细胞,并在递送后1小时通过荧光显微镜分析。DAPI核复染(蓝色)显示细胞核。向递送溶液中添加MgCl
图12A-C是显示递送平台技术对Jurkat细胞(图12A)、人外周血单核细胞(PBMC)(图12B)和HSC(图12C)的存活力的影响的一组图。将无载荷物的递送溶液(SBO)递送到Jurkat细胞、人PBMC或HSC。膜联蛋白V和碘化丙锭分析显示了每种细胞类型的活性的最小的降低。UT=未处理。
图13是比较Neon电穿孔仪和递送平台的存活力和效率的一组图。(图13A)比较使用递送平台相比于Neon递送后Jurkat细胞与的存活力。(图13B)使用递送平台相比于Neon电穿孔仪将BSA-FITC递送到Jurkat细胞的效率。
图14A和B是显示递送平台技术对Jurkat细胞活化的影响的一组图。没有载荷物的递送溶液(SBO)被递送到Jurkat细胞。对照细胞未经处理(UT)。分别在PHA刺激后4hr或24hr分析CD69(图14A)和CD25(图14B)的表达。
图15是示例性Cas9蛋白(SEQ ID NO:4)的氨基酸序列,其从N末端到C末端包括化脓性链球菌蛋白(SEQ ID NO:1),接着是核定位信号(SEQ ID NO:2),接着是人流感血凝素(HA)表位标签(SEQ ID NO:3)。核定位信号和HA表位标签用下划线标出。
图16是显示Cas9蛋白递送到PBMC和Beas-2B细胞的一组蛋白质印迹。Cas9蛋白被递送到PBMC和Beas-2B细胞,并通过细胞裂解物的蛋白质印迹检测。
图17是说明递送溶液对基因编辑工具功能的影响的电泳琼脂糖凝胶的图像。泳道1为在无核糖核酸酶水中的Cas9 600ng/μl(阳性对照);泳道2为在喷雾无核糖核酸酶水的Cas9 600ng/μl,泳道3位在递送溶液中的Cas9 600ng/μl;泳道4为喷雾的递送溶液中Cas9600ng/μl;泳道5位递送溶液中的复合物Cas9/gRNA;泳道6为喷雾的递送溶液中的复合物Cas9/gRNA;泳道7为加入EtOH后立即产生的递送溶液中的复合物Cas9/gRNA;泳道8为包含多于4倍S缓冲液的递送溶液中的Cas9;泳道9为在递送溶液中的标记的Alexa 488Cas9;泳道10为阴性对照(递送溶液中的Cas9,未添加gRNA);不切割DNA的阴性对照。在除样品7之外的所有样品中观察到编辑,其中EtOH与所有其它组分一起加入(没有预先10分钟温育)。对于所有其他样本,片段化DNA的条带出现在未切割的DNA附近。结果表明,无论是递送溶液还是喷雾都没有阻碍Cas9和RNP的切割活性。琼脂糖凝胶是包含CyberSafe染料的1.0%凝胶,在100伏下用凝胶加载染料跑胶60分钟。
图18是表示Cas9纯化的图像。较大的标记Cas9与未结合的标记物分离,并首先从柱中洗脱。
图19是说明葡聚糖凝胶(Sephadex)柱上分离后标记的Cas9洗脱物的图像。在透照器上观察四个等分试样。第二洗脱物的强度(从左)表示最浓,游离染料(最右)用作参考。
图20是表示通过递送平台技术(绿点)递送到A549细胞的Alexa 488标记的Ca9的摄取的图像。细胞核(蓝色)用DAPI染色,以便于可视化细胞。
图21是在光学显微镜下观察的显微载玻片上(2μl)描绘递送溶液中未标记的Cas9500ng/μl(0.5μl Cas9 10μg/μl、2.5μl S缓冲液、2.5μl乙醇、MGW至10μl)的图像。图片中可见的团簇(箭头)表示加入25%乙醇后立即从溶液中沉降的Cas9蛋白的聚集体。
图22是描述递送溶液中乙醇浓度对Cas9沉淀的影响的图像。前三幅图像显示了放大倍数为10倍的递送溶液(25% S缓冲液、25%乙醇、水至10μl)中的标记Cas9 93ng/μl,后三幅图像显示的放大倍数为20倍。在荧光显微镜下观察载玻片上的2μl样品。1%、5%和10%未观察到聚集和沉淀。在15%乙醇时,形成并沉淀小聚集体(箭头),在20%和25%乙醇时,形成并从溶液中分离蛋白质大团簇(箭头)。在乙醇加入到包含Cas9的溶液和递送溶液的除乙醇之外的所有组分后10秒记录图像。
图23是描述递送溶液中乙醇浓度对Cas9沉淀的影响的图像。描绘了放大20倍的递送溶液(25% S缓冲液、25%乙醇、水至10μl)中的标记Cas9 93ng/μl。12%乙醇是引发蛋白质沉淀的最低浓度。
图24是描绘从柱洗脱的没有乙醇情况下的Cas9和蛋白质的图像。描述了洗脱的Cas9和S缓冲液中的Cas9(放大40倍),以显示在加入乙醇之前没有显著聚集或沉淀,并显示S缓冲液对蛋白质溶解度没有不利影响。
图25是电泳琼脂糖凝胶的图像,说明在包含不同量乙醇的递送溶液中制成的RNP复合物的Cas9浓度。样品与表9中报告的那些相对应。样品10代表阴性对照,其中不存在RNP工具,因此DNA靶是完整的。在所有其他样品中,编辑由代表由RNP工具切割的靶DNA的两个片段的两条前带的存在来确认。琼脂糖凝胶是在100伏下运行的1.5%凝胶。
图26A-26C是描述NDSB-201对Cas9沉淀的影响的图像。图26A是在光学显微镜下观察的显微载玻片上(2μl)描绘递送溶液中未标记的Cas9 500ng/μl(0.5μl Cas9 10μg/μl、2.5μl S缓冲液、2.5μl乙醇、MGW至10μl)的图像。图片中可见的团簇表示加入25%乙醇后立即从溶液中沉降的Cas9蛋白的聚集体;图26B是描绘在包含0.5M NDSB的递送溶液中的RNP(3:1gRNA/Cas9)的图像,沉淀现象显著减少;图26C是描绘包含20%甘油的递送溶液中的RNP(3:1gRNA/Cas9)的图像,未观察到可见沉淀。
图27是描绘Cas9:gRNA对HPRT DNA的编辑效率的凝胶(编辑效率=带2/(带1+带2))。编辑效率归一化为0%乙醇样品。每个条件重复两次。样品如下,泳道1:0%乙醇,泳道2:25%乙醇,泳道3:山梨醇(4%w/v),泳道4:D-甘露醇(2%w/v),泳道5:α-晶体蛋白(100μM),泳道6:α-晶体蛋白(220μM),泳道7:NDSB(0.2M),泳道8:海藻糖(26.4mM),泳道9:海藻糖(26.4mM)/α-晶体蛋白(100μM),泳道10:用TF-乙醇(0.7%)代替乙醇,泳道11:未切割HPRTDNA。
图28是描绘Cas9:gRNA对HPRT DNA的编辑效率的凝胶(编辑效率=带2/(带1+带2))。编辑效率归一化为0%乙醇样品。每个条件重复两次。样品如下,泳道1:1:0%乙醇,泳道2:25%乙醇,泳道3:D-山梨醇(4%w/v)(在S缓冲液中),泳道4:D-山梨醇,泳道5:α-晶体蛋白(220μM),泳道6:α-晶体蛋白(220μM)/D-山梨醇(4%w/v)(在S缓冲液中),泳道7:明胶A(1%w/v),泳道8:明胶B(1%w/v),泳道9:甘氨酸(0.25M),泳道10:脯氨酸(75mM),泳道11:L-精氨酸(50mM),泳道12:组氨酸(12.8mM),泳道13:肌醇)(1%w/v),泳道14:吐温20(0.1%v/v),泳道15:未切割HPRT DNA。
图29A和29B是表示Cas9:gRNA对HPRT DNA的编辑效率的柱状图(编辑效率=带2/(带1+带2))。编辑效率归一化为0%乙醇样品。每个条件重复两次。图29A描绘了指示添加剂的效果的柱状图,所述添加剂包括0% EtOH、25% EtOH、D-山梨醇、D-甘露醇、α-晶体蛋白(100μM)、α-晶体蛋白(221μM)、NDSB、海藻糖、α-晶体蛋白/海藻糖、7%TF-EtOH和阴性对照。图29B描绘了指示添加剂的效果的柱状图,所述添加剂包括0%EtOH、25% EtOH、D-山梨醇(缓冲液)、D-山梨醇(添加剂)、α-晶体蛋白、α-晶体蛋白/山梨醇(缓冲液)、明胶A、明胶B、甘氨酸、脯氨酸、L-精氨酸、L-组氨酸、肌醇、吐温20和阴性对照。
图30是描述具有和不具有添加剂的MSC细胞的CRISPR诱导的hprt编辑的百分比的柱状图。
图31是说明具有和不具有添加剂的U2OS细胞的CRISPR诱导的hprt编辑的百分比的柱状图(2个命中)。
图32是说明较小孔(24孔板、48孔板和96孔板)中编辑效率的柱状图。较小孔中接种的细胞中编辑效率提高。
图33是描述增加Cas9浓度对编辑效率的影响的图。增加Cas9的数量增加了MSC中编辑的百分比。
图34是描述Cas9:gRNA比率对编辑效率的影响的柱状图。改变Cas9与引导RNA的比率不影响编辑效率。
图35是描述RNP递送剂量对编辑效率的影响的柱状图。增加剂量的数量增加了MSC中编辑的百分比。
图36是描述MgCl
图37是描述递送平台技术和电穿孔之间编辑效率的比较的柱状图。与电穿孔相比,当通过递送平台技术递送Cas9 RNP时,MSC中的编辑效率更高。
图38是描述RNP递送后细胞存活力的比较的柱状图:递送平台技术与电穿孔。在通过递送平台技术或电穿孔处理的细胞中观察到相当的存活力。
图39是描述RNP递送后细胞功能的比较的图像:递送平台技术与电穿孔。MSC的功能不受递送平台技术的影响。显示了来自未处理组(未处理)的分化的MSC、由递送平台技术组(递送平台)递送的Cas9 RNP和由电穿孔组(电穿孔)递送的Cas9 RNP的三个代表性图像。未处理的细胞和递送平台处理的细胞之间没有差别。然而,电穿孔处理的细胞中分化受到抑制。
图40是描绘粘附细胞系和悬浮细胞系以及原代细胞中编辑效率的柱状图。粘附(MSC,U2OS)和悬浮(T细胞,Jurkat)、细胞系(U2OS,Jurkat)和原代细胞(MSC,T细胞)的编辑效率数据。在细胞系和原代人细胞中观察到成功的编辑。
图41是描述使用Cas9 mRNA的编辑效率的柱状图。由递送平台技术递送Cas9 mRNA和引导RNA诱导了MSC中的编辑
各种图中相同的附图标记表示相同的元素。
具体实施方式
迄今为止,CRISPR-Cas9系统最常见是以质粒DNA的形式递送到细胞中,质粒DNA编码这三种组成,或者是单独的或者是组合的质粒。还使用了Cas9的mRNA编码(Liang等人,(2015)J Biotechnol 20;208:44-53)。Cas9蛋白已被用作Cas9编码质粒或mRNA的替代物。有趣的是,最近据报道Cas9核糖核蛋白(包括Cas9的RNP,其中纯化的Cas9蛋白在递送到细胞中之前与gRNA复合)提供了优于质粒和mRNA方法的多种益处,例如,如以下文献中描述的:Cho SW,Lee J,Carroll D,Kim JS,Lee J.Heritable gene knockout inCaenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins.Genetics.2013.195(3):1177-80;Sung YH,Kim JM,Kim HT,Lee J,Jeon J,Jin Y,ChoiJH,Ban YH,Ha SJ,Kim CH,Lee HW,Kim JS.Highly efficient gene knockout in miceand zebrafish with RNA-guided endonucleases.Genome Res.2014.24(1):125-131;KimS,Kim D,Cho SW,Kim J,Kim JS.Highly efficient RNA-guided genome editing inhuman cells via delivery of purified Cas9ribonucleoproteins.GenomeRes.2014.24(6):1012-9(下文中为“Kim等人”);Lin S,Staahl BT,Alla RK,DoudnaJA.Enhanced homology-directed human genome engineering by controlled timingof CRISPR/Cas9 delivery.eLife.2014.15;3:e04766:Zuris JA,Thompson DB,Shu Y,Guilinger JP,Bessen JL,Hu JH,Maeder ML,Joung JK,Chen ZY,Liu DR.Cationiclipid-mediated delivery of proteins enables efficient protein-based genomeediting in vitro and in vivo.Nat Biotechnol.2015.33(1):73-80:Liang X,PotterJ,Kumar S,Zou Y,Quintanilla R,Sridharan M,Carte J,Chen W,Roark N,RanganathanS,Ravinder N,Chesnut JD.Rapid and highly efficient mammalian cell engineeringvia Cas9 protein transfection.J Biotechnol.2015.20;208:44-53(下文中为“Lin等人”);以及Schumann K,Lin S,Boyer E,Simeonov DR,Subramaniam M,Gate RE,Haliburton GE,Ye CJ,Bluestone JA,Doudna JA,Marson A.Generation of knock-inprimary human T cells using Cas9ribonucleoproteins.Proc Natl Acad Sci U SA.2015.18;112(33):10437-42,其每一篇的全部内容通过引用并入本文。
编辑核酸酶的基因,包括ZFN,其已经在Bhakta等人,Genome Research 23:530-538;2013,and Beerli,R.et al.,Proc.Natl.Acad.Sci v.95pp 14628-14633;1998中描述,TAL,其已经在Cermak,T.等人,Nucleic Acids Research 2011,v.39,no.12,Miller,J.等人,Nature Biotechnology vol.29no.2;2011,Christian,M.等人,Genetics 186:757-761;2010,Deng,D.等人,Science 2012:v.335p.720,以及Boch,J.等人,Science2009:v.326p.1509中描述,其每一篇的全部内容通过引用并入本文。另外,Cre已经在Chevalier,B.等人,Nucleic Acids Research 2001,v.29no.18中描述,其全部内容通过引用并入本文。MegaTal已经在Sather,B.等人Sci Transl Med 7(307)2015,Ibarra,G.等人,Molecular Therapy-Nucleic Acids(2016)5,e352,Osborn,M.等人,MolecularTherapyv.24no.3,570-581(2016);Wang,Y.等人,Nucleic Acid Research 2014;v.42,6463-6475;以及Gaj,T.等人,Cold Spring Harbor Perspectives in Biology 2015中描述,其每一篇通过引用并入本文。Cas9已经在Wiedenheft,Bl.等人,Nature 2012;v.482;331,Fineran,P.等人,Virology 2012;433;202-209,以及Hsu,P.等人,Cell 2014;v.157;1262-1278中描述,其每一篇的全部内容通过引用并入本文。
质粒和mRNA在某些细胞类型中可能会沉默,而其它细胞类型可能很难实现DNA转染。RNP已在多项研究中显示,与质粒转染相比,RNP产生既更快又更有效的编辑。这可能是由于直接引入了完全形成的复合物,该复合物可以在细胞中立即起作用,并且不需要像质粒一样转录、翻译和装配。据报道,RNP的毒性也低于质粒DNA。这可能是因为外源DNA本身对一些细胞有毒,或者因为RNP的半衰期明显短于质粒的半衰期,质粒产生这些外源mRNA和蛋白质的长期表达。重要的是,与质粒相比,RNP的较短表达时间也与较少的脱靶编辑相关,这可能是因为Cas9蛋白的活性持续时间缩短。质粒也可以随机整合到基因组或Cas9产生的位点。虽然后一个事件至少可以预测和监控,但前一个事件可能难以检测,因此可能更成问题。不管整合的位点如何,这些外源序列都会引起宿主免疫反应,这给基因编辑干细胞或原代细胞在临床应用中的使用带来了挑战。此外,用于临床应用的质粒转染的细胞被监管当局认为是遗传修饰的,因此受到冗长且昂贵的监管程序。
本主题提供了跨质膜的载荷物的无载体(例如,无病毒载体)递送。在一些实施方式中,递送不涉及表达载体。特别地,已经发现,通过使细胞(和/或细胞群)与包括醇和递送材料(例如载荷物)的水溶液接触,可以实现材料的细胞内递送。醇起到渗透膜的作用,以允许载荷物跨膜转位。但是当接触细胞的水溶液的体积太大和/或暴露时间太长时,可能发生对细胞的永久或严重(例如不可逆)损伤(不利地影响细胞存活力)。相反,当接触细胞的水溶液体积太小和/或暴露时间太短时,不能实现材料的细胞内递送。因此,为了在维持细胞存活力的同时实现载荷物跨质膜的递送,可以施加适当体积的水溶液和/或可以控制暴露的时长。
与细胞群接触的水溶液的适当体积可以基于预期的应用而变化,例如基于(例如,随以下变化)群中细胞的数量、暴露的细胞表面积、细胞大小、水溶液的组成、载荷物、水溶液与细胞群接触的技术等。
递送平台用于将Cas9 RNP递送到细胞,从而实现基因组编辑。
为了确定是否可以通过使用递送平台技术递送Cas9 RNP来实现基因组编辑,设计了从人肺细胞系A549中的目标基因中删除区域的策略(图1)。Cas9介导的切割之后是非同源末端连接(NHEJ)。设计位于靶区侧翼的PCR引物和用作参比内部探针和脱落探针。如果编辑成功,ddPCR分析将导致微滴分离,描述在编辑的基因中,脱落探针无法结合,而只有参考探针结合的位置。
两种引导RNA方法采用两部分引导RNA和单个引导RNA。对于两部分引导RNA方法,设计了42bp CRISPR RNA(crRNA)分子来靶向靶基因中的选定区域。还设计了一种69bp的转录激活crRNA(tracrRNA或trRNA)。crRNA和tracrRNA分子是化学合成的,tracrRNA分子用FAM标记。在实验中,tracrRNA与crRNA一起温育,形成两部分引导RNA分子。对于单个引导RNA方法,从DNA模板体外转录对应于靶序列的sgRNA。
Cas9 RNP是通过将Cas9蛋白与两部分引导RNA或单个引导RNA温育而形成的。
一般定义和一般技术
除非另有特别限定,本文所用的所有技术和科学术语应被认为具有与本领域普通技术人员通常理解的相同含义(例如,在细胞培养、分子遗传学和生物化学中)。
在以上说明书和权利要求中,可以出现诸如“至少一个”或“一个或多个”的短语,其后面连接元素或特征的列表。术语“和/或”也可以出现在两个或多个元素或特征的列表中。除非与使用该短语的上下文隐含或明确矛盾,否则该短语旨在表示单独列出的任何元素或特征,或与任何其他列举的元素或特征组合的任何列举的元素或特征。例如,短语“A和B中的至少一个;”“A和B中的一个或几个;”以及“A和/或“B”各自意在表示“A单独、B单独或A和B一起。”类似的解释也适用于包括三个或更多项目的列表。例如短语“A、B和C中的至少一个;”“A、B和C中的一个或几个;”以及“A、B和/或C”各自意在表示“A单独、B单独、C单独、A和B一起、A和C一起、B和C一起、或者A和B和C一起。”此外,上文和权利要求中使用的术语“基于”意在表示“至少部分基于”,使得未列举的特征或元素也是可允许的。
如本文所用,在数值或范围的上下文中,术语“约”是指所列举或要求保护的数值或范围的±10%,除非上下文需要更有限的范围。
应当理解,在提供参数范围的情况下,该范围内的所有整数及其十分之一也由本发明提供。例如,“0.2-5mg”是0.2mg、0.3mg、0.4mg、0.5mg、0.6mg等,最高至且包括5.0mg的公开。
引导RNA(gRNA)可以以两种形式使用:crRNA和tracrRNA可以以双链RNA(或“双RNA)的形式使用,或者可以被设计成单引导RNA(sgRNA)分子。本文所用的“gRNA”是指CRISPR-Cas系统引导RNA。术语“gRNA”可以指crRNA与tracrRNA(双RNA)或包括crRNA和tracrRNA序列(sgRNA)二者的单个RNA分子的组合。
如本文所用,复合物是指通过非共价相互作用的分子缔合。例如,复合物可以包括彼此流体静力缔合的两种或更多分子,例如通过氢键。复合物的非限制性实例是包括Cas蛋白和gRNA的RNP。
如本文所用,“基因编辑蛋白”是切割DNA分子的糖-磷酸骨架的蛋白或以足够亲和力结合靶DNA以降低基因表达的蛋白质。以足够亲和力结合靶DNA以降低基因表达的蛋白质的非限制性实例是核酸酶失活Cas9(dCas 9),即不能切割DNA的Cas9。dCas 9的实例是具有SEQ ID NO:1所示氨基酸序列的Cas 9蛋白的D10A和H840A双突变体。
如本文所用,“表达载体”是能够实现一种或多种多核苷酸表达的DNA或RNA载体。优选地,表达载体也能够在宿主细胞内复制。表达载体可以是原核的或真核的,并且通常是质粒。本发明的表达载体包括在本发明宿主细胞中起作用(即直接基因表达)的任何载体,包括在本文描述的原核或真核细胞之一,例如革兰氏阳性、革兰氏阴性、致病、非致病、共生、球菌、芽孢杆菌或螺旋形细菌细胞;古细菌细胞;或原生动物、藻类、真菌、酵母、植物、动物、脊椎动物、无脊椎动物、节肢动物、哺乳动物、啮齿动物、灵长类或人类细胞中起作用的那些。本发明的表达载体包含调节序列,诸如转录控制序列、翻译控制序列、复制起点和与宿主细胞相容和控制多核苷酸表达的其它调节序列。特别地,本发明的表达载体包括转录控制序列。转录控制序列是控制转录起始、延伸和终止的序列。特别重要的转录控制序列是控制转录起始的那些,例如启动子、增强子、操纵子和阻抑子序列。合适的转录控制序列包括可在本发明的至少一种细胞中起作用的任何转录控制序列。各种这样的转录控制序列是本领域技术人员已知的在优选实施方式中,方法不包括使用病毒载体诸如腺病毒递送核酸分子或构建体。
可以纯化和/或分离载荷物组合物,诸如多核苷酸、多肽、复合物或其它剂。具体而言,如本文所用,“分离的”或“纯化的”化合物、核酸分子、多核苷酸、多肽或蛋白质,或包括例如蛋白质和多核苷酸的复合物,在其天然存在的情况下,基本上不含其它细胞材料,或当通过重组技术产生时,基本上不含培养基,或当化学合成时,基本上不含化学前体或其它化学物质。纯化的化合物是至少60重量(干重)%的目标化合物。优选地,制剂是至少75重量%,更优选至少90重量%,最优选至少99重量%的目标化合物。例如,纯化化合物是至少90%、91%、92%、93%、94%、95%、98%、99%或100%(w/w)所需化合物重量的化合物。纯度通过任何合适的标准方法测量,例如通过柱色谱、薄层色谱或高效液相色谱(HPLC)分析。纯化或分离的多核苷酸(核糖核酸(RNA)或脱氧核糖核酸(DNA))不含在其天然存在状态下侧翼的基因或序列。分离或纯化的核酸分子的实例包括:(a)DNA,其是天然存在的基因组DNA分子的一部分,但其没有侧接两侧的核酸序列,该核酸序列位于在天然存在的生物体的基因组中该分子的那部分侧翼;(b)以一种方式掺入载体或原核生物或真核生物基因组DNA中的核酸,使得所得分子与任何天然存在的载体或基因组DNA不相同;(c)单独的分子,诸如cDNA、基因组片段、通过聚合酶链式反应(PCR)产生的片段或限制性片段;以及(d)作为杂交基因的一部分的重组核苷酸序列,即编码融合蛋白的基因。根据本发明的分离的核酸分子还包括合成产生的分子,以及化学改变的和/或具有修饰的骨架的任何核酸。
A549细胞(A549欧洲细胞培养物保藏中心(ECACC)细胞通过Sigma-Aldrich公司(分类编号86012804)(St.Louis,MO,美国)和Beas-2B(Sigma-Aldrich公司,分类编号95102433)购买,并在补充了5%胎牛血清和2mM L-谷氨酰胺(Gibco)的DMEM(Gibco)中常规培养。Jurkat细胞(欧洲细胞培养物保藏中心;ECACC)在37℃、5%CO
递送系统包括递送设备、装置或仪器和渗透递送溶液(图6描绘递送机构的图示)。递送装置包括保持在板定位器平台上的可调节x-y-z轴线上的雾化器。雾化器通过管道与5巴压缩机相连。定位环允许喷雾在孔上方居中,板定位器平台允许雾化器下方的可重复的孔定位。载荷物被吸移至位于雾化器顶部的递送口,并使用喷雾致动器按钮产生喷雾。优选的递送溶液包括在分子级水中(Sigma-Aldrich)的32mM蔗糖、12mM氯化钾、12mM乙酸铵、5mMHEPES和25%乙醇),以及任选的2.5mM MgCl
为了递送到粘附细胞,将细胞接种在48孔或96孔培养板(Nunc)(在其它实例中,可以使用具有选自1、6、9、12、24、48、96、384和1536的样品孔的培养板)中,密度达到在递送时70-95%的汇合度。将上清液从目标孔中移出,将板放置在板定位器平台上,并使用x-y-z轴线将雾化器定位到从喷头向细胞单层的31mm距离处。将10μl递送溶液喷雾到细胞上。室温下2分钟后,使用微量吸管将100μl 0.5×-PBS加到细胞上,并在室温下温育细胞30秒(对于48孔培养板);如果使用96孔板,则将50μl 0.5×-PBS添加到细胞上。取出0.5×-PBS溶液,加入100μl新鲜培养基。3小时后,每孔加入100μg/ml青霉素和100μg/ml链霉素。在双喷雾的情况下,在从第一次喷雾处理温育1-5小时(例如,2小时或优选4小时)后重复该程序。然后在加入抗生素前温育细胞3小时。
为了递送至悬浮细胞,将1.0×10
sgRNA的体外转录按照制造商的指南,使用安捷伦(Santa Clara,CA)Sure Guide体外转录试剂盒进行。
重组Cas9蛋白购自ToolGen公司和IDT-DNA。引导RNA是根据制造商的指南使用SureGuide IVT试剂盒(安捷伦)生成的。pCas9-GFP-382a2-384质粒用作分析中的阳性对照。该质粒包含CAS9-GFP和两个gRNA。将Cas9蛋白(1ug-20ug)与体外转录的gRNA(1ug-20ug)预混合,并将复合物在室温下温育10分钟。将RNP复合物添加到递送溶液(1×S缓冲液25% EtOH)中,并使用递送平台递送到细胞中。对于质粒介导的RGEN表达,使用Lipofectamine 2000用pCas9-GFP-382a2-384转染1×10
将细胞刮在冰冷的1×PBS溶液中,以2200rpm离心5min。将细胞团在放射免疫沉淀(RIPA)缓冲液(Sigma,Dublin)中裂解,并将25mg蛋白质加载到4-20%聚丙烯酰胺凝胶上。凝胶电泳后,将蛋白质转移到硝酸纤维素膜上,并在5%牛奶/1×TBS 0.1%吐温20(Sigma,Dublin)中封闭。用一级抗体在4℃下探测膜过夜,然后在室温下施加与辣根过氧化酶偶联的二级抗体1小时。使用ECL检测系统(Gbox,Syngene,Cambridge UK)进行凝胶可视化。使用的一级抗体包括抗Cas9(Abcam,Cambridge UK)、GAPDH(Sigma,Dublin)。
使用1/200稀释度的Millipore抗Cas9抗体进行免疫荧光。
用于PCR的反应混合物是:H2O 30.5μl,缓冲液5×10μl,MgCl
使用35个循环:
95℃:120秒(s)
------------
95℃:30s
55℃:30s ×35
72℃:45s
-----------
72℃:5分钟
根据制造商的方案,用膜联蛋白V和碘化丙啶(PI)凋亡检测试剂盒(eBioscience)分析Jurkat细胞的活性和凋亡标记物的存在。细胞在1×结合缓冲液中洗涤一次,再以1-5×10
在24孔板中用5μg/ml PHA刺激Jurkat细胞4小时和24小时。通过流式细胞术分析活化标记物CD25和CD69的表面标记物表达。收集细胞,用PBS洗涤两次,并以1×10
在体外编辑分析中,递送溶液组成对Cas9 RNP活性的影响
首先在体外试验中分析递送溶液组成对Cas9蛋白活性的影响。Cas9蛋白(来自Toolgen;Seoul,South Korea)与安捷伦试剂盒中提供的供应gRNA和对照质粒以及试剂盒缓冲液(溶液A,表1)一起温育。观察到切割产物(图2)。
接下来确定用递送溶液(不含乙醇)替换试剂盒缓冲液的效果(溶液B,表1)。未观察到切割产物(图3)。
进行实验以确定递送溶液的效果。使用递送溶液:试剂盒缓冲液的各种不同比例。发现切割发生在特定比例(最高达1.5μl S缓冲液:0.5μl试剂盒缓冲液)的递送溶液中(表2和图4)。因此,考察了向递送溶液/反应缓冲液中加入MgCl
考察了包含15%乙醇并补充2.5mM MgCl
因此,对于随后的研究,Cas9 RNP是在包含15%乙醇和2.5mM MgCl
Cas9通常以编码Cas9蛋白的DNA形式递送。然而,DNA可以在细胞中持续数天至数月,这可能导致酶在细胞中非特异性位点切割的脱靶效应。已经提出递送Cas9蛋白形式将导致酶表达持续时间的缩短,并因此导致离靶效应降低。是否可以使用递送平台将纯化的不含gRNA的重组Cas9蛋白递送到A549细胞中。还考察了与质粒介导的表达相比的表达持续时间。将Cas9蛋白(5μg)递送至A549细胞,并通过细胞裂解物的蛋白质印迹分析考察其表达。在递送后1小时在细胞中检测到Cas9蛋白,此后水平下降(图9)。相反,当编码Cas9的质粒被脂质体转染到细胞中时,在6hr之前没有观察到表达,并且在24小时观察到强水平的表达(图9)。
Cas9蛋白也成功地递送至BEAS-2B细胞、Jurkat细胞、人PBMC和HSC(表3)。
在CRISPR/Cas9病毒递送的情况下,很难控制递送的编辑工具的剂量。因此,考察了递送平台技术是否能够递送递增剂量的Cas9蛋白。
当增加浓度的Cas9蛋白被递送到A549和Jurkat细胞时,看到剂量响应(图10A和B)。
由于Cas9 RNP在细胞核中起作用,因此确定了Cas9蛋白和Cas9 RNP在用递送平台递送后的亚细胞定位。用Cy3标记的Cas9和FAM标记的双RNA通过荧光显微镜检查Cas9和RNP的定位。此外,通过使用抗Cas9抗体的免疫荧光检测Cas9。
使用递送平台将1ug Cas9-Cy3蛋白递送到A549细胞中。递送后1小时,在细胞的整个细胞质和细胞核中观察到Cas9(图11A)。
使用递送平台将10ug未标记的Cas9蛋白递送到A549细胞中,并使用抗Cas9抗体通过免疫荧光检测Cas9。递送后1小时,在细胞的整个细胞质和细胞核中观察到Cas9(图11B)。染色图案与Cas9-Cy3观察到的相似。
使用递送平台将100μM sgRNA1-FAM递送到A549细胞中。递送后1小时,在细胞的整个细胞质和细胞核中观察到sgRNA1-FAM(图11C)。
接下来,在递送到A549细胞后检查RNP的定位。未标记的Cas9与sgRNA1-FAM复合,并使用递送平台递送。递送后1小时,在细胞的整个细胞质和细胞核中观察到sgRNA1-FAM(图11D)。染色图案比图11C中所示的sgRNA1-FAM出现更多的“斑点”,表明sgRNA确实与Cas9复合。
然后使用抗Cas9抗体通过免疫荧光检查RNP的定位。未标记的Cas9与sgRNA1-FAM复合,并使用递送平台递送。递送后1小时,在细胞的整个细胞质和细胞核中观察到Cas9(图11E)。如预期的,染色图案类似于图11D所示。
对于临床应用来说,细胞在递送和转染方案后保持高存活力和功能性是至关重要的。考察了递送平台技术对Jurkat细胞、PBMC和HSC活性的影响。为了分析平台介导递送的效果,将没有载荷物的递送溶液递送到Jurkat细胞、人PBMC和HSC。通过膜联蛋白V/碘化丙啶(PI)染色检测细胞递送后的存活力。与未处理的对照相比,细胞的存活力降低程度最小(图12)。此外,GFP mRNA被递送到原代NK细胞(图7),细胞具有96.6%的存活力。
电穿孔已用于体外将Cas9 RNP递送至细胞(Kim等人,(2014)Genome Res 24(6):1012-9;Lin等人,(2014)eLife 15;3:e04766;Liang等人,(2015)J Biotechnol 20;208:44-53;Schumann等人,(2015)Proc Natl Acad Sci USA 18;112(33):10437-42)。然而,电穿孔已知会影响临床相关细胞(包括T细胞和干细胞)的存活力和功能。因此,将平台介导的递送效果与电穿孔的效果进行比较。使用Neon电穿孔,这已经被其他团体使用(Liang等人,(2015)J Biotechnol 20;208:44-53;Schumann等人,(2015)Proc Natl Acad Sci USA 18;112(33):10437-42)。
将牛血清白蛋白-FITC(BSA-FITC)递送至Jurkat细胞,并在递送后24hr测定存活力。在递送平台的情况下,接受BSA-FITC的细胞的存活力从未处理对照的81%降低到65%(图13A)。相反,通过电穿孔将BSA-FITC递送到细胞时,存活力水平下降到8%。当仅递送缓冲液(没有BSA-FITC)时,存活力也出现类似的降低。
喷雾后24小时用流式细胞术检测BSA-FITC的递送效率。在递送平台的情况下,在65%存活的细胞中,34%是BSA-FITC阳性的(图13)。在电穿孔的情况下,在8%存活的细胞中,62%是BSA-FITC阳性的。因此,在最初的起始群体中,BSA-FITC在平台介导的递送后存在于22%的细胞中,在电穿孔后存在于5%的细胞中。
递送平台技术介导的递送对原代Jurkat细胞功能的影响
接下来,考察平台介导的递送对Jurkat细胞功能的影响。递送平台用于仅向细胞递送喷雾缓冲液。CD69和CD25分别是T细胞活化的早期和晚期细胞表面标志物。
使用递送平台将不含载荷物的递送溶液(仅喷雾缓冲液,SBO)递送到细胞。用PHA刺激细胞,分别在刺激后4小时和24小时用流式细胞术测量CD69和CD25的表达。在处理过的细胞中观察到CD69和CD25的上调(图14)。
基因编辑的Crispr Cas9系统已被广泛采用。然而,大多数用户将Cas9的DNA质粒形式与引导RNA结合使用。递送RNP形式的系统是令人感兴趣的,因为这更加短暂,因此应该导致更少的脱靶效果。必需的工具(重组Cas9核酸酶、单引导物、两部分引导物)最近已经在市场上获得。定制引导RNA序列是基于文献报道的相应质粒序列设计的。进行验证分析以检查试剂的质量。为了评估基因编辑工具,开发了基于安捷伦SureGuide Cas9可编程核酸酶试剂盒的体外编辑测试的分析。在该分析中,Cas9蛋白与引导物(单个或两部分)一起温育以形成RNP。然后将该RNP与包含靶位点的纯化长度的DNA(~250至750bp)温育。如果编辑成功,它将产生两条DNA链,一旦在琼脂糖DNA凝胶上分离,这将是显而易见的。这种分析有两个目的:第一,作为质量控制分析,检查基因编辑工具的功能;第二,用于确定递送溶液对基因编辑工具的影响,特别是对Cas9/RNP活性的影响。为了后一个目的,在递送溶液中制备Cas9或RNP的溶液,喷雾,然后收集喷雾溶液的等分试样并用于编辑分析。
安捷伦Sure Guide Cas9核酸酶试剂盒:该试剂盒提供Cas9核酸酶、对照DNA靶、对照gRNA、10×Cas9消化缓冲液(安捷伦)和无RNase水。对照反应的优化体积列于表4中。各种gRNA和DNA靶由IDT-DNA Cas9(IDT-DNA)合成或提供。本文描述了递送溶液。
该分析基于安捷伦Sure Guide Cas9核酸酶试剂盒。每个组分的优化体积列于表4中。试剂盒中提供了DNA靶、gRNA和Cas9,以验证程序。将储存在冰上的所有组分按表4中所述的比例加入适合于热循环仪的试管中。然后将管转移到在30℃预热的热循环仪中。运行了以下程序:
·30℃下30min
·65℃下15min
·保持在4℃
然后通过凝胶电泳分析消化的样品,其中通过存在额外的DNA条带(其大小对应于预期的DNA片段)来确认编辑成功。该分析证实了这些研究中使用的重组Cas9蛋白和各种引导RNA是有功能的。
表4:IVE反应的组分体积*
这里列出了测试的所有gRNA和相应的靶DNA:具有pCMV EGFR靶DNA的sun sgGFPRNA、具有EGFP靶DNA的sgRNA EGFP4、具有作为靶的HRT1 DNA的sgHPRT1、具有作为靶DNA的AAVS1的sgAAVS1 T1、sgAAVS1 T2和AAVS1 T3
在该分析中,IVE方案适用于研究递送溶液和喷雾程序对Cas9和RNP功能的影响。为此,首先在递送溶液中制备单独的Cas9或RNP(Cas9与gRNA复合),然后将其用于IVE,或喷雾然后收集用于IVE。如果发生编辑,这证实了RNP的组分能够切割目标DNA序列,因此它们是有功能的,尽管递送溶液中存在乙醇和其他化学物质。
在递送溶液中Cas9或RNP的溶液的制成如表5中所报告。RNP通过在加入S缓冲液和水之前将Cas9与gRNA温育15分钟而形成。此外,为了进行比较,在水中制成了相应的样品。为了制成复合物,使用1:1摩尔比的Cas9/gRNA。在仅有Cas9溶液的情况下,在进行IVE反应时提供gRNA(见表6)。
将除乙醇之外的所有组分加入试管中并温育10分钟。然后加入乙醇。根据表6和表7,对于每种溶液,将1μl(Cas9溶液)或2μl(RNP)的等分试样转移到试管中进行IVE测试。为了测试喷雾工艺对RNP活性的影响,将其它10μl等分试样喷雾到48孔板的空孔中,喷雾后收集1或2μl溶液并转移到PCR试管中。对于IVE,见表6和表7。如上所述消化样品,然后通过琼脂糖凝胶电泳进行分析。凝胶的典型图像在图17中提供。
为了利用递送平台技术分析Cas9在递送后是否进入细胞,探索将蛋白质与荧光标记缀合。必须避免针对胺基基团的Cas9标记方法,因为由于Cas9的物理化学特性,它们导致Cas9沉淀。因此,通过半胱氨酸的巯基(每分子蛋白质2个半胱氨酸残基),用荧光标签Alexa-488马来酰亚胺标记Cas9。这种反应没有改变生理pH下蛋白质的总电荷。所遵循的实验方案是基于先前的工作(Zuris、JA等人,Nat Biotechnol 2015;33(1)73-80)。
Cas9是从Toolgen基因组工程公司(韩国)或IDT集成DNA技术公司(美国)购买的。TCEP(三(2-羧乙基)膦)、Hepes、甘油和KCl为分析级,购自Sigma Aldrich。Alexa 488马来酰亚胺(Mw 720.66g/mol)是从Thermo Fisher Scientific购买的,以粉末形式接收,1mg。通过向粉末中加入139μl DMSO制备10mM原液。原液储存在-20℃。Alexa 488马来酰亚胺溶液必须加入到Cas9溶液中,使得DMSO的最终浓度不高于10%,以避免蛋白质沉淀。对于12.5μM Cas9,20倍摩尔过量对应于250μM Alexa模具,这又对应于250μl中的6.25μl 10mM原液的体积。为了使用所有制成的250μl蛋白质溶液,将6.25μl模具原料加入到250μl Cas9溶液中以产生256.25μl最终体积,其中DMSO为2.5%,远低于诱导蛋白质沉淀的10%极限)。对于尺寸排阻凝胶层析,使用Sephadex G-25(Sigma-Aldrich)。洗脱缓冲液包含250mM Kcl,20mM Hepes,pH 7.8。
将2mg/ml Cas9溶液(12.5μM)溶解在197μl标记缓冲液(20mM Hepes pH 7.8;250mM KCl;50μl甘油(63.1mg);3μlTCEP 10mM(摩尔过量10倍))。溶液用氩气去氧。在上下移液搅拌的同时,逐滴加入20摩尔过量的马来酰亚胺在DMSO中的溶液。混合物在4℃在Ar气氛中温育过夜(Zuris等人Nat Bitoechnol2015 33(1):73-80)。注:最终250μl体积的KCl浓度为200mM,盐KCl和甘油有助于将Cas9保持在溶液中。TCEP是生物化学和分子生物学应用中常用的还原剂。TCEP能阻止半胱氨酸形成二硫键,与二硫苏糖醇和β-巯基乙醇不同,它不会像马来酰亚胺残基那样容易反应。
第二天,在检查小瓶中不存在可见沉淀后,将蛋白质溶液加载到Sephadex G25柱上并洗脱,以纯化标记的Cas9并将其与游离标记分离。微弱的条带几乎立即分离,表明标记成功(图18)。洗脱的蛋白质被显著稀释。但是,荧光是肉眼可见的。用绿色荧光的透照器确认荧光(见图19)。纯化后的Cas9浓度用二辛可宁酸分析法评价。更浓缩的等分试样用于RNP研究。浓度在500-700ng/μl之间,收集约1ml体积。
在标记过程中,Cas9蛋白易于聚集和沉淀。其他人也报告了Cas9在各种溶液中的溶解度问题(Burger,A等人2016.Development2016 143(11):2025-37),当将蛋白质添加到递送溶液中时,也观察到Cas9沉淀。因为乙醇可以促进蛋白质沉淀,所以假设导致Cas9沉淀的主要因素之一是递送溶液中乙醇的存在。评估了乙醇浓度的增加对Cas9沉淀的影响。
根据下面描述的研究,超过15%的乙醇诱导蛋白质快速沉淀。根据这些信息,决定尝试通过1)降低递送溶液中的乙醇浓度,和2)用赋形剂/分子伴侣保护蛋白质来增加Cas9的溶解度。
荧光显微镜下观察标记的Cas9,溶液呈绿色,带有少量绿色“斑点”。当将递送溶液(25%乙醇)加入到Cas9中时,这些斑点的大小和数量大大增加。为了确保沉淀不是由蛋白质上荧光探针的存在引起的,在显微镜下观察含有25%乙醇的递送溶液中的未标记Cas9溶液。图21中示出了该图,可见蛋白质聚集体。在显微镜下观察载玻片时,蛋白质的聚集和从溶液中沉降是实时发生的。
在上述初步步骤之后,制备包含浓度增加的乙醇的用荧光染料标记的Cas9溶液,并在加入乙醇后立即在荧光显微镜下分析。将内部标记和纯化的Alexa 488Cas9 133ng/μl加入S缓冲液和水中,如表8所示。就在对该样品进行显微镜分析之前,将乙醇加入试管中。
加入乙醇后,立即将5μl溶液转移到载玻片上,并在荧光显微镜下观察。首先,对样品1-6进行研究,结果如图22所示。在对结果进行第一次评价后,加入样品7、8和9(图23)。由于Alexa 488Cas9的浓度较低,在10μl中可递送的最大量为931ng,约为编辑实验正常递送量(5gμg)的五分之一。
从图22清楚地看出,在具有15%乙醇的样品中出现聚集,并且在20%和25%时该现象变得显著。对于1、5和10%乙醇,没有观察到明显聚集。为了比较,图24中报告了从柱上洗脱的不含乙醇的Cas9和蛋白质的。
由于低于10%的乙醇含量导致细胞对载荷物不良地吸收,而高于15%的乙醇含量导致蛋白质显著沉淀,因此对10%至15%范围内的乙醇进行了更仔细的研究,见图23。当乙醇达到12%时,在载玻片上的微滴边缘观察到聚集,表明10%乙醇是促进载荷物吸收和避免蛋白质沉淀的最佳折衷方案。
最后,重要的是要强调,随着时间的推移(1-2h),在所有样品中观察到蛋白质聚集(除了1%乙醇,2h后进一步检查)。这些观察表明,Cas9和含乙醇的溶液必须在制备后几分钟内使用。
值得注意的是,Cas9的浓度低于编辑实验通常使用的浓度,因为溶解度测试是用市售Cas9制成的Alexa 488Cas9进行的。标记的蛋白质一旦纯化,就会经历显著稀释。用于这些分析的递送溶液旨在保持在用于单次递送的10μl体积中的25%乙醇和25% S缓冲液的比例,这意味着可用于添加Cas9的体积最高达5μl。由于标记蛋白比亲本蛋白稀释得更多,难以在可用的5μl体积中递送5μg。
递送溶液最合适的乙醇含量是10%。在这样的含量的情况下,细胞膜的透化仍然是可能的,如果在几分钟内制备和使用溶液,就避免了Cas9的沉淀。然而,在乙醇浓度低于25%的情况下,诸如mRNA的大载荷物的递送效率降低,因此优选保持25%的乙醇浓度,并避免或减少Cas9沉淀。
进行体外编辑测试以评估Cas9沉淀对基因编辑工具编辑效率的影响。如表9所示,针对不同的乙醇含量,制成了几种具有不同Cas9量的溶液。RNP复合物是通过加入除乙醇以外的所有试剂(Cas9和gRNA)和递送溶液组分(S缓冲液,MGW)而制成。温育10分钟后和喷雾前加入乙醇。Cas9/gRNA的比例为1:1摩尔比。将cas9以1:1摩尔比加入gRNA(384b,5.3M,223.7ng/μl原料)中,温育15分钟,然后加入S缓冲液和MGW,温育混合物10分钟。最后,就在喷洒前加入乙醇。
加入乙醇后,立即将10μl溶液喷雾入48孔板的空孔中。然后将2μl喷雾的溶液转移到适合于热循环仪的试管中并用于IVE测试,如表10中所报告的,以评估复合物的功能。将保存在冰中的组分加入适合于热循环仪的反应管中,也保存在冰中。然后将管转移到在30℃预热的热循环仪中。运行了以下程序:
·30℃下30min
·65℃下15min
·保持在4℃
然后用1.5%凝胶的凝胶电泳分析消化的样品。
如图25所示,在一定程度上编辑了所有DNA靶。样品1-5没有显示出差异,表明Cas9蛋白的用量比编辑所需的量大得多,因此尽管沉淀,溶液中残留的Cas9足以编辑靶。乙醇效应仅在最低Cas9浓度时才变得明显。在样品7(50ng/μl Cas9,20%乙醇)中,编辑的条带较弱,未切割的DNA信号更强。这表明乙醇诱导的Cas9在最低浓度(和最高乙醇)下的沉淀剥夺了可编辑的功能性Cas9系统。在较高浓度下,残留在溶液中的蛋白质可能足以切割靶DNA。
Cas9要求在溶液中保持较高的盐浓度,最重要的公布中使用的浓度为约200-250mM KCl。为了防止添加乙醇后出现沉淀,在递送溶液中制成了Cas9或RNP溶液,其中水被0.5M KCl或1M KCl替代。如前所述,加入除乙醇之外的所有组分,然后在室温下温育溶液15分钟,最后加入乙醇。在所有情况下,观察到蛋白质或复合物的大量沉淀。
已知甘油可改善蛋白质溶解度。在该实验中,将5、20或20%甘油加入到递送溶液的配方中。在室温温育15分钟后,再次将乙醇加入其余组分中。在包含Cas9的样品中,只有乙醇的加入仍然导致沉淀,但没有形成大的聚集体。相反,即使在加入乙醇后,在含有20%甘油的递送溶液中也没有观察到RNP沉淀(图25)。对于含有5%和10%甘油的递送溶液中的RNP,很难确定是否形成沉淀,因为所看到的颗粒看起来像一些载玻片上存在的背景污垢。当然,蛋白质的典型的大团簇没有出现(图26A-26C)。然而,当含甘油的溶液喷雾到细胞上时,观察到细胞损伤。因此,甘油在这种情况下不是合适的赋形剂。
已知NDSB-201可改善蛋白质溶解度。Cas9单独或与RNP一起在包含0.05(D'Astolfo DS等人,Cell 161(3):674-90)0.2和0.5M NDSB的递送溶液中制成。再次,将除乙醇之外的所有组分一起加入,将溶液温育15分钟,然后加入乙醇。当加入乙醇时,仅包含Cas9的所有样品都形成沉淀。在0.5M NDSB情况下的RNP(核糖核蛋白,Cas9与引导RNA的复合物)未见沉淀(图26A-26C)。对于在0.05和0.2M NDSB情况下的RNP,观察到很少沉淀或没有沉淀(由于背景噪声而难以分辨)。
考虑到上述结果,指定了一系列实验来评估已知保护剂/分子伴侣/赋形剂“保护”Cas9蛋白免受乙醇诱导沉淀的能力。热休克蛋白(HSP)对于多种多样的客户蛋白质的正确折叠和成熟以及保护蛋白质免于应激诱导的解折叠和聚集是必不可少的(Morimoto,R.I.,2008:Genes&Development,v.22,p.1427-1438;Richter,K等人2010:MolCell,v.40,p.253-266)。小HSP(sHSP)是主要的“保持”分子伴侣,将解折叠蛋白质保持在适于随后再折叠的构象中,从而防止它们不可逆聚集(Eyles,S.J.,and L.M.Gierasch,2010:PNAS,v.107,p.2727-2728;Stengel,F等人,2010:PNAS,v.107,p.2007-2012)。α-晶体蛋白是小热休克蛋白家族的重要成员,也是晶状体的重要结构成分(Reddy,等人2006:Iubmb Life,v.58,p.632-641)。
使用以下组分的原料浓度:Cas9(10μg/μl)(IDT,1074182)、Cas9消化缓冲液(Sureguide试剂盒,安捷伦)、HPRT(IDT)的gRNA(21.8μM)、D-山梨醇(Sigma,S3889)、D-甘露醇(Sigma,M4125)、来自牛眼晶状体的α-晶体蛋白(Sigma,C4163)、NDSB(Sigma,804)、明胶A(Sigma,G1890)、明胶B(Sigma,G9391)、甘氨酸(Sigma,G7126)、脯氨酸(Sigma,P5607)、吐温20(Sigma,P9416)、L-组氨酸(Sigma,H8000)、肌醇(Sigma,15125)和海藻糖(Sigma,90210)。
添加剂的选择是根据目前关于稳定蛋白质和防止聚集的文献来决定的。所选择的添加剂的最终列表显示在表11中。测试添加剂,看看它们是否能在乙醇存在下保持Cas-9核酸酶功能。将原料Cas9在不含核酸酶的H
使用Genetools程序(Syngene),在暴露于有或没有添加剂的不同递送溶液后,测量Cas9:gRNA的编辑效率。获得条带2(编辑的HPRT)相对于条带1的强度,并以百分比表示。将所有值归一化为不含乙醇的样品。
为了测试这些保护剂具有的对细胞内基因编辑的影响,通过递送平台技术将Cas9RNP与一些上述保护剂一起递送到细胞。将骨髓来源的间充质干细胞(MSC)以每孔9×10
表11:本研究中使用的添加剂
体外编辑实验显示,添加剂可以在乙醇存在下保持Cas9核酸酶的功能(图28和图19-29B)。α-晶体蛋白和山梨醇的加入尤其提高了Cas-9:gRNA暴露于递送溶液后的核酸酶活性。Cas-9:gRNA的体外编辑效率平均从无添加剂对照的49%分别提高到山梨醇和α-晶体蛋白样品的98%和103%效率。将α-晶体蛋白和NDSB添加到递送溶液中,以改进MSC中hprt的CRISPR-Cas9编辑(图30)。α-晶体蛋白添加到最终浓度为220μM时,hprt编辑从11.09%(无添加剂对照)增加到16.34%。将NDSB添加到递送溶液中对MSC的hprt编辑没有影响。α-晶体蛋白在U2OS细胞中最大限度地编辑hprt方面也是比山梨醇更好的添加剂(图31)。
已知乙醇在改变蛋白质结构的蛋白质位点置换水(Dwyer,1999Biopolymers49(7):635-45)。这可能导致疏水核暴露,从而导致蛋白质聚集。最近,Ferns等人进行的一项研究(Ferns,2012:Neurochemical Research,v.37,p.244-252)发现乙醇诱导的GAPDH聚集被α-晶体蛋白阻止。因此,α-晶体蛋白可能阻止了Cas-9的聚集。
递送平台技术影响粘附细胞编辑的能力受到靶细胞单层的大小、Cas9的浓度、蛋白质与引导RNA的比例以及该技术处理细胞的次数的影响。Cas9蛋白与引导RNA可以有一个最佳比例,它可以是细胞和基因靶二者特异性的
所用材料包括:U2OS细胞(骨肉瘤细胞系-ECACC)、骨髓来源的间充质干细胞(MSC;lonza)、Jurkat 6.1细胞(T细胞系-ECACC)、DMEM低葡萄糖(Sigma-Aldrich)、FBS(Sigma)、RPMI-1640(Life Technologies)、胰蛋白酶/EDTA(Gibco)、Cas9核酸酶(IDT-DNA)、crRNA(IDT-DNA)、trRNA(IDT-DNA)、递送S缓冲液、α-晶体蛋白(Sigma)、乙醇、Hex标记的脱落探针(IDT-DNA)、FAM标记的参比探针(IDT-DNA)和ddPCR Supermix(BioRad)。
U2OS细胞以5×10
在第一个实验中,评估了添加MgCl
在第二个实验中,在蛋白质与引导物比例为1:1的情况下,Cas9的量在0.1μg至24μg之间变化。递送载荷物,细胞被如上所述处理。在第三个实验中,细胞被重复处理。如上所述,将5μgCas9以1∶6的引导物比例递送到细胞。然后胰蛋白酶消化细胞并重新接种到相同的孔中,然后培养过夜。第二天早上,在相同条件下再次处理细胞。将细胞在培养基中放置4小时,然后在相同条件下进行第三次处理。细胞再次培养过夜。第二天早上,在相同条件下对细胞进行第四次处理,并放置4小时,然后进行胰蛋白酶消化并接种到48孔板的孔中,并培养72小时。然后收集细胞,并制备用于通过微滴数字PCR和脱落分析进行基因编辑分析。
使用微滴式数字PCR(ddPCR)分析编辑的存在,ddPCR是一种基于水-油乳液微滴技术进行数字PCR的方法。样品被分成20,000个微滴,每个单独微滴中都会发生模板分子的PCR扩增。在该方法中,由于Cas9 RNP对基因组的作用,该技术被用于检测HPRT基因中编辑的存在。这种分析是专门针对DNA双链切割的非同源末端连接修复而建立的。正向和反向引物产生~150bp的扩增子。使用两个内部探针;一种用作参比探针,应该结合所有扩增的DNA链,另一种针对Cas9切割位点,当没有编辑发生时结合,但当编辑受到影响时不能结合
在这个实验中,使用MagNA纯紧凑系统从样品中提取DNA。ddPCR组分是根据制造商指南制备的。将组分转移到DG8盒中,用QX200微滴发生器产生微滴。然后将微滴转移到96孔PCR板上并密封。进行PCR,并使用QX200微滴读取器分析微滴。
i)细胞单层尺寸对编辑效率的影响
通过在较小的孔中接种细胞,当细胞单层的直径减小时,编辑效率增加,从而使单层的直径与喷雾的直径更好地匹配(图32)。
将U2OS细胞接种在24、48或96孔板中并培养过夜。第二天,两剂Cas9 RNP(5μg;1:6比例的HPRT gRNA)通过递送平台技术递送,细胞在收获前再培养72小时。通过ddPCR分析编辑效率,表明接种在最小孔中的细胞产生最大的编辑效率
ii)增加Cas9浓度对编辑效率的影响
通过在引导RNA情况下以1:1的比例递送Cas9而实现的编辑百分比随着Cas9浓度的增加而增加,稳定在约5μg Cas9(图33)。
将MSC接种到96孔板中并培养过夜。第二天,通过递送平台技术递送在HPRT引导RNA的情况下,以1:1比例的不同浓度的Cas9 RNP(0.1、0.5、1.0、5.0、10.0、15.0、24.0μg),细胞在收获前再培养72小时。通过ddPCR分析编辑效率,表明编辑效率随着Cas9蛋白量的增加而增加,并且增加稳定在约5μgCas9。
iii)Cas9:gRNA比例对编辑效率的影响
以不同比例(1:1、1:3或1:6)递送5ug Cas9和引导RNA并没有改变MSC细胞中编辑的效率(图34)。
将MSC接种到96孔板中并培养过夜。第二天,通过递送平台技术递送在HPRT引导RNA情况下1:1、1:3或1:6摩尔比的Cas9 RNP(5gμg),细胞在收获前再培养72hr。通过ddPCR分析编辑效率,并表明,对于这些细胞以及这种特定的靶和引导RNA,测试的蛋白质:引导RNA的比例在编辑效率上没有差异
iv)递送的RNP剂量数量对编辑效率的影响
增加细胞通过递送过程(Cas9-5μg,比例1:6)的次数增加了在HPRT基因中检测到的编辑百分比(图35)。
将MSC接种到96孔板中并培养过夜。在接下来的3天内,1、2、3或4剂Cas9 RNP(5g;1:6比例的HPRT gRNA)通过递送平台技术递送,在最后一剂递送后,在收获前,细胞再培养72小时。通过ddPCR分析编辑效率,表明编辑效率随着细胞接收的Cas9 RNP剂量的数量增加而增加。*p<0.05,独立均值的学生t检验。
如本文中显而易见的,平台递送技术可以将一系列分子递送到细胞,同时保持它们的存活力和功能性。该特性允许在不同时间点多次定量给予相同分子或可能地不同分子的方案(图35)。这不同于电穿孔,在电穿孔中,由于导致存活力大大降低的对细胞的有害影响,所以不能实现多次定量给予。因此,为了递送质粒形式的Cas9,潜在的出路,允许细胞表达蛋白质一段时间,然后递送引导RNA,当其结合时,会影响编辑。
v)MgCl
基于以前的体外编辑研究,观察到添加MgCl
与蛋白质递送的行业标准电穿孔进行比较,评估递送效率、存活力和功能性。
与电穿孔相比,在通过本文提供的递送技术递送Cas9的情况下,在MSC中编辑效率更高(图37)。
将MSC接种到96孔板中并培养过夜。在接下来的3天内,4剂Cas9 RNP(5g;1:6比例的HPRT gRNA)通过递送平台技术递送,在最后一剂递送后,在收获前,细胞再培养72小时。为了电穿孔比较,MSC在缓冲液中制备,并按照制造商的说明进行处理。根据递送平台处理的细胞,递送相同量的Cas9和引导物。收获细胞并通过ddPCR分析编辑效率。当通过任何一种方法递送Cas9时,观察到成功的编辑,然而,在通过递送平台技术递送Cas9 RNP的情况下,效率显著更高。*p<0.05,独立均值的学生t检验。
细胞存活力在通过递送平台技术或电穿孔递送的Cas9 RNP编辑过的细胞之间是相当的(图38)。递送最终剂量Cas9 RNP后24小时,通过EBAO计数分析细胞的存活力。这两个组之间的存活力没有观察到显著差异。
MSC的功能不受递送平台技术的影响(图39)。测试了来自未处理组(未处理)的分化的MSC、由递送平台技术组(递送平台)递送的Cas9 RNP和由电穿孔组(电穿孔)递送的Cas9 RNP的三个代表性图像。Cas9 RNP(5μg;1:6比例HPRT gRNA)递送后一天,细胞以1×10
将MSC和U2OS细胞接种到96孔板中并培养过夜。在接下来的3天内,4剂Cas9 RNP(5μg;1:6比例的HPRT gRNA)通过递送平台技术递送,细胞再培养72小时。对于悬浮细胞,将1.5×10
评估了影响Cas9 mRNA递送后细胞中的编辑的能力。在Cas9 mRNA和引导RNA以相同的载荷递送的情况下,在细胞中检测到基因编辑(图41)。将MSC接种到96孔板中并培养过夜。第二天,Cas9 mRNA(4μg)和引导RNA(相当于1:6的比例;HPRT gRNA)通过递送技术递送,细胞在收获前再培养72小时。通过ddPCR分析编辑效率,并在这些细胞中检测到编辑。
基因编辑蛋白向细胞和组织的无载体递送
Crispr Cas9系统在基因组编辑领域迅速变得突出。Crispr Cas9借助于引导RNA识别基因组中的特定序列,并在DNA中诱导双链切割。细胞可以通过易错非同源末端连接过程修复这种切割,从而编辑基因。评估了本文提供的递送平台技术向细胞递送基因编辑工具以影响编辑的能力。
为了评估这些工具(重组Cas9蛋白和市售的引导RNA)是否诱导DNA靶切割,基于安捷伦SureGuide Cas9可编程核酸酶试剂盒建立了体外编辑分析。递送平台递送溶液对Cas9和引导RNA功能的影响也使用这种非限制性分析进行评估。发现Cas9影响编辑的能力并没有被一个示例性递送溶液所取消。此外,当将Cas9蛋白添加到递送溶液中,喷雾入孔中,收集,然后在体外编辑测试中进行功能性评估,以复制整个递送过程时,Cas9活性得以保留。为了证实Cas9在递送后被靶细胞吸收,通过半胱氨酸氨基酸的巯基(每分子蛋白2个半胱氨酸残基)用荧光标签Alexa-488马来酰亚胺标记Cas9蛋白。蛋白质的成功标记使得蛋白质在细胞内可视化。
Cas9是一种细菌蛋白质,并不自然存在于哺乳动物细胞中。Cas9在某些条件下有从溶液中沉淀出来的趋势,这将影响其功能。对于大多数细胞类型来说,使用递送平台递送技术将货物最佳递送到细胞中是在乙醇浓度最高达25%(v/v)的情况下实现的,并且观察到Cas9在接触超过15%的乙醇时易于沉淀。为了减少乙醇诱导的Cas9沉淀,使用体外编辑分析和随后递送至细胞来评估一系列赋形剂。虽然一些赋形剂,诸如甘油和非洗涤剂磺基甜菜碱(NDSB)防止乙醇诱导的沉淀,但是这些赋形剂在包括在喷雾递送实验中时造成一些细胞损伤。然而,向递送溶液中添加热休克蛋白α-晶体蛋白似乎提高了Cas9在体外编辑分析中的编辑能力,并且也与递送至细胞相容。当Cas9 RNP在α-晶体蛋白存在下被递送到细胞时,通过微滴数字PCR检测细胞的有效基因组编辑。将这种赋形剂添加到递送溶液中,使得随后能够优化递送平台技术介导的Cas9 RNP基因组编辑在贴壁细胞和悬浮细胞二者,和原代细胞以及细胞系中的方案。作为实例,这种优化已经在50%的原代人MSC中产生了基因编辑。此外,已经证明,递送平台技术介导的MSC中编辑水平高于电穿孔实现的水平,并且与电穿孔相比,使用本文提供的递送技术编辑的MSC的存活力和功能性更好
其他实施方式
本文引用的所有美国专利和已公布或未公布的美国专利申请通过引用并入本文。本文引用的所有公布的外国专利和专利申请通过引用并入本文。本文引用的所有其他公布的参考文献、文件、手稿、登录号和科学文献通过引用并入本文。
根据期望的配置,本文描述的主题可以体现在系统、设备、方法和/或物品中。前面描述中阐述的实施方案并不代表与本文描述的主题一致的所有实施方案。相反,它们仅仅是与所描述的主题相关的方面相一致的一些实例。尽管上面已经详细描述了一些变化,但是其他修改或添加也是可能的。特别地,除了本文阐述的那些特征和/或变化之外,还可以提供其它特征和/或变化。例如,以上描述的实施方案可以涉及所公开的特征的各种组合和子组合和/或以上公开的若干另外特征的组合和子组合。此外,附图中描述和/或本文中描述的逻辑流程不一定需要所示的特定顺序或相继顺序来获得期望的结果。其它实施方案可以在所附权利要求的范围内。
- 含有脂肪间充质干细胞外泌蛋白提取物的干细胞组合物及提取物的制备方法及组合物的应用
- 基因编辑蛋白和组合物向细胞和组织的无载体递送
- 含苯硼酸修饰的高分子材料及其在基因编辑核糖核蛋白复合物胞内递送中的应用