一种单细胞多组学文库构建方法及应用
文献发布时间:2024-04-18 20:01:23
技术领域
本发明涉及生物技术领域,具体涉及一种单细胞多组学文库构建方法及其应用。
背景技术
作为一种复杂系统疾病,肿瘤的发生发展不仅表现在细胞组织的多个维度上的变异和紊乱,还体现在肿瘤微环境的建立及癌细胞与人体周围正常细胞和免疫系统的密切交流上。无数临床和科研领域的工作者在癌症领域进行了数年的广泛跨学科研究,但迄今为止,我们对这一疾病的了解仍然远远不够。肿瘤的诊治周期长,花费高,恶性肿瘤预后的效果差,造成了社会民生和医疗投入的持续增加,给社会和家庭带来较大经济和心理上的负担。
肿瘤的发生发展是多时期多阶段的,对于常见的实体瘤,治疗的最佳时期是在原初肿瘤发生淋巴侵袭和周边组织侵袭之前,此时多数患者可以通过低风险手术切除病灶,达到较好的治疗效果(Curtius,K.,N.A.Wright,and T.A.Graham,An evolutionaryperspective on field cancerization.Nat Rev Cancer,2018.18(1):p.19-32.);部分已经发生远端转移的和切除后复发的病人,往往会进一步辅助化疗或放疗;对药物不敏感或者产生耐药的个体再进一步根据组织活检的结果选择其他个性化治疗方式,如联合靶向用药和免疫治疗。对肿瘤进展阶段的准确判断、对于治疗策略的准确选择以及对于后期肿瘤转移及复发的及时发现是肿瘤治疗的最关键要点,成为了提高肿瘤病人治疗效果、相对生存率及生存质量的重要突破点。
近年来,基于下一代测序的单细胞测序近年来进展迅速,2009年开展了首例单细胞mRNA测序实验,2011年开展了首例人类癌细胞单细胞DNA测序实验,2012年开展了首例单细胞外显子组测序实验(Tang,F.,et al.,mRNA-Seq whole-transcriptome analysis ofa single cell.Nat Methods,2009.6(5):p.377-82.)(Navin,N.,et al.,Tumourevolution inferred by single-cell sequencing.Nature,2011.472(7341):p.90-4.)。单细胞测序在我们对恶性肿瘤细胞和免疫细胞的人类生物学的理解方面,明显优于以往的测序技术,它能够以单细胞分辨率探测细胞和微环境异质性,它彻底改变了我们深入研究数千个单个细胞的转录、基因组、表观基因组和代谢特征的能力,可以对肿瘤病灶内的细胞进行无偏倚的分析(Kashima,Y.,et al.,Single-cell sequencing techniquesfromindividual to multiomics analyses.Exp Mol Med,2020.52(9):p.1419-1427.)。单细胞测序技术的出现最大限度的解决了肿瘤组织间具有较大异质性的问题,稀有的细胞间差异信息得以从混杂的组织样品中被筛选出来,是肿瘤进展监测、治疗方法选择及肿瘤转移复发检测一个极具前景的方向。
迄今为止,单细胞测序已经被较多的应用于对肿瘤的研究中,然而,目前对于各个癌症的单细胞研究多局限于单一组学层面的研究,且多局限于对癌症单细胞转录组的揭示,能提供的信息非常有限(Lei,Y.,et al.,Applications of single-cell sequencingin cancer research:progress and perspectives.J Hematol Oncol,2021.14(1):p.91.)。实际上,癌症进展过程中除了会发生转录组层面的变化,表观遗传组层面上的染色质重塑也是癌症进展的重要驱动因素。染色质的重塑常常影响DNA物理可及性的动态变化,造成基因开放状态的极大改变(Wu,C.Y.,et al.,Integrative single-cell analysis ofallele-specific copy number alterations and chromatin accessibility incancer.Nat Biotechnol,2021.39(10):p.1259-1269.)。染色质可及性的变化多能反应出增强子、启动子与染色质结合因子的作用情况,染色体上开放区域与对应转录因子或其他调控蛋白的结合将直接影响细胞内基因复制及转录行为的发生。最近,已有较多研究对染色质开放性检测在肿瘤研究中的重要意义进行了充分论证。使用临床肿瘤样本进行测序,研究者发现肿瘤的发生伴随着异常染色质开放状态的改变(Pereira,R.M.,et al.,Transcriptional and epigenetic regulation of T cell hyporesponsiveness.JLeukoc Biol,2017.102(3):p.601-615.)(Scott-Browne,J.P.,et al.,Dynamic Changesin Chromatin Accessibility Occur in CD8(+)T Cells Responding to ViralInfection.Immunity,2016.45(6):p.1327-1340.)。针对皮肤T细胞淋巴瘤的研究发现,淋巴瘤的发生与异常的染色质开放性改变密切相关,该研究还揭示了表观遗传组学变化在肿瘤治疗中的干预意义(Qu,K.,et al.,Chromatin Accessibility Landscape ofCutaneous T Cell Lymphoma and Dynamic Response to HDAC Inhibitors.CancerCell,2017.32(1):p.27-41.e4.)。此外,研究人员使用ATAC-seq测序技术检测了多种肿瘤类型的全基因组染色质开放性,发现染色质开放性的异常改变与肿瘤的产生与发展具有较强相关性(Corces,M.R.,et al.,The chromatin accessibility landscape of primaryhuman cancers.Science,2018.362(6413).)。因此,实现癌症病人染色质开放性的精确检测在临床指导及机制研究中都有着至关重要的意义。
迄今为止,研究者已经开发了几种用于单细胞转录组及表观遗传组染色质开放性多组学测序的方法,但都存在一定的技术局限及弊端。市面上的10x genomics测序技术,仍然存在通量低、批次效应、不同组学建库方式不兼容的难题。同时,已报道的可以实现高通量单细胞染色质开放性捕获的技术,例如SHARE-seq技术,未针对更加复杂的肿瘤组织进行优化,且文库结构无法适用于常用PE150测序,造成测序成本显著提升(Chen,S.,B.B.Lake,and K.Zhang,High-throughput sequencing of the transcriptome and chromatinaccessibility in the same cell.Nat Biotechnol,2019.37(12):p.1452-1457.)(Ma,S.,et al.,Chromatin Potential Identified by Shared Single-Cell Profiling ofRNA and Chromatin.Cell,2020.183(4):p.
1103-1116.e20.)。
因此,亟需开发一种相对高通量、高精度的单细胞多组学测序技术。
发明内容
本发明旨在至少在一定程度上解决相关技术中的技术问题之一。
为此,本发明的一个目的在于提供一种基于单细胞的多组学建库方法,所述方法包括:
(a)获得目标样本的细胞悬液,并对所述细胞悬液进行固定处理;
(b)对步骤(a)中固定后的细胞进行透化处理及ATAC转座;
(c)用带有亲和标签的RT引物对ATAC转座后的细胞进行逆转录;
(d)将步骤(c)获得的逆转录后的细胞以此进行第一轮、第二轮、第三轮Barcode;
(e)用磁珠分离步骤(d)标记后的细胞,分别得到ATAC文库及RNA文库,
其中,
所述第一轮的Barcode标记的引物包括Round_1_linker和barcode1,所述第二轮的Barcode标记的引物包括Round_2_linker、barcode2,所述第三轮的Barcode标记的引物包括Round_3_linker、barcode3,
所述Round_3_linker为与Read2互补的序列,所述barcode3包括PE150测序P7端的index。
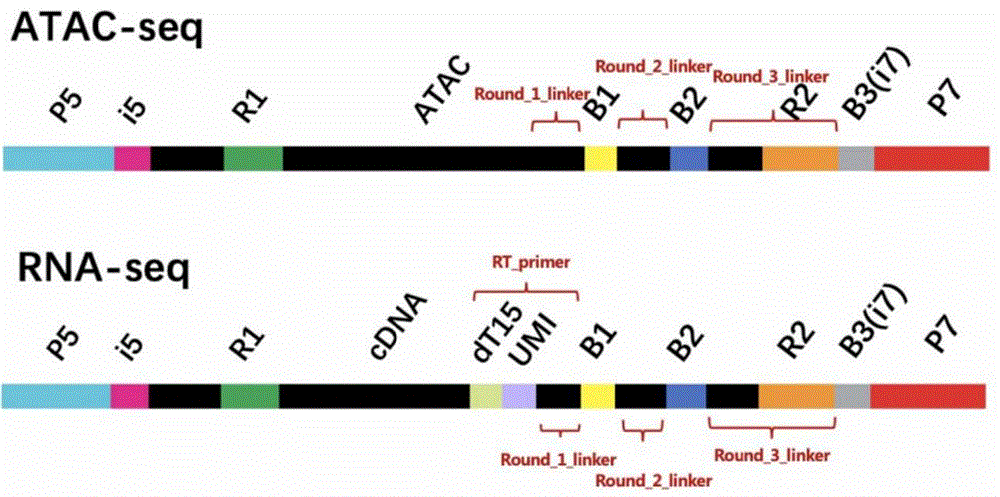
本申请发明人通过改进文库结构,改变了Read2的引入顺序及位置,在Tn5转座时不对Read2进行引入,而将Read2序列包含在Round_2引物的尾部及Round_3引物的头部,位置处于barcode2与barcode3之间。Round_3引物最终包含三部分:Read2的尾部序列、barcode3及P7序列。barcode1仍然包含在Round_1引物中。从而通过这种设计可以达到将barcode1、barcode2包含在Read2序列内部的效果,barcode3充当Read2向后测读的正常index(8bp)。这种设计能够很好的适应目前市面上的illumina平台测序仪器,不用对测序模式进行文库特异性调整,可以与其他样本进行正常的混样测序,大大降低了样本测序的成本。
在一些具体实施方案中,步骤(a)中所述固定处理时的试剂为质量浓度百分比为0.02%-0.12%的甲醛溶液。
在一些具体实施方案中,所述甲醛溶液质量浓度百分比为0.05%-0.1%。
在一些具体实施方案中,所述透化处理及ATAC转座在同一体系內进行。
在一些具体实施方案中,在步骤(b)之前,进一步包括对所述ATAC转座反应用到的Tn5酶进行组装标记,获得两端含有Read1、磷酸化A2及与ME互补的部分的Tn5酶,其中,所述A2为与所述Round_1_linker至少部分互补的序列。
在一些具体实施方案中,步骤(c)中,逆转录过程设置了50℃与42℃的温度循环体系。
在一些具体实施方案中,步骤(c)中,所述亲和标签包括选自生物素标签、链球菌标签中的任意一种,优选为生物素标签。
在一些具体实施方案中,所述barcode3包括PE150测序P7端的5-12bp的index。
在一些具体实施方案中,所述barcode3包括PE150测序P7端的8bp的index。
本发明第二方面提供上述方法构建的文库,所述包括ATAC文库及RNA文库。
本发明第三方面提供上述文库在单细胞ATAC测序中的应用。
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
本发明的上述和/或附加的方面和优点从结合下面附图对实施例的描述中将变得明显和容易理解,其中:
图1显示了本发明一个实施方案中REACT-seq建库流程示意图;
图2显示了本发明实施例1中逆转录程序对RNA文库长度的捕获效果,其中,图2中的A图为现有技术捕获到的RNA文库效果,B图为优化的逆转录程序1捕获到的RNA文库效果,C图为优化的逆转录程序2捕获到的RNA文库效果;
图3显示了本发明实施例1中分离出的ATAC-seq及RNA-seq序列示意图图;
图4显示了本发明实施例3中Tn5酶用量梯度测试结果,其中,图4中的A为2ul酶量酶切后的RNA文库效果,图4中的B为1ul酶量酶切后的RNA文库效果,图4中的C为1ul酶量酶切后的RNA文库效果;
图5显示了本发明实施例4中应用本发明REACT-seq的质量控制结果。
具体实施方式
下面详细描述本发明的实施例。下面描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。
需要说明的是,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者更多个该特征。进一步地,在本发明的描述中,除非另有说明,“多个”的含义是两个或两个以上。
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
为了更容易理解本发明,以下具体定义了某些技术和科学术语。除显而易见在本文件中的它处另有明确定义,否则本文中使用的所有其它技术和科学术语都具有本发明所属领域的一般技术人员通常理解的含义。
在本文中,术语“包含”或“包括”为开放式表达,即包括本发明所指明的内容,但并不排除其他方面的内容。
在本文中,术语“任选地”、“任选的”或“任选”通常是指随后所述的事件或状况可以但未必发生,并且该描述包括其中发生该事件或状况的情况,以及其中未发生该事件或状况的情况。
在本文中,术语“染色质可及性”,真核生物中的核小体是染色质的基本结构单位。DNA与组蛋白结合后形成核小体,核小体再进一步折叠压缩后最终形成染色质。DNA的复制和转录都需要将染色质紧密结构打开,从而允许调控因子结合DNA。这部分被打开的染色质,就叫开放染色质(Accessible-Chromatin)。开放染色质允许调控因子结合的特性称为染色质的可及性(Chromatin Accessibility)。简而言之,染色质可及性/开放性是指某个时间点同时进行转录的DNA区域。
在本文中,术语“PE150测序”是指将DNA样品分别进行两次测序,生成两个长度为150bp的reads,这两个reads之间存在一定的空隙,可以利用这个空隙进行序列比对和拼接,从而得到更长的序列信息。PE150适合于高精度的基因组组装和SNP检测等应用。
在本文中,术语“细胞通透化处理”是指在不造成细胞裂解以及不破坏细胞内部有机结构的情况下改变细胞壁和细胞膜的通透性,使得小分子物质和一些较大分子物质能够自由进出细胞。细胞通过透化处理后在提高通透性的同时,整体结构保持完整,对胞内酶仍具有相当的保护作用,可保证胞内酶催化作用的充分发挥,并延长酶的使用寿命。
本发明基于已发表的SHARE-seq技术,通过不同建库流程测试、文库结构优化,建立起高通量、高精度且适用于癌症组织的单细胞多组学(ATAC/RNA)测序技术REACT-seq,具体流程如图1所示:使用消化酶组合对复杂肿瘤组织进行适应性的组织消化,将组织裂解为单细胞后进行合适浓度的细胞固定,将核小体与DNA链进行交联;在对基因组进行转座打断后,对细胞中的RNA进行逆转录操作;之后使用基于Split-pool的方式为每个细胞加上特异性的三轮barcode,以对每个单细胞进行区分;解交联后,使用链霉亲和素磁珠对DNA和cDNA进行分离,分别扩增完成两种组学的建库,其中,逆转录时,相较于现有逆转录程序,本申请发明人在程序设计中加入50度与42度的温度循环体系,获得了较好的长片段RNA捕获效果,REACT-seq可以实现对临床肿瘤样本的低成本、超高通量的大规模组织单细胞测序。
在一些具体实施方案中,发明人将临床新鲜组织样品剪碎为小片后使用酶解法消化为单细胞,使用红细胞裂解缓冲液去除红细胞后进行流式细胞分选,筛选活细胞进行测序文库的构建。采用多次添加barcode短DNA接头连接的方式实现超高通量单细胞唯一标签的添加,使用链霉亲和素磁珠实现cDNA及DNA的分离,从而分别进行单细胞转录组及染色质开放性基因组学的文库构建。多步质量控制步骤将被用于对组织样本建库效果的评估,以保证癌症高异质性组织建库效果的均一性。单细胞测序将对肿瘤组织样本涵盖的所有细胞类型进行揭示,以超高分辨率最大程度的反应癌症个体不同阶段细胞类型比例的变化。
本发明可以以极低的成本实现对人类复杂组织所有细胞类型单细胞转录组及染色质可及性图谱的建立。REACT-seq技术在基因组学、转录组学和表观组学研究中有广泛的应用,如研究癌症发生发展机制、解析人类基因调控机制、探索神经发育机制等,可以更精确地描述转录组及其调控机制在人体内的变化,从而为人类疾病的生物学机制的研究和治疗提供新的思路和方法。此外,随着技术的发展,REACT-seq技术也可以用于基因编辑、药物研发和药物疗效监测等方面,从而为医学领域的发展和进步提供有效的支持。
在一些具体实施方案中,本申请发明人经过多次测试后,选用200ul小体系在PCR管中进行固定操作。本申请中使用100ulPBSI对100,000细胞进行重悬,经过测试发现,对比大体系,在PCR管中进行的小体系操作能在之后的多次离心中实现更好的弃去上清、留下细胞沉淀的效果,从而将可能的细胞损失降低到最小,实现高比率投入细胞到建库细胞量的回收。另外,为了规避小体积高浓度甲醛加入后在局部形成的过高固定强度可能带来的负面影响,本申请首先使用PBSI预先配置质量浓度百分比为0.2%甲醛溶液,实验时将100ul细胞悬液与低浓度甲醛溶液进行等体积混合,获得更好的整体固定效果。
在一些具体实施方案中,逆转录步骤的充分与否极大的决定了测序文库中对较长片段RNA捕获的全面性。该步骤的意义在于通过逆转录的方式利用引物对生物素标记进行引入,从而实现下游中DNA文库及RNA文库的分离。在兼顾标记引入成功的同时,还应该保障逆转录正常延伸的完全性。RNA存在未解开的复杂二级结构、RNA与蛋白交联过强、逆转录时间不充足等多种原因均会导致RNA在全长逆转录前停止,影响后期文库对RNA的完整无偏捕获。为了尽可能提高RNA的完整捕获效率,在设计技术逆转录反应时,发明人进行了如下考虑:在程序设计中加入50摄氏度与42摄氏度温度体系的循环。由于部分RNA具有二级结构,容易出现位阻现象,导致链的延伸提前终止。50摄氏度温度条件下可以对二级结构进行破坏,继而再进行42摄氏度逆转录反应,循环多次以提高延伸效率。经过设计,本技术构建了一套新的RNA逆转录体系(终浓度为1xRT缓冲液、0.4U/ul Enzymatics RNA酶抑制剂、500uMdNTP、10uM带有亲和标签的RT引物、15% PEG 6000和25U/ul Maxima H Minus逆转录酶)及两套逆转录程序:样品在50℃加热10分钟,在42℃加热90分钟(程序1)或30分钟(程序2),然后经历10个热循环(50℃2分钟,42℃2分钟),最后在50℃温育5分钟。使用该逆转录体系,本技术获得了较好的长片段RNA捕获效果。
在一些具体实施方案中,发明人对RNA逆转录程序进行优化,原SHARE-seq逆转录程序在测试中发现不适应于人类癌症组织的RNA逆转录,容易造成不充分逆转录的现象,重新优化后使用样品在50℃加热10分钟,在42℃加热30分钟,然后经历10个热循环(50℃2分钟,42℃2分钟),最后在50℃温育5分钟的程序进行逆转录,获得了较好的RNA文库效果。
不同组织在组学建库中适用的组织消化、固定条件、通透条件等均需要根据不同组织的特点进行调整建立。本申请针对肿瘤组织进行了多实验步骤的摸索,建立起一套与SHARE-seq不同的,适用于肿瘤复杂组织的多组学建库方法。本发明通过对不同建库流程测试、文库结构优化,建立起高通量、高精度且适用于癌症组织的单细胞多组学(ATAC/RNA)测序技术REACT-seq,可以实现人不同类型组织的单细胞转录组及表观遗传组染色质开放性多组学匹配测序(现有技术未针对更加复杂的人类组织进行优化,如SHARE-seq仅能进行细胞系及小鼠肺、脑、皮肤多组学测序)。
本发明提供的REACT-seq特有的文库结构、文库序列及新的Read1、Read2引入方式,可以适应于常规PE150测序,对比现有技术实现极大的成本缩减,REACT-seq实验创新细节,可以实现ATAC及RNA的同等高质量文库构建。
由于单细胞多组学技术需要同时实现对转录组及染色质开放性信息的建库捕获,实验所需时间相对较长,难以实现单天内建库,基于此情况,发明人对实验暂停点进行了测试及设置。实验经历组织解离、固定、转座、逆转录、杂交连接、解交联及亲和下拉、单细胞ATAC-seq文库构建、单细胞RNA-seq文库构建等步骤,经过不同阶段暂停优劣情况的调研考虑及实验测试,发明人选取了杂交连接前的实验反应体系进行暂停点设置。在此节点设置暂停点,由于已对RNA已进行了初步的逆转录反应,可以大大降低暂停点设置造成的可能RNA大量降解情况。同时,该暂停点的设置还较好的提升了ATAC文库构建的质量,完整标记细胞数和启动子区域单细胞片段读数获得了较为明显的提升,单细胞聚类分群能力获得了较大的进步。
在一些具体实施方案中,发明人针对不同的人类癌症类型组织,测试了不同类型消化酶组合、不同消化酶浓度、组织解离时间、组织解离条件(摇床/振荡/旋转等),最终建立起对于每种癌症的特异性消化方式,能够同时实现保证组织消化所得的细胞活率及不同组织中各细胞类型的最大程度原貌保留。
在一些具体实施方案中,对于组织解离细胞与细胞系细胞的固定条件是不同的,本申请的发明人在固定步骤中使用细胞系及多种癌症类型测试固定条件,发现在固定强度过强时易造成RNA文库质量的下降及长RNA捕获的困难。最终发现,对于组织解离细胞,使用质量浓度为0.02%-0.12%的甲醛进行固定可以获得高质量的ATAC及RNA文库数据,优选甲醛质量浓度为0.05%-0.1%;对于细胞系细胞,使用质量浓度为0.05%的甲醛进行固定可以获得高质量的ATAC及RNA文库数据。
在一些具体实施方案中,发明人测试了解交联的时间及条件,发现过短时间的解交联容易造成解交联不充分,RNA模版转换受阻,影响长片段RNA捕获测序的问题;过长时间的解交联容易造成RNA降解问题,影响RNA文库质量。本申请发明人最终选取在解交联过程中加入RNA酶抑制剂,同时实行在55℃解交联2小时,之后-80℃解交联过夜的流程,同时保证了解交联的充分性及RNA较好的文库质量。
在一些具体实施方案中,发明人在对RNA文库的Tn5酶打断建库流程中,对酶切使用酶量进行了独立测试,发现酶量使用过大会造成最终打断片段的过短,在测序后无法获得充足的RNA信息;过少的酶量又容易造成cDNA打断不充分。最终使用的酶投入量为0.02U/ul,即50ul体系中含1ul 1U/ul Tn5酶在55℃打断5分钟时能获得最适宜的测序文库长度(300-400bp)。
在一些具体实施方案中,Tn5酶二聚体里面的双链的ME识别序列Oligo使用的是通用Nextera Tn5binding site,本申请的发明人对Tn5酶二聚体两端伸出的单链序列进行了改造设计,一端为illumina平台测序Nextera tn5 read1,但另一端为与Round_1_linker一端互补配对的序列,以便进行后续的Round_1引物连接。
在一些具体实施方案中,在原有技术如share-seq中,转座步骤Tn5两端组装的序列直接为测序使用的Read1及Read2,之后再通过杂交连接步骤在Read2后连接Round_1、Round_2、Round_3引物,对三轮barcode进行引入,这种引入方法带来的最大弊端是用于单细胞区分的标记barcode1、barcode2、barcode3被排除在Read2外部,需要通过定制化的测序模式对文库结构进行测序(在Read2后向后测99bp的index长度),定制化的测序大大提升了小量多批次样本测序的成本。在本申请中,发明人通过改进文库结构,改变了Read2的引入顺序及位置,在Tn5转座时不对Read2进行引入,而将Read2序列包含在Round_2引物的尾部及Round_3引物的头部,位置处于barcode2与barcode3之间。Round_3引物最终包含三部分:Read2的尾部序列、barcode3及P7序列。barcode1仍然包含在Round_1引物中。从而通过这种设计可以达到将barcode1、barcode2包含在Read2序列内部的效果,barcode3充当Read2向后测读的正常index(8bp)。这种设计能够很好的适应目前市面上的illumina平台测序仪器,不用对测序模式进行文库特异性调整,可以与其他样本进行正常的混样测序,大大降低了样本测序的成本。
下面将结合实施例对本公开的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本公开,而不应视为限定本公开的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例1样品准备
1.组织解离及固定
使用1mg/ml胶原酶2(购自Thermo Fisher,货号17101015)、1mg/ml胶原酶4(购自Thermo Fisher,货号17104019)、1mg/ml分散酶2(购自Roche,货号4942078001)及DMEM1640培养基(购自GIBCO,货号C11995500BT)配置10ml消化液,将胃癌组织样本用组织箭剪碎至1立方毫米左右,转移到配置好的消化液中,在37℃摇床150rpm的条件下消化30分钟。
将组织解离细胞置于离心机中500g离心5分钟后转移至含有0.1U/ul EnzymaticsRNase Inhibitor的PBS中重悬,至100000个细胞/ml,并使用终浓度为0.1%的甲醛进行固定,固定即指在室温下孵育5分钟。加入甘氨酸、Tris-HCL及7.5%BSA混合液进行终止固定,室温孵育5分钟后500g离心5分钟。去除上清液,细胞沉淀用1ml PBSI洗涤两次,两次沉淀之间以500g离心5分钟,将细胞重悬于PBSI中以用于转座。
2.转座
使用的Tn5酶(购自强微特,货号M0221)通过与等体积含有Read1、具有5’端具有磷酸化修饰的与Round_1_linker一端互补配对的序列A2,及等量ME接头序列混合的oligo进行退火组装,获得ATAC文库的两端带有Read1及5’端具有磷酸化修饰的与Round_1_linker一端互补配对的序列A2接头Tn5酶。对于各转座反应,取10000-20000个细胞投入转座缓冲体系(5xLM缓冲液,18.8%DMF,0.1%NP-40,0.47%蛋白酶抑制剂混合物,和0.8U/ul RNA酶抑制剂,0.1%Digitonin,0.02%Tween-20,Mgcl2),样品在37度下500rpm振荡30分钟后在1000g离心3分钟,用1ml核分离缓冲液(NIB)洗涤两次,然后进行逆转录。
本申请通过测试后,采用将细胞通透化处理与ATAC转座同时进行的一个复合体系对细胞进行一次性处理。使用通透化处理剂为0.1%NP-40、0.01%Digitonin及0.02%Tween-20,同时体系内包括5xLM缓冲液、DMF及Tn5酶对细胞进行转座反应。现有技术需要先对细胞进行通透化处理,再进行转座反应,本申请流程缩减了通透化处理所需要的额外时间,可以在一个体系内同时进行打断和通透,经测试,该优化能在缩减实验步骤、降低实验操作难度及时间的同时,实现很好的细胞转座效果,对多种不同的细胞类型适用性较好。具体实验反应体系如表1所示。
表1
3.逆转录
在逆转录过程中,本申请的发明人对现有技术中的逆转录程序进行了优化:
现有技术逆转录程序:
样品在50℃加热10分钟,然后经历3个梯度升温热循环(8℃加热12秒,15℃加热45秒,20℃加热45秒,30℃加热30秒,42℃加热2分钟,50℃加热3分钟),最后在30℃温育5分钟,其中,RNA文库长度捕获效果如图2中的A图所示;
本申请优化的逆转录程序1:
样品在50℃加热10分钟,在42℃加热90分钟,然后经历10个热循环(50℃2分钟,42℃2分钟),最后在50℃温育5分钟,其中,RNA文库长度捕获效果如图2中的B图所示;
本申请优化的逆转录程序2:
样品在50℃加热10分钟,在42℃加热30分钟,然后经历10个热循环(50℃2分钟,42℃2分钟),最后在50℃温育5分钟,其中,RNA文库长度捕获效果如图2中的C图所示。
由图2可知,优化的逆转录条件能达到更优的片段长度情况。其中,程序2达到了更好的效果,可能由于程序1运行过程时间较长,造成了RNA相对更强的降解。综上结果,我们使用优化体系及程序2进行单细胞RNA的逆转录过程实现了较好的RNA全长捕获。
逆转录时,将转座细胞与逆转录混合液混合(终浓度为1xRT缓冲液、0.4U/ulEnzymatics RNA酶抑制剂、500uM dNTP、10uM带有UMI序列、生物素标签的RT引物、15% PEG6000和25U/ul Maxima H Minus逆转录酶)。样品在50℃加热10分钟,在42℃加热30分钟,然后经历10个热循环(50℃2分钟,42℃2分钟),最后在50℃温育5分钟。加入200ulNIB,1000g离心3分钟,去除上清液,用200ul NIB洗涤细胞沉淀,1000g离心3分钟,-80℃冰冻过夜。
4.杂交和连接
将细胞重悬于4608ul杂交混合液中(1×T4连接缓冲液、0.32U/ul RNA酶抑制剂、0.05U/ul SUPERaseRI、10% NP-40、NIB-RI)。将杂交混合液中的细胞(40ul)加入到第一轮的条形码平板中,条形码平板中预先分有10ul不同的96个DNA条形码。加入细胞后的第一轮条形码平板在室温下轻轻振荡(300rpm)孵育30分钟。之后加入10ul第一轮的封闭寡核苷酸在室温下轻轻振荡(300rpm)孵育30分钟以封闭之前连接的链,避免出现之后的错误杂交连接。第二轮和第三轮分别将50ul和60ul合并细胞加入到相应轮到条形码平板中,其余操作同第一轮。加入第三轮封闭寡核苷酸后,合并所有孔的细胞,在1000g离心3分钟,去上清,用1mlNIB-RI洗涤两次。将细胞重悬于连接混合物(1xT4连接缓冲液,0.32U/ul RNA酶抑制剂,20U/ul T4 DNA连接酶,0.1% Triton X-100,NIB),在室温下轻轻振荡(300rpm)孵育30分钟。用50ulNIB-RI重悬细胞,计数,以20000细胞每管等分至PCR管中,其中第一、二、三轮条形码对应的的引物序列分别如表2、3、4所示,其中表2中的Round1、表3中的Round2的5’端均带有磷酸化修饰。
表2
/>
表3
/>
/>
表4
/>
/>
5.解交联及亲和下拉
在50ul细胞体系中,加入解交联缓冲液(100mM Tris pH 8.0、100mM NaCl、0.04%SDS、2ul 20mg/ml蛋白酶K,和1ul SUPERase RI),混合后在55℃孵育2小时,之后将样品放入-80℃解交联过夜。在解交联后样品中加入5ul 100mM PMSF以终止解交联,室温孵育10分钟。对于每份样品,吸取10ul Dynabeads MyOne Streptavidin C1(购自ThermoFisher,货号65001)在1x B&W-T缓冲液(5mM Tris pH8.0,1M NaCl,0.5mM EDTA,0.05% Tween-20)中洗涤两次,在添加2U/ul SUPERase RI的1xB&W-T缓冲液中洗涤一次,最后重悬于100ul2xB&W缓冲液(10mM Tris pH 8.0,2M NaCl,1mM EDTA,和4U/ul SUPERase RI)。将每份样品与重悬后的Dynabeads MyOne Streptavidin C1混合,在室温下以10rpm的转速在旋转器上旋转60分钟,之后将样品管置于磁力架上分离上清及Dynabeads MyOne Streptavidin C1。其中,带有亲和素的cDNA附着在Dynabeads MyOne Streptavidin C1上,其余DNA残留在上清液中。
原SHARE-seq方法由于文库结构限制,无法适用于常用的PE150测序,造成了测序成本的显著提升。如表5所示,本方法对文库结构重新进行了设计,其中,重新设计的引物序列为:RT引物序列、用于第一轮barcode引入的linker序列、用于第三轮barcode引入的linker序列。在表5所示的引物中,其中与Round_1_linker一端互补配对的序列A2及RT_primer的5’端带有磷酸化修饰,RT_primer中dT的5’端修饰有生物素标签iBiodT。
表5
其中,N为A/T/C/G中的任意一种,N的个数在4-20之间,V为A/C/G中的任意一种。
本发明调整了Read2、i7的文库位置及Read1、Read2在建库中的引入方式,对于ATAC文库,Round_1_linker直接通过与ATAC引入的3’端接头互补配对进行连接,从而继续引入barcode1,继续通过ligation的方式引入Round_2_linker、barcode2、Round_3_linker、barcode3,比较特殊的是,发明人调整后的文库结构实际上使用Round_3_linker的互补配对序列充当了Read2序列,从而将barcode3作为PE150测序P7端8bp正常index,进而将barcode1、barcode2都包含在测序Read2内部,规避了现有技术需要通过修改测序程序向Read2外P7端读取数十bp index的弊端。RNA文库与ATAC文库barcode引入方式相同,不同的是Round_1_linker通过与RT_primer右侧序列互补配对引入。使新REACT-seq的文库结构适应于市面上的常规PE150测序,在测序中可以进行任意数据量的样品混合上机,极大的降低了测序的成本。分离出的ATAC-seq及RNA-seq序列如图3所示,其中,B1、B2指barcode1、barcode2,Round_1_blocker、Round_2_blocker、Round_3_blocker为Round_1_linker、Round_2_linker、Round_3_linker的互补配对序列,位置与linker序列相同。
实施例2scATAC-seq文库制备
用1.2x VAHTSDNA Clean Beads(购自Vazyme,货号N411-01)纯化上清液,并洗脱至20ul去离子水中。加入到PCR反应体系中(2x KAPA Mix,0.5uM文库特异性Ad1引物和0.5uM P7引物)。在PCR仪中逆行以下反应:72℃5分钟,98℃30秒,之后98℃20秒,65℃30秒,72℃30秒运行15个循环,72℃孵育3分钟。反应结束后使用1.2x VAHTSDNA Clean Beads结合体系中的DNA来纯化反应体系,洗脱至20ul去离子水中,测量DNA浓度。
实施例3scRNA-seq文库制备
对于实施例1中分离出的Dynabeads MyOne Streptavidin C1(购自ThermoFisher,货号65001),使用含有1U/ul SUPERase抑制剂的1x B&W-T缓冲液洗涤磁珠三次,用STE缓冲液洗涤一次(10mM Tris pH8.0,50mM NaCl和1mM EDTA,1U/ul SUPERase抑制剂)。在室温下以10rpm的速度在旋转器上旋转30分钟,在42℃下以300rpm的速度振荡90分钟进行模版转换,振荡期间每30分钟吹吸重悬beads一次。模版转换后将样品置于磁力架上去除上清,用200ul STE缓冲液清洗beads。之后将beads重悬于55ul PCR混合液中(2xKAPA、400nM P7引物、400nM的RNA PCR引物)。将管子置于PCR仪中进行以下反应:98℃ 3分钟,然后在98℃ 15秒、67℃ 20秒和72℃ 6分钟下进行22个循环,之后在72℃下孵育5分钟。扩增后的cDNA产物使用0.8x的VAHTSDNA Clean Beads(购自Vazyme,货号N411-01)进行纯化,洗脱到20ul去离子水中,测量cDNA浓度。
在转座过程中,发明人对Tn5酶(购自强微特,货号M0221)具体用量进行了梯度测试,统计cDNA的投入量为50ng的条件下,使用2ul、1ul、0.5u的Tn5酶,55℃打断5分钟时能获得最适宜的测序文库长度(300-400bp)。分别评估了打断后片段分布情况。结合二代测序PE150读长情况,理想的文库长度应该在300bp左右呈现峰近似正态分布的效果。如图4所示,其中,图4中的A显示使用2ul酶量时,峰面积出现向左的略微偏移,提示可能出现酶切过度情况,片段长度过短;图4中的C显示使用0.5ul酶量时,峰在300bp右侧呈现更多的富集,提示部分片段酶切不足;图4中的B显示使用1ul Tn5酶进行切割时,获得了较为理想的切割效果。
对于每个样品,取50ng cDNA进行Tn5打断及接头连接,以便进行Read1的引入。将50ng cDNA投入打断体系(5x MF缓冲液、0.5ul Tn5酶、去离子水),在55℃下打断5分钟。打断后产物使用0.8xVAHTSDNA Clean Beads(购自Vazyme,货号N411-01)纯化,以便筛选合适大小的DNA进行扩增建库。洗脱到10ul去离子水中。纯化后产物加入到扩增PCR体系中(2xKAPA、0.5uM文库特异性Ad1引物和0.5uM P7引物)。将管子置于PCR仪中进行以下反应:72℃5分钟,98℃ 30秒,之后98℃ 10秒,65℃ 30秒,72℃ 1分钟运行7个循环,72℃孵育5分钟。反应结束后使用0.8x VAHTSDNA Clean Beads(购自Vazyme,货号N411-01)纯化反应体系,洗脱至20ul去离子水中,测量DNA浓度。
实施例4REACT-seq质量控制结果
将本申请实施例1中的癌症样本一分为二,分别进行REACT-seq建库及Bulk ATAC建库。对本申请获得的REACT-seq进行质控,结果如图5所示,其中,图5中的A显示了本申请的REACT-seq技术与大量细胞构建Bulk ATAC信号在活跃表达基因窗口的重复性的对比结果,结果显示利用REACT-seq获取的单细胞数据,在两次独立实验间重复性较高,并且与利用大量细胞构建的文库信号具有较高一致性。图5中的B显示REACT-seq文库DNA片段长度分布,其中横坐标表示片段长度,纵坐标表示片段密度,结果显示.本申发明构建的ATAC文库读段长度呈现符合核小体排列的周期性分布特征。图5中的C显示REACT-seq文库TSS区域读段富集倍数,其中横坐标表示与TSS区域的距离,纵坐标表示TSS富集倍数,结果显示本发明文库读段数在TSS区域相比左右2000bp的区域有超过40倍的富集,表明该技术具有高灵敏度以及准确度,可以在癌症组织中获取高质量的数据。
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、“一些实施方案”或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。