利用寡核苷酸合成废液制备4,4′-双甲氧基三苯甲基氯的方法
文献发布时间:2023-06-19 18:37:28
技术领域
本发明涉及资源循环技术领域,具体而言,涉及一种利用寡核苷酸合成废液制备4,4'-双甲氧基三苯甲基氯的方法。
背景技术
随着生物制药工业的发展,小核酸药物的研究和应用越来越广泛,寡核苷酸作为小核酸药物的一种,指的是2~10核苷酸残基以磷酸二酯键连接而成的线性多核苷酸片段,其合成通过采用固相亚磷酰胺化学合成法,在该合成方法的脱保护过程中采用三氯乙酸(TCA)或二氯乙酸(DCA)从延长的核苷酸链的5’端末端移除二甲氧三苯甲基(DMT)基团,并产生一个5’端活性羟基基团,结束后,产生大量的副产物4,4'-二甲氧基三苯甲基(DMT)阳离子和二氯甲烷,使得寡核苷酸制备过程原子利用率低,造成极大的浪费。
为了回收利用脱保护废液中的4,4'-二甲氧基三苯甲基(DMT)阳离子,传统的方法为:先将游离三芳甲基保护基与碱反应生成三芳基甲醇,再将三芳基甲醇与三氯化铝、二氯亚砜等卤化剂进行氯化,获得DMT-Cl,在回收过程中首先要除去废液中二氯甲烷,通常采用加热蒸发的方式进行,热量需求高,能量损耗大,且二氯甲烷的蒸发具有一定的操作危险性,其次采用强腐蚀性的氯化剂进行氯代,对设备要求高,增加了生产成本和操作危险性。
因此,有必要开发一种条件温和,操作简单,不需要进行溶剂置换,且安全环保的回收方法。
发明内容
基于此,本发明直接利用废液中的二氯甲烷作为溶剂,通过硅烷还原和氯代回收制备获得4,4'-双甲氧基三苯甲基氯,该过程中反应条件温和,后处理简单,工艺能耗低,安全环保。
本发明提供了一种利用寡核苷酸合成废液制备4,4'-双甲氧基三苯甲基氯的方法,具体包括:
(a)收集寡核苷酸合成脱保护过程产生的含有4,4'-二甲氧基三苯甲基阳离子的废液;
(b)将所述废液与部分取代硅烷混合反应,经洗涤、干燥获得4,4'-二甲氧基三苯甲烷溶液;
(c)将所述4,4'-二甲氧基三苯甲烷溶液与偶氮类自由基引发剂和氯代试剂混合反应,制备4,4'-双甲氧基三苯甲基氯。
在其中一个实施例中,所述部分取代硅烷的结构为R-SiH3、(R)2-SiH2、(R)3-SiH;进一步的,所述R各自独立地为C1-C6的烷基或芳基。
在其中一个实施例中,所述废液中4,4'-二甲氧基三苯甲基阳离子与部分取代硅烷的摩尔比为1:2-10。
在其中一个实施例中,所述步骤(b)洗涤、干燥过程具体包括:
将反应后的溶液采用碱性盐溶液进行多次洗涤,获得有机相;再加入无机干燥剂进行干燥获得4,4'-二甲氧基三苯甲烷溶液。
在其中一个实施例中,所述碱性盐溶液为饱和碳酸钾水溶液,饱和碳酸氢钠水溶液、饱和碳酸钠水溶液、氢氧化钠水溶液或氢氧化钾水溶液中的任意一种。
在其中一个实施例中,所述4,4'-二甲氧基三苯甲烷溶液中的4,4'-二甲氧基三苯甲烷与氯代试剂、偶氮类自由基引发剂的摩尔比为1:2-10:0.05-1。
在其中一个实施例中,所述步骤(b)中反应温度为20-30℃,反应时间为0.5-4h。
在其中一个实施例中,所述步骤(c)中反应的温度为20-80℃,反应时间为2-6h。
在其中一个实施例中,所述氯代试剂为N-氯代丁二酰亚胺、1,3-二氯-5,5-二甲基乙内酰脲和苄基三甲基四氯硼酸铵中的任意一种。
在其中一个实施例中,其特征在于,所述偶氮类自由基引发剂为过氧化苯甲酰、偶氮二异丁酸二甲酯、偶氮二异庚腈、偶氮二异丁腈中的任意一种。
在其中一个实施例中,所述步骤(c)中混合反应后还包括后处理过程,包括:
反应后加入惰性溶剂析出粗产物,并将所述粗产物进行洗涤后溶解进行柱层析纯化,经浓缩、干燥获得4,4'-双甲氧基三苯甲基氯。
在其中一个实施例中,所述惰性溶剂为C3-C10的烷烃或环烷烃。
在其中一个实施例中,所述柱层析纯化过程的流动相为石油醚:乙酸乙酯=5:1。
在其中一个实施例中,所述干燥过程的温度为40-50℃,干燥时间为10-15h。
本发明的有益效果:
本发明的回收制备4,4'-双甲氧基三苯甲基氯的方法首先采用部分取代的硅烷还原剂对4,4'-二甲氧基三苯甲基阳离子进行还原,该过程可以以二氯甲烷为溶剂,不需要进行溶剂的置换,且反应条件温和、后处理纯化过程操作简单;然后将反应获得的产物采用偶氮类自由基引发剂和温和的卤代试剂进行自由基氯代反应,该过程反应条件温和,副产物少,产率高,经济环保,便于扩大生产。
附图说明
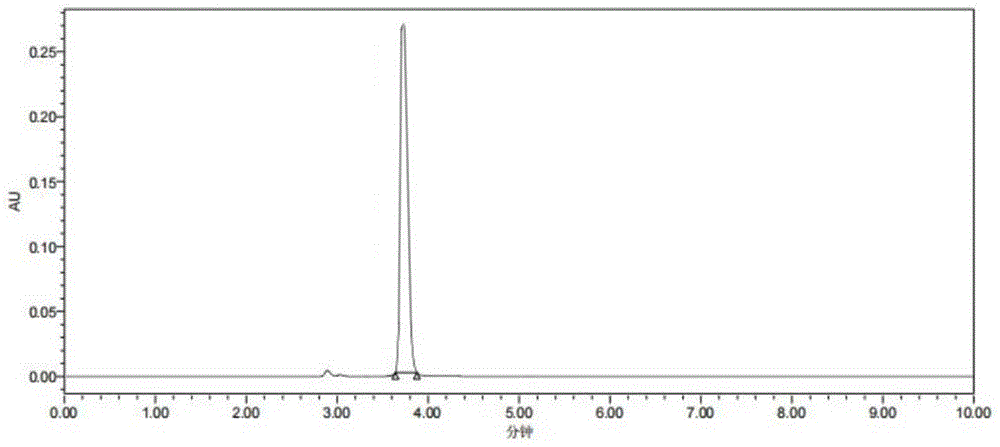
图1为本发明实施例1获得的4,4'-双甲氧基三苯甲基氯的HPLC谱图。
具体实施方式
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合附图对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施例的限制。
此外,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括至少一个该特征。在本发明的描述中,“多个”的含义是至少两个,例如两个,三个等,除非另有明确具体的限定。
在本发明中,除非另有明确的规定和限定,术语“安装”、“相连”、“连接”、“固定”等术语应做广义理解,例如,可以是固定连接,也可以是可拆卸连接,或成一体;可以是机械连接,也可以是电连接;可以是直接相连,也可以通过中间媒介间接相连,可以是两个元件内部的连通或两个元件的相互作用关系,除非另有明确的限定。对于本领域的普通技术人员而言,可以根据具体情况理解上述术语在本发明中的具体含义。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
本发明提供了一种利用寡核苷酸合成废液制备4,4'-双甲氧基三苯甲基氯的方法,具体包括:
(a)收集寡核苷酸合成脱保护过程产生的含有4,4'-二甲氧基三苯甲基阳离子的废液;
(b)将所述废液与部分取代硅烷混合反应,经洗涤、干燥获得4,4'-二甲氧基三苯甲烷溶液;在该过程中,4,4'-二甲氧基三苯甲基阳离子经部分取代硅烷还原获得4,4'-二甲氧基三苯甲烷;
(c)将所述4,4'-二甲氧基三苯甲烷溶液与偶氮类自由基引发剂和氯代试剂混合反应,制备4,4'-双甲氧基三苯甲基氯。在该过程中,氯代试剂经引发剂引发产生氯自由基和丁儿酰亚胺自由基,丁儿酰亚胺自由基与4,4'-二甲氧基三苯甲烷反应,由于DMT基团上的甲基由于p-π共轭,对自由基电荷的分散效果好,所以该自由基稳定,与氯自由基结合主要生产4,4'-双甲氧基三苯甲基氯,副产物少。
在具体的示例中,所述部分取代硅烷的结构为R-SiH
在具体的示例中,所述废液中4,4'-二甲氧基三苯甲基阳离子与部分取代硅烷的摩尔比为1:2-10。进一步的,所述4,4'-二甲氧基三苯甲基阳离子与部分取代硅烷的摩尔比为1:2、1:3、1:5、1:6、1:8、1:10。
在具体的示例中,所述步骤(b)洗涤、干燥过程具体包括:
将反应后的溶液采用碱性盐溶液进行多次洗涤,获得有机相;再加入无机干燥剂进行干燥获得4,4'-二甲氧基三苯甲烷溶液。
在具体的示例中,所述碱性盐溶液为饱和碳酸钾水溶液,饱和碳酸氢钠水溶液、饱和碳酸钠水溶液、氢氧化钠水溶液或氢氧化钾水溶液中的任意一种。
在具体地示例中,无机干燥剂可替换成活性氧化铝,3A分子筛,CaSO4,NaSO4,MgSO4,CaCl2等常用干燥剂。
在具体的示例中,所述4,4'-二甲氧基三苯甲烷溶液中的4,4'-二甲氧基三苯甲烷与氯代试剂、偶氮类自由基引发剂的摩尔比为1:2-10:0.05-1。
在具体的示例中,所述步骤(b)中反应温度为20-30℃,反应时间为0.5-4h。
在具体的示例中,所述步骤(c)中反应的温度为20-80℃,反应时间为2-6h。
在具体的示例中,所述氯代试剂为N-氯代丁二酰亚胺、所述氯代试剂为N-氯代丁二酰亚胺、1,3-二氯-5,5-二甲基乙内酰脲、苄基三甲基四氯鹏酸铵和次氯酸钠。优选N-氯代丁二酰亚胺
在具体的示例中,其特征在于,所述偶氮类自由基引发剂为过氧化苯甲酰、偶氮二异丁酸二甲酯、偶氮二异庚腈、偶氮二异丁腈中的任意一种。
在具体的示例中,所述步骤(c)中混合反应后还包括后处理过程,包括:
反应后加入惰性溶剂析出粗产物,并将所述粗产物进行洗涤后溶解进行柱层析纯化,经浓缩、干燥获得4,4'-双甲氧基三苯甲基氯。
在具体的示例中,所述惰性溶剂为C3-C10的烷烃或环烷烃。所述柱层析纯化过程的流动相为石油醚:乙酸乙酯=5:1。
在具体的示例中,所述干燥过程的温度为40-50℃,干燥时间为10-15h。
在具体的示例中,上述从产生4,4'-二甲氧基三苯甲基阳离子到回收制备4,4'-双甲氧基三苯甲基氯的化学反应式如下式(1)-(3)所示:
在本发明实施例中,废液中的4,4'-二甲氧基三苯甲烷(DMT)是以游离的DMT阳离子形式存在,加入部分硅烷为还原剂搅拌反应后,将DMT阳离子还原成DMT。加入氯代试剂NCS,在自由基引发剂引发下产生氯自由基,DMT基团上的甲基由于p-π共轭,对自由基电荷的分散效果好,所以自由基越稳定,因此主要生产甲基氯代产物,副产物少。
以下结合具体实施例详细描述本发明,这些实施例用于理解而不是限定本发明。
实施例1
采用AKTA 100合成仪,FineLINE 35合成柱,3%(w/v)三氯乙酸-二氯甲烷脱保护试剂,按合成仪上通用合成程序合成寡核苷酸。合成过程中单独收集脱保护步骤废液。并取上述废液稀释10倍,测量498nm吸光度,得到废液中4,4'-二甲氧基三苯甲基阳离子的浓度为3.3mM。
取1L废液,加入3.8g三乙基硅烷,室温条件下搅拌反应1h,溶液由橙红色变成淡黄色混合溶液。采用1L饱和碳酸氢钠溶液洗涤淡黄色混合溶液三次,然后再加入50g无水硫酸钠干燥过夜,过滤除去固体,并在35℃条件下真空浓缩至50ml,获得4,4'-二甲氧基三苯甲烷溶液。
将4,4'-二甲氧基三苯甲烷溶液加入4.4g N-氯代丁二酰亚胺(NCS),0.5g偶氮二异丁腈在60℃回流反应1h,再将50mL正庚烷滴加入至上述反应液中,10min中滴加完,析出固体室温下搅拌30min,过滤取固体,再向固体中加入10mL正庚烷打浆,过滤后固体用10mL正庚烷淋洗;固体进行硅胶柱层析纯化,流动相石油醚:乙酸乙酯=5:1,固体在45℃条件下干燥12h获得4,4'-双甲氧基三苯甲基氯0.68g。
实施例2
采用AKTA 100合成仪,FineLINE 35合成柱,3%(w/v)三氯乙酸-二氯甲烷脱保护试剂,按合成仪上通用合成程序合成寡核苷酸。合成过程中单独收集脱保护步骤废液。并取上述废液稀释10倍,测量498nm吸光度,得到废液中4,4'-二甲氧基三苯甲基阳离子的浓度为3.3mM。
取1L废液,加入3.8g三乙基硅烷,室温条件下搅拌反应1h,溶液由橙红色变成淡黄色混合溶液。采用1L饱和碳酸氢钠溶液洗涤淡黄色混合溶液三次,然后再加入50g无水硫酸镁干燥过夜,过滤除去固体,并在35℃条件下真空浓缩至50ml,获得4,4'-二甲氧基三苯甲烷溶液。
将4,4'-二甲氧基三苯甲烷溶液加入4.4g N-氯代丁二酰亚胺(NCS),0.5g偶氮二异丁腈在室温条件下反应6h,再将50mL正己烷滴加入至上述反应液中,10min中滴加完,析出固体室温下搅拌30min,过滤取固体,再向固体中加入10mL正己烷打浆,过滤后固体用10mL正己烷淋洗;固体采用流动相溶解进行硅胶柱层析纯化,流动相为石油醚:乙酸乙酯=5:1,浓缩后的产物在45℃条件下干燥12h,获得4,4'-双甲氧基三苯甲基氯0.61g。
实施例3
采用AKTA 100合成仪,FineLINE 35合成柱,3%(w/v)三氯乙酸-二氯甲烷脱保护试剂,按合成仪上通用合成程序合成寡核苷酸。合成过程中单独收集脱保护步骤废液。并取上述废液稀释10倍,测量498nm吸光度,得到废液中4,4'-二甲氧基三苯甲基阳离子的浓度为3.3mM。
取1L废液,加入3.8g三乙基硅烷,室温条件下搅拌反应1h,溶液由橙红色变成淡黄色混合溶液。采用1L饱和碳酸氢钠溶液洗涤淡黄色混合溶液三次,然后再加入50g无水硫酸钠干燥过夜,过滤除去固体,并在35℃条件下真空浓缩至50ml,获得4,4'-二甲氧基三苯甲烷溶液。
将4,4'-二甲氧基三苯甲烷溶液加入4.4g N-氯代丁二酰亚胺(NCS),0.5g偶氮二异丁腈在室温条件下反应6h,再将50mL石油醚滴加入至上述反应液中,10min中滴加完,析出固体室温下搅拌30min,过滤取固体,再向固体中加入10mL石油醚打浆,过滤后固体用10mL石油醚淋洗;固体采用流动相溶解进行硅胶柱层析纯化,流动相为石油醚:乙酸乙酯=5:1,浓缩后的产物在45℃条件下干燥12h,获得4,4'-双甲氧基三苯甲基氯0.64g。
实施例4
采用AKTA 100合成仪,FineLINE 35合成柱,3%(w/v)三氯乙酸-二氯甲烷脱保护试剂,按合成仪上通用合成程序合成寡核苷酸。合成过程中单独收集脱保护步骤废液。并取上述废液稀释10倍,测量498nm吸光度,得到废液中4,4'-二甲氧基三苯甲基阳离子的浓度为3.3mM。
取1L废液,加入4.29g三苯基硅烷,室温条件下搅拌反应1h,溶液由橙红色变成淡黄色混合溶液。采用1L饱和碳酸氢钠溶液洗涤淡黄色混合溶液三次,然后再加入50g无水硫酸镁干燥过夜,过滤除去固体,并在35℃条件下真空浓缩至50ml,获得4,4'-二甲氧基三苯甲烷溶液。
将4,4'-二甲氧基三苯甲烷溶液加入4.4g N-氯代丁二酰亚胺(NCS),0.5g偶氮二异丁腈在室温条件下反应6h,再将50mL正己烷滴加入至上述反应液中,10min中滴加完,析出固体室温下搅拌30min,过滤取固体,再向固体中加入10mL正己烷打浆,过滤后固体用10mL正己烷淋洗;固体采用流动相溶解进行硅胶柱层析纯化,流动相为石油醚:乙酸乙酯=5:1,浓缩后的产物在45℃条件下干燥12h,获得4,4'-双甲氧基三苯甲基氯0.55g。
实施例5
采用AKTA 100合成仪,FineLINE 35合成柱,3%(w/v)三氯乙酸-二氯甲烷脱保护试剂,按合成仪上通用合成程序合成寡核苷酸。合成过程中单独收集脱保护步骤废液。并取上述废液稀释10倍,测量498nm吸光度,得到废液中4,4'-二甲氧基三苯甲基阳离子的浓度为3.3mM。
取1L废液,加入3.8g三乙基硅烷,室温条件下搅拌反应1h,溶液由橙红色变成淡黄色混合溶液。采用1L饱和碳酸氢钠溶液洗涤淡黄色混合溶液三次,然后再加入50g无水硫酸镁干燥过夜,过滤除去固体,并在35℃条件下真空浓缩至50ml,获得4,4'-二甲氧基三苯甲烷溶液。
将4,4'-二甲氧基三苯甲烷溶液加入1.63g 1,3-二氯-5,5-二甲基乙内酰脲,0.5g偶氮二异丁腈在室温条件下反应6h,再将50mL正己烷滴加入至上述反应液中,10min中滴加完,析出固体室温下搅拌30min,过滤取固体,再向固体中加入10mL正己烷打浆,过滤后固体用10mL正己烷淋洗;固体采用流动相溶解进行硅胶柱层析纯化,流动相为石油醚:乙酸乙酯=5:1,浓缩后的产物在45℃条件下干燥12h,获得4,4'-双甲氧基三苯甲基氯0.48g。
实施例6
采用AKTA 100合成仪,FineLINE 35合成柱,3%(w/v)三氯乙酸-二氯甲烷脱保护试剂,按合成仪上通用合成程序合成寡核苷酸。合成过程中单独收集脱保护步骤废液。并取上述废液稀释10倍,测量498nm吸光度,得到废液中4,4'-二甲氧基三苯甲基阳离子的浓度为3.3mM。
取1L废液,加入0.95g三乙基硅烷,室温条件下搅拌反应4h,溶液由橙红色变成淡黄色混合溶液。采用1L饱和碳酸氢钠溶液洗涤淡黄色混合溶液三次,然后再加入50g无水硫酸钠干燥过夜,过滤除去固体,并在35℃条件下真空浓缩至50ml,获得4,4'-二甲氧基三苯甲烷溶液。
将4,4'-二甲氧基三苯甲烷溶液加入4.4g N-氯代丁二酰亚胺(NCS),0.5g偶氮二异丁腈在60℃回流反应1h,再将50mL正庚烷滴加入至上述反应液中,10min中滴加完,析出固体室温下搅拌30min,过滤取固体,再向固体中加入10mL正庚烷打浆,过滤后固体用10mL正庚烷淋洗;固体采用流动相溶解进行硅胶柱层析纯化,流动相为石油醚:乙酸乙酯=5:1,浓缩后的产物在45℃条件下干燥12h,获得4,4'-双甲氧基三苯甲基氯0.51g。
将上述实施例获得的4,4'-双甲氧基三苯甲基氯配置成溶液,进行高效液相检测,采用标准4,4'-双甲氧基三苯甲基氯标定标准曲线,获得各实施例产物的出锋时间以及含量。
采用标准4,4'-双甲氧基三苯甲基氯配置成一系列标准溶液,(75、150、300、600、1200ug/mL浓度的乙腈溶液),进行高效液相检测,所述液相色谱检测的条件为:Waters2695-2424,Waters XBridge C18色谱柱3.5um 4.6x100mm,柱温25℃,流速1ml/min,检测波长为260nm。流动相A为乙腈,流动相B为甲醇,洗脱梯度为0-10min,20%B,获得标准曲线为:y=115459x–863604,R
回收率=[(峰面积+863604)/115459*1000]/(3.3*10
表1各实施例结果
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准,说明书及附图可以用于解释权利要求的内容。