伏诺拉生中间体的制备方法
文献发布时间:2024-04-18 19:58:21
技术领域
本发明属于药物合成技术领域,具体涉及一种伏诺拉生中间体的制备方法。
背景技术
伏诺拉生(Vorinostat,又称沃诺拉赞),是一种钾离子竞争性酸阻滞剂,用于治疗糜烂性食管炎、胃溃疡、十二指肠溃疡、幽门螺杆菌根除等,最早由日本武田药品工业株式会社开发,并于2014年在日本上市,其结构式如下:
富马酸伏诺拉生是伏诺拉生的富马酸盐,具有更好的稳定性和溶解性,日本武田药品工业株式会社在WO2007026916和CN101300229中公开了伏诺拉生及其富马酸盐的制备路线如下:
该方案以2-氟苯乙酮为起始原料,引入氰基乙酸乙酯后关环,得到中间体2-氯-5-(2-氟苯基)-1H-吡咯-3-羧酸乙酯,再经过加氢还原、氧化、芳香亲电加成后与富马酸反应后制得富马酸伏诺拉生,收率74%。
随后该公司又在2010年申请的专利WO2010098351和CN102421753中提出了制备富马酸伏诺拉生的新路线,以2-((2-氟苯基)-2-氧代乙基)丙二腈为起始原料,制得中间体2-(2-氟苯基)-1H-吡咯-3-腈,再经过加氢还原、氧化、加成,最终经富马酸成盐,制得富马酸伏诺拉生。
山东金城医药股份有限公司在2016年申请的专利CN106366071中公开了富马酸伏诺拉生如下合成方法:2-氟苯乙酮首先与烯丙基胺进行缩合反应,然后在铜催化剂催化下进行关环反应,得到中间体3-甲基-5-(2-氟苯基)-1H-吡咯,再经过磺酰胺化、溴代,胺化反应后,与富马酸成盐,得到富马酸伏诺拉生。
上述制备方法中,均使用了中间体2-(2-氟苯基)-1H-吡咯或5-(2-氟苯基)-1H-吡咯关键中间体,但制备该关键中间体的路线比较复杂,副产物多,转化不充分,不利于产品的收率和纯度的提升。
日本化学药品株式会社在WO2019131695和CN111527067中公开了富马酸伏诺拉生的一种制备方法,以邻氟苯衍生物为原料,经钯催化剂催化,与吡咯反应得到中间体2-(2-氟苯基)四氢吡咯,再对吡咯环的氮原子进行保护,进一步甲酰化得到吡咯-3-甲醛衍生物,之后脱保护得到5-(2-氟苯基)-1H-吡咯-3-甲醛,然后在碱存在下与吡啶-3-磺酰氯或其盐反应,与甲胺还原胺化后,得到1-[5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1H-吡咯-3-基]-N-甲基甲胺,最后与富马酸成盐,得到富马酸伏诺拉生。该路线对原有路线进行了简化,但需要用到昂贵的钯催化剂催化。
因此,需要提供一种针对上述现有技术不足的改进技术方案。
发明内容
本发明的目的在于提供一种伏诺拉生中间体的制备方法,用来获得伏诺拉生的中间体2-(2-氟苯基)-1H-吡咯,具有合成路线短、易纯化、收率高的优点,以解决现有的合成路线副产物多、成本较高的问题。
为了实现上述目的,本发明提供如下技术方案:
一种伏诺拉生中间体的制备方法,包括如下步骤:
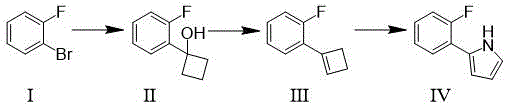
S1、以邻溴氟苯为起始原料,与镁试剂或锂试剂反应后,再与环丁酮反应,得到式Ⅱ所示化合物;
S2、化合物Ⅱ与对甲苯磺酸反应后使用碳酸氢钠淬灭,得到式Ⅲ所示化合物;
S3、化合物Ⅲ与乙酸铵反应,得到式Ⅳ所示的伏诺拉生中间体;
反应过程如下:
优选的,步骤S1中,将邻溴氟苯溶解在四氢呋喃中,然后与镁试剂或锂试剂反应。
优选的,步骤S1中,所述镁试剂包括异丙基氯化镁-氯化锂或镁屑中的至少一种,所述锂试剂为正丁基锂。
优选的,步骤S1中,在≤-5℃的温度下滴加环丁酮,然后停止冷却。
优选的,步骤S1中,反应时间不低于1小时。
优选的,步骤S1中,反应结束后,使用饱和氯化铵水溶液淬灭,然后经过萃取、洗涤、干燥、过滤、浓缩后柱色谱分离,得到化合物Ⅱ。
优选的,步骤S2中,将化合物Ⅱ溶解在甲醇中,加入对甲苯磺酸一水合物,待化合物Ⅱ反应完全后,添加饱和碳酸氢钠水溶液淬灭,然后经过萃取、洗涤、干燥、过滤后真空浓缩,得到化合物Ⅲ。
优选的,步骤S3中,将化合物Ⅲ溶解在有机溶剂中,加入Co(acac)
所述有机溶剂选自二甲醚、甲苯、二甲苯、1,4-二氧六环中的至少一种。
优选的,步骤S3中,所述Co(acac)
优选的,步骤S3中,反应温度为60~100℃,反应时间为3~8小时。
有益效果:
本发明的目的在于提供一种伏诺拉生关键中间体的合成方法,采用商业化的原料,只需三步反应即可得到中间体2-(2-氟苯基)-1H-吡咯,相对于现有的合成路线更短,且每一步反应的产物产率均可达到80%以上,反应过程中无需使用贵金属催化剂,可显著降低伏诺拉生的制备成本,具有良好的经济效益,相对现有方法具有明显的经济优势,适合工业化生产。
附图说明
构成本申请的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。其中:
图1为本发明的伏诺拉生中间体合成路线图。
图2为本发明实施例1制备的化合物Ⅱ的核磁共振氢谱谱图。
图3为本发明实施例3制备的化合物Ⅳ的核磁共振氢谱谱图。
图4为本发明实施例3制备的化合物Ⅳ的质谱图。
图5为本发明实施例3制备的化合物Ⅳ的HPLC检测结果。
图6为本发明实施例4制备的化合物Ⅴ的核磁共振氢谱谱图。
图7为本发明实施例5制备的化合物Ⅵ的核磁共振氢谱谱图。
图8为本发明实施例6制备的富马酸伏诺拉生的核磁共振氢谱谱图。
具体实施方式
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员所获得的所有其他实施例,都属于本发明保护的范围。
下面将结合实施例来详细说明本发明。需要说明的是,在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。
本发明针对目前伏诺拉生合成方法存在的合成路线长、副产物多的问题,提供一种伏诺拉生中间体的制备方法,包括如下步骤:
S1、以邻溴氟苯为起始原料,与镁试剂或锂试剂反应后,再与环丁酮反应,得到式Ⅱ所示化合物;
S2、化合物Ⅱ与对甲苯磺酸反应后使用碳酸氢钠淬灭,得到式Ⅲ所示化合物;
S3、化合物Ⅲ与乙酸铵反应,得到式Ⅳ所示的伏诺拉生中间体;
反应过程如图1所示。
本发明方法以邻溴氟苯为起始原料,经过镁试剂或锂试剂活化,再与环丁酮反应得到1-(2-氟苯基)环丁烷-1-醇,然后进一步缩合获得碳-碳双键,最后通过与乙酸铵反应,使环丁烯直接转化为吡咯环,反应过程中不需要经过关环反应,也不需要用到昂贵的钯催化剂,适合工业化生产。
本发明提供的伏诺拉生中间体的制备方法,
本发明优选实施例中,步骤S1中,将邻溴氟苯溶解在四氢呋喃中,然后与镁试剂或锂试剂反应。
本发明优选实施例中,步骤S1中,镁试剂包括异丙基氯化镁-氯化锂或镁屑中的至少一种,锂试剂为正丁基锂。
本发明优选实施例中,步骤S1中,在≤-5℃(例如-5℃、-6℃、-7℃、-13℃、-8℃、-9℃、-10℃、-11℃)的温度下滴加环丁酮,然后停止冷却。
本发明优选实施例中,步骤S1中,反应时间不低于1小时,优选为1~3小时(例如1小时、1.5小时、2.0小时、2.5小时、3.0小时)。
本发明优选实施例中,步骤S1中,反应结束后,使用饱和氯化铵水溶液淬灭,然后经过萃取、洗涤、干燥、过滤、浓缩后柱色谱分离,得到化合物Ⅱ。
本发明优选实施例中,步骤S2中,将化合物Ⅱ溶解在甲醇中,加入对甲苯磺酸一水合物,待化合物Ⅱ反应完全后,添加饱和碳酸氢钠水溶液淬灭,然后经过萃取、洗涤、干燥、过滤后真空浓缩,得到化合物Ⅲ。
本发明优选实施例中,步骤S3中,将化合物Ⅲ溶解在有机溶剂中,加入Co(acac)
有机溶剂选自二甲醚、甲苯、二甲苯、1,4-二氧六环中的至少一种。
本发明优选实施例中,步骤S3中,Co(acac)
本发明优选实施例中,步骤S3中,反应温度为60~100℃(例如61℃、65℃、70℃、75℃、80℃、85℃、90℃、95℃、99℃),反应时间为3~8小时(例如3.5小时、4.0小时、4.5小时、5.0小时、5.5小时、6.0小时、6.5小时、7.0小时、7.5小时)。
下面通过具体实施例对本发明伏诺拉生中间体的制备方法进行详细说明。
下列实施例中,所使用的药品来源如下:
邻溴氟苯,购自 上海毕得医药科技股份有限公司;
四氢呋喃,购自上海泰坦科技股份有限公司;
异丙基氯化镁-氯化锂,购自上海泰坦科技股份有限公司;
环丁酮,购自上海泰坦科技股份有限公司;
氯化铵,购自上海泰坦科技股份有限公司,使用去离子水配制成浓度为1-5M的水溶液;
乙酸乙酯,购自上海泰坦科技股份有限公司;
无水硫酸镁,购自上海泰坦科技股份有限公司;
甲醇,购自上海泰坦科技股份有限公司;
对甲苯磺酸一水合物,购自上海泰坦科技股份有限公司;
碳酸氢钠,购自上海泰坦科技股份有限公司;
二甲醚,购自上海泰坦科技股份有限公司;
Co(acac)
DPEphos,又称双(2-二苯基磷苯基)醚,购自上海泰坦科技股份有限公司;
乙酸铵,购自上海泰坦科技股份有限公司;
TMSiA,又称叠氮三甲基硅,购自上海泰坦科技股份有限公司;
4-甲基四氢吡喃,购自上海泰坦科技股份有限公司;
1,3-二甲基-2-咪唑啉酮,购自上海泰坦科技股份有限公司;
氢化钠,购自上海泰坦科技股份有限公司;
三异丙基氯硅烷,购自上海泰坦科技股份有限公司;
Vilsmeier试剂,购自上海泰坦科技股份有限公司;
环己烷,购自上海泰坦科技股份有限公司;
DMAP,购自上海泰坦科技股份有限公司;
3-磺酰氯吡啶,购自上海毕得医药科技股份有限公司;
N,N-二异丙基乙胺,购自上海泰坦科技股份有限公司;
乙腈,购自上海泰坦科技股份有限公司;
甲胺-甲醇溶液,纯度30%,生产商山东道合药业有限公司;
硼氢化钠,生产商山东道合药业有限公司;
二氯甲烷,购自上海泰坦科技股份有限公司;
富马酸,购自上海泰坦科技股份有限公司;
柱色谱,固定相为200-300目硅胶,流动相为石油醚和乙酸乙酯。
实施例1
1-(2-氟苯基)环丁烷-1-醇的合成:
将邻溴氟苯(1eq)溶于四氢呋喃中,在-5到-8℃的温度下搅拌20分钟,随后加入异丙基氯化镁-氯化锂(1.5eq),在-5到-8℃的温度下继续搅拌10分钟,随后滴加环丁酮(2eq),然后移至常温环境,搅拌1.5小时,TLC监测反应,待邻溴氟苯反应完全后,滴加饱和氯化铵水溶液淬灭,再用乙酸乙酯萃取,合并有机相,有机相经饱和食盐水洗涤,再用无水硫酸镁干燥后过滤,将滤液在真空中浓缩,通过柱色谱分离得到无色油状液体,即为目标化合物Ⅱ,产率87%。
使用核磁共振对制备的化合物Ⅱ进行检测,如图2所示。
实施例2
1-(环丁-1-烯-1-基)-2-氟苯的合成:
将1-(2-氟苯基)环丁烷-1-醇(1eq)溶解在甲醇中,加入对甲苯磺酸一水合物(2eq),反应3个小时,TLC检测到化合物Ⅱ反应完全后,滴加饱和碳酸氢钠水溶液进行淬灭反应,并用乙酸乙酯萃取,合并有机相,再经饱和食盐水洗涤,无水硫酸镁干燥、过滤,滤液在真空中浓缩,得到无色油状液体,即为目标化合物Ⅲ,产率93%。
本实施例制备的化合物Ⅲ无需进一步纯化,可直接进行后续反应。
实施例3
2-(2-氟苯基)-1H-吡咯的合成:
将实施例2制备的1-(环丁-1-烯-1-基)-2-氟苯(1eq)溶解在二甲醚中,加入0.1eqCo(acac)
化合物Ⅳ的核磁共振、质谱、HPLC检测结果分别如图3、4、5所示。
化合物Ⅳ可用于进一步制备伏诺拉生及其富马酸盐,使用化合物Ⅳ制备富马酸伏诺拉生的方法,可参考如下实施例4~6,也可按照现有的其它制备方法进行。
实施例4
5-(2-氟苯基)-1H-吡咯-3-甲醛的合成:
将2-(2-氟苯基)-1H-吡咯(1eq)溶于4-甲基四氢吡喃中,随后加入1,3-二甲基-2-咪唑啉酮(3eq),冰浴下搅拌3分钟后,加入氢化钠(1.2eq),继续冰浴搅拌5分钟,随后滴加三异丙基氯硅烷(1.1eq),冰浴搅拌,直到TLC监测到2-(2-氟苯基)-1H-吡咯点消失后,加入Vilsmeier试剂,逐渐加热到70℃,反应5小时,随后撤除加热,恢复常温后,加入2M氢氧化钠水溶液脱保护,再经过饱和食盐水洗涤,乙酸乙酯萃取,合并有机相后,减压蒸馏,随后残渣继续加入适量乙酸乙酯,解热溶解大部分残渣后,加入环己烷,冰浴搅拌至固体析出,过滤即得目标化合物,白色固体,产率67%。
实施例5
5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1H-吡咯-3-甲醛的合成:
将5-(2-氟苯基)-1H-吡咯-3-甲醛(1eq)、DMAP(0.15eq)、3-磺酰氯吡啶溶解在四氢呋喃溶液中,随后加入N,N-二异丙基乙胺(0.15eq),升温到40℃,搅拌2小时。然后将温度冷却至常温,使用1M盐酸调节pH至4-5之间,加入去离子水,出现固体析出后,继续搅拌1小时并过滤,滤饼用乙腈-水混合溶液洗涤,减压干燥后即得目标产物,产率87%。
实施例6
富马酸伏诺拉生的合成:
将5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1H-吡咯-3-甲醛(1eq)使用甲醇溶解,然后将甲胺-甲醇溶液(1.3eq)滴入反应液中,常温下反应1小时,随后降温至0-10℃,向反应液中加入硼氢化钠固体(0.45eq),继续低温反应1小时,待反应完全,加入5%盐酸溶液调pH值至8-9,饱和氯化钠洗涤,二氯甲烷萃取,合并有机相,减压浓缩后得粗品伏诺拉生,随后直接加入70%的甲醇水溶液溶解粗品,升温至70℃,加入富马酸,搅拌半小时后在0℃下继续搅拌2小时,最后经抽滤、滤饼真空干燥,即得富马酸伏诺拉生固体,收率92.57%。
使用核磁共振对制备的富马酸伏诺拉生样品进行检测,结果如图8所示。
本发明提供的伏诺拉生中间体的合成方法,只需三步反应即可得到中间体2-(2-氟苯基)-1H-吡咯,相对于现有的合成路线更短,且每一步反应的产物产率均可达到80%以上,反应过程中无需使用贵金属催化剂,可显著降低伏诺拉生的制备成本,具有良好的经济效益,适合在产业上应用。
以上所述仅为本发明的优选实施例,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种富马酸伏诺拉生中间体的制备方法
- 一种富马酸伏诺拉生中间体的制备方法