一种基于生物电子等排体搜索平台的数据处理方法
文献发布时间:2023-06-19 19:35:22
技术领域
本发明涉及药物设计技术领域,具体涉及一种基于生物电子等排体搜索平台的数据处理方法。
背景技术
根据相关文献的阅读,可以得知为了帮助研究人员选择生物电子等排体,目前国内外已经开发了多种用于查询生物电子等排体的程序、数据库和网页服务器等计算工具。然而,虽然目前已有的多种生物电子等排体搜索工具所使用的技术以及数据库各不相同,但用户利用这些工具查询某些化学基团的生物电子等排体时,能够查询到数量很少或几乎没有。
此外,随着药物研发成本的增加,对生物电子等排体的需求也越来越多,然而目前关于生物电子等排体的信息仍然存在不足。在过去,确定合适的生物电子等排基团并非易事,需要大量的试验。由于计算工具提供的只是可能的生物电子等排体,所以能够有效的提供越多的生物电子等排体,则工具越有效。
发明内容
本发明的目的在于提供一种基于生物电子等排体搜索平台的数据处理方法,以能够促进药物研发中先导化合物的优化进程加快,减少药物研发时间和成本。
本发明解决上述技术问题的技术方案如下:
本发明提供一种基于生物电子等排体搜索平台的数据处理方法,所述基于生物电子等排体搜索平台的数据处理方法包括:
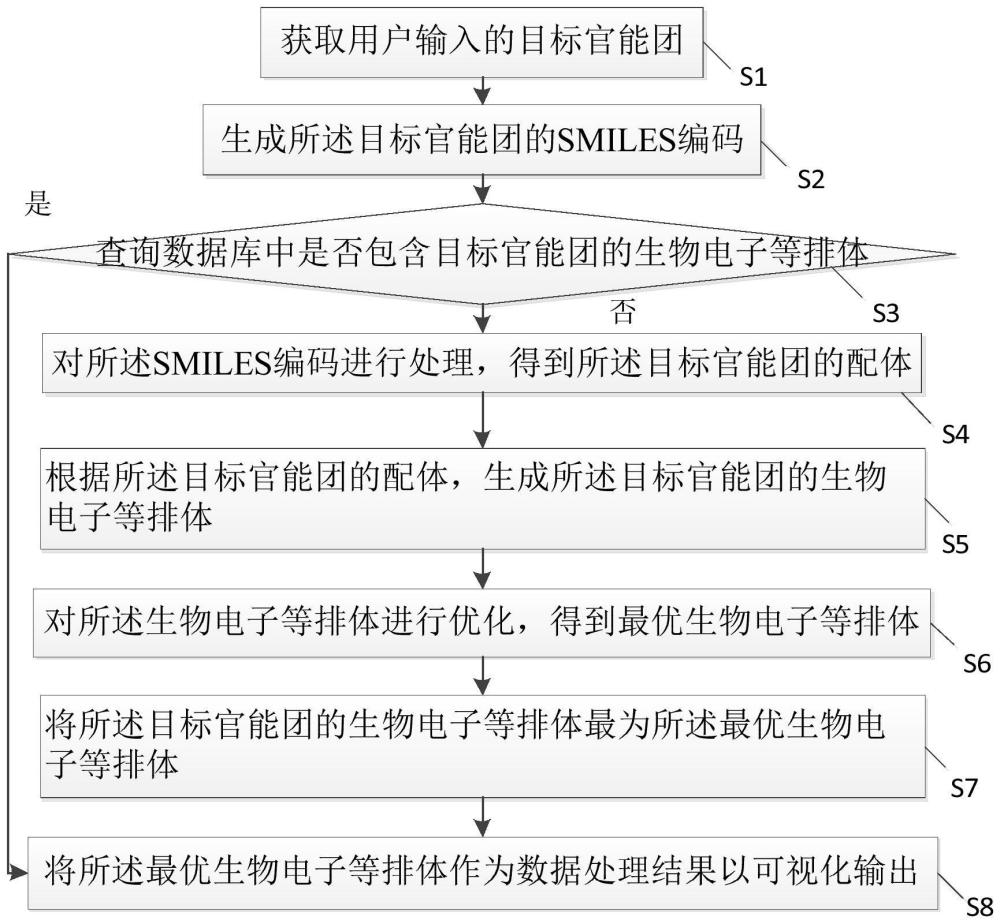
S1:获取用户输入的目标官能团;
S2:生成所述目标官能团的SMILES编码;
S3:根据所述SMILES编码查询存放生物电子等排体的数据库中是否包含目标官能团的生物电子等排体,若是,进入步骤S8,否则,进入步骤S4;
S4:对所述SMILES编码进行处理,得到所述目标官能团的配体;
S5:根据所述目标官能团的配体,生成所述目标官能团的生物电子等排体;
S6:对所述生物电子等排体进行优化,得到最优生物电子等排体;
S7:将所述目标官能团的生物电子等排体最为所述最优生物电子等排体;
S8:将所述最优生物电子等排体作为数据处理结果以可视化输出。
可选择地,所述步骤S1中,所述目标官能团包括需要查询生物电子等排体的化学基团。
可选择地,所述步骤S4包括:
S41:将所述SMILES编码中的通配符*替换为R,得到替换后的SMILES编码;
S42:将所述替换后的SMILES编码转换为mol格式并做加氢处理,得到新的SMILES编码;
S43:从所有配体中查询所述新的SMILES编码的配体;
S44:将所述新的SMILES编码的配体作为所述目标官能团的配体。
可选择地,所述步骤S5包括:
S51:将所述目标官能团的配体所存在的蛋白确定为参考蛋白;
S52:对所述参考蛋白进行预处理,得到预处理后的参考蛋白子链;
S53:将所述预处理后的参考蛋白子链的FASTA序列输入Blastp工具中,与蛋白质序列数据库进行比较,得到与所述预处理后的参考蛋白子链相似的相似蛋白子链;
S54:重叠所述参考蛋白子链和所述相似蛋白子链,得到重叠后的结构;
S55:以所述重叠后的结构的各个原子为中心,获取指定半径内相似蛋白的原子,即为生物电子等排体。
可选择地,所述S52包括:
S521:判断所述参考蛋白的分辨率,保留分辨率高于
S522:利用序列对比算法对所有所述分辨率高于
S523:两两比对所有可用参考蛋白子链的FASTA序列的一致性,如果相同,保留分辨率数值小参考蛋白子链,删除分辨率数值大的参考蛋白子链;若不同,则全部保留,得到预处理后的参考蛋白子链。
可选择地,所述S6包括:利用ShaEP计算目标官能团与所述生物电子等排体叠加后的相似性指数,并设置阈值进行筛查,以得到最优生物电子等排体。
可选择地,在所述S8之前,所述基于生物电子等排体搜索平台的数据处理方法还包括:
对所述最优生物电子等排体进行后处理。
可选择地,所述后处理包括:
A1:将所述最优生物电子等排体进行格式转换操作,得到转换后的数据;
A2:判断转换后的数据是否完整,若是,进入A4,否则,进入A3;
A3:对所述转换后的数据进行数据清洗并进入A4;
A4:计算所述转换后的数据的分子指纹,并根据所述分子指纹计算分子间的相似性,得到相似性矩阵;
A5:根据所述相似性矩阵,利用机器学习无监督聚类算法对所述转换后的数据进行数据聚类,得到聚类结果;
A6:将所述聚类结果作为所述数据处理结果以可视化输出。
可选择地,所述A4包括:
利用RDKit计算所述分子指纹和分子间的相似性。
可选择地,所述A5包括:
根据所述相似性矩阵,对所述机器学习无监督聚类算法进行参数调整,以得到最优超参数;
根据所述最优超参数确定聚类数;
根据所述聚类数对所述转换后的数据进行数据聚类,得到聚类结果。
本发明具有以下有益效果:
(1)本发明可以使药物化学家不再需要通过反复试验来获取少量的生物电子等排体,仅通过搜索便能得到大量的生物电子等排体信息,从而能够促进药物研发中先导化合物的优化进程加快,减少药物研发时间和成本;
(2)本发明能够查询到较多的生物电子等排体结果,得到对应的信息也更多,而且提供了多种无监督聚类方法对搜索得到的生物等排体进行聚类,为用户提供了更加广阔的思考空间和角度。
附图说明
图1为本发明基于生物电子等排体搜索平台的数据处理方法的流程图;
图2为生物电子等排体无监督聚类算法参数优化结果示意图;
图3为生物电子等排体无监督聚类的可视化结果示意图。
具体实施方式
以下结合附图对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
本发明提供一种基于生物电子等排体搜索平台的数据处理方法,参考图1所示,所述基于生物电子等排体搜索平台的数据处理方法包括:
S1:获取用户输入的目标官能团;
本发明所述的目标官能团即为需要查询的生物电子等排体的化学基团。
S2:生成所述目标官能团的SMILES编码;
作为一种实施方式,本领域技术人员可在Django(开放源代码的Web应用框架)前端嵌入的化学分子编辑器JSME中绘制需要查询生物电子等排体的化学基团,Django自行生成目标官能团的SMILES编码。
S3:根据所述SMILES编码查询存放生物电子等排体的数据库中是否包含目标官能团的生物电子等排体,若是,进入步骤S8,否则,进入步骤S4;
S4:对所述SMILES编码进行处理,得到所述目标官能团的配体;
可选择地,所述S4包括:
S41:将所述SMILES编码中的通配符*替换为R,得到替换后的SMILES编码;
S42:将所述替换后的SMILES编码转换为mol格式并做加氢处理,得到新的SMILES编码;
S43:从所有配体中查询所述新的SMILES编码的配体;
S44:将所述新的SMILES编码的配体作为所述目标官能团的配体。
S5:根据所述目标官能团的配体,生成所述目标官能团的生物电子等排体;
可选择地,所述S5包括:
S51:将所述目标官能团的配体所存在的蛋白确定为参考蛋白;
即从蛋白质数据库中获取含有参考配体的所有蛋白质PDB文件,将含有参考配体的蛋白质定义为参考蛋白。
S52:对所述参考蛋白进行预处理,得到预处理后的参考蛋白子链;
这里,预处理包括:
S521:判断所述参考蛋白的分辨率,保留分辨率高于
其中在结构解析中,分辨率对应最小可观察特征的距离,在X射线晶体学中,分辨率对应在衍射图中分辨的晶格面之间的最小距离。较大的分辨率数值表示较低的分辨率,反之较小的分辨率数值表示较高的分辨率。总而言之,分辨率是衡量蛋白、核酸晶体数据质量的一种度量指标。
S522:利用序列对比算法对所有所述分辨率高于
具体利用利用Needleman-Wunsch序列比对算法进行对比,对比命令为:needle-asequence seq1.fasta-bsequence seq2.fasta-outfileout.needle-gapopen10-gapextend 0.5-sprotein。除非指定,否则needle可能根据IUPAC字母猜测比对的是核酸还是蛋白,IUPAC是系统命名化学物质的方法,所以命令使用“-sprotein”标志来表示正在比对的是蛋白质序列,防止抛出错误,命令执行完毕后得到并分析序列比对结果文件,选择性保留数据。对两条蛋白质子链经过全局比对后得到的文件,需要查看文件中Identity的数值是否相同,不同的话两条子链均保留,相同的话保留一条链的数据即可。
S523:两两比对所有可用参考蛋白子链的FASTA序列的一致性,如果相同,保留分辨率数值小参考蛋白子链,删除分辨率数值大的参考蛋白子链;若不同,则全部保留,得到预处理后的参考蛋白子链。
同样的,本发明利用Needleman-Wunsch序列比对算法两两对比所有可用参考蛋白子链的FASTA序列的一致性。
S53:将所述预处理后的参考蛋白子链的FASTA序列输入Blastp工具中,与蛋白质序列数据库进行比较,得到与所述预处理后的参考蛋白子链相似的相似蛋白子链;
S54:重叠所述参考蛋白子链和所述相似蛋白子链,得到重叠后的结构;
S55:以所述重叠后的结构的各个原子为中心,获取指定半径内相似蛋白的原子,即为生物电子等排体。
即将处理后的蛋白质单链FASTA序列输入Blastp工具中与蛋白质序列数据库进行比较,检测具有高度相似性的蛋白质链。将输入Blastp前后的相似蛋白链利用US-align工具进行重叠比对,在重叠后的结构中以目标官能团的各个原子为中心,获取指定半径范围内的所有原子,该指定半径范围内的所有原子即为生物电子等排体。
S6:对所述生物电子等排体进行优化,得到最优生物电子等排体;
即:利用ShaEP计算目标官能团与所述生物电子等排体叠加后的相似性指数,并设置阈值进行筛查,以得到最优生物电子等排体。
S7:将所述目标官能团的生物电子等排体最为所述最优生物电子等排体;
S8:将所述最优生物电子等排体作为数据处理结果以可视化输出。
可选择地,在所述S8之前,所述基于生物电子等排体搜索平台的数据处理方法还包括:
对所述最优生物电子等排体进行后处理。
可选择地,所述后处理包括:
A1:将所述最优生物电子等排体进行格式转换操作,得到转换后的数据;
即将SMILES编码格式转换为SDF格式。
A2:判断转换后的数据是否完整,若是,进入A4,否则,进入A3;
Sdf文件具有标准的格式,通过编写判定代码来检查是否符合标准格式,从而判断数据是否完整。
A3:对所述转换后的数据进行数据清洗并进入A4;
这里,数据清洗包括无效数据的删除和重复数据的删除。作为一种实施方式,本发明通过计算流程获得对应的2228个分子数据后,需要对2228个数据进行数据清洗,首先通过编写代码删除掉无效数据后得到1236个数据点,再删除重复数据得到1157个可用数据。
A4:计算所述转换后的数据的分子指纹,并根据所述分子指纹计算分子间的相似性,得到相似性矩阵;
本发明利用RDKit计算所述分子指纹和分子间的相似性。
使用RDKit方法计算这些数据点的2048位长度的摩根分子指纹(MFF),并根据MFF计算各个分子的“Tanimoto相似性系数”以此来获得分子间相似性矩阵
A5:根据所述相似性矩阵,利用机器学习无监督聚类算法对所述转换后的数据进行数据聚类,得到聚类结果;
即以1157个数据的相似性系数组成分子相似性矩阵作为模型输入,通过机器学习无监督聚类算法对数据进行自动分类。
需要说明的是,机器学习无监督聚类算法包括需要调整超参数聚类数目的类型和无需调整超参数的类型。需要调整超参数的算法类型以K-means为例进行说明:K-means算法需要指定超参数“聚类数目”(n_components)”,因此需要对此超参数在一定范围内进行搜索来获得最佳值,用来评估该算法聚类效果的指标称为轮廓系数(SilhouetteCoefficient),在参数空间2~150范围内进行超参数搜索,获得超参数与轮廓系数的关系,依据“手肘法则”选取图中斜率急剧变化区域对应的超参数范围5~20如图2(a),之后进一步选择参数如图2(b),通过计算可知选择超参数为6,即聚类数为6时,无监督聚类的分类效果最好。
因此,若本领域技术人员选择需要调整超参数的算法类型,则:
根据所述相似性矩阵,对所述机器学习无监督聚类算法进行参数调整,以得到最优超参数;
根据所述最优超参数确定聚类数;
根据所述聚类数对所述转换后的数据进行数据聚类,得到聚类结果。
而需调整超参数的算法类型以DBSCAN为例进行说明:DBSCAN依据数据密度进行聚类,在进行DBSCAN无监督聚类时只需设定初始参数(eps=1.5,min_samples=3)无需设定超参数聚类数目,之后便可以直接进行无监督聚类,聚类结果显示当聚类数为142类时,达到最好的聚类效果,此时轮廓系数为0.216。
在通过无监督聚类算法进行聚类后,还需对数据进行可视化以便给用户直观展现聚类结果。分别使用TSNE和PCA对2048位的分子指纹降维至2维和3维,之后通过matplotlib对降维后的数据进行二维和三维图的绘制,图中分别用不同的颜色表示不同的类别,如图3。
A6:将所述聚类结果作为所述数据处理结果以可视化输出。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种基于大数据的网络舆情数据采集、处理方法及监测平台
- 一种基于区块链的数据处理平台及数据处理方法
- 一种基于连通器方式的一体化A/O膜生物反应器以及污水处理方法
- 一种基于教育云平台的缓存数据的方法及电子设备
- 一种基于深度搜索的税务智能咨询平台的数据处理方法
- 一种基于搜索引擎的数据处理方法及平台