抗肿瘤活性分子苯并b氮杂卓衍生物及其合成方法和应用
文献发布时间:2024-04-18 19:58:53
技术领域
本发明属于苯并[b]氮杂卓衍生物的合成,具体涉及一种抗肿瘤活性分子苯并[b]氮杂卓衍生物的合成方法。
背景技术
苯并氮杂卓分子片段在一系列生物学和药理学上重要的化合物中作为基本结构元素发挥着至关重要的作用(Le Diguarher,T.;Ortuno,J.-C.;Sh anks,D.;Guilbaud,N.;Pierré,A.;Raimbaud,E.;Fauchère,J.-L.;Bioorg*.Med.Chem.Lett.2004,14,767-771;Kondo,K.;Ogawa,H.;Shinohara,T.;Kurimura,M.;Tanada,Y.;Kan,K.;Yamashita,H.;Nakamura,S.;Hira no,T.;Yamamura,Y.;Mori,T.;Tominaga,M.;Itai,A*.J.Med.Chem.2000,43,4388-4397;Failli,A.A.;Shumsky,J.S.;Steffan,R.J.;Caggiano,T.J.;Wi lliams,D.K.;Trybulski,E.J.;Ning,X.;Lock,Y.;Tanikella,T.;Hartmann,D.;Chan,P.S.;Park,C.H.Bioorg.Med.Chem.Lett.2006,16,954-959.)。值得注意的是,苯并[b]氮杂卓分子片段是各种抗抑郁药物的核心组成部分,如贝那普利、莫扎普坦、依他平、达伦西平和二苯并氮杂卓(Hou,F.F.;Zhang,X.;Zhang,G.H.;Xie,D.;Chen,P.Y.;Zhang,W.R.;Jiang,J.P.;Liang,M.;Wang,G.B.;Liu,Z.R.;Geng,R.W.N.Engl.J.Med.2006,354,131-140;Decaux,G.;Soupart,A.;Vassart,G.The Lancet 2008,371,1624-1632;Groth,C.;Alvord,W.G.;Quinones,O.A.;Fortini,M.E.;Mol.Pharmaco l.2010,77,567-574;C.M.;Allan,C.E.;Ashworth,D.M.;Barne,J.;Bax ter,A.J.;Broadbridge,J.D.;Franklin,R.J.;Hampton,S.L.;Hudson,P.;Horton,J.A.;Jenkins,P.D.;Penson,A.M.;Pitt,G.R.W.;Riviere,P.;Ro bson,P.A.;Rooker,D.P.;Semple,G.;Sheppard,A.;Haigh,R.M.;Roe,M.B.J.Med.Chem.2008,51,8124-8134.)。目前发展的方法主要存在合成步骤繁琐、反应条件苛刻且底物结构受限等问题。
发明内容
由于上述已知方法的局限性,以及苛刻的反应条件,亟需开发一种简单高效,反应条件温和的实用型方法。针对现有技术的不足,本发明提供一种通用、简便、高效的一步合成苯并[b]氮杂卓及其衍生物活性分子的合成方法。该方法通过单电子转移和氢原子转移相结合的分子内自由基接力串联环化反应,在温和的条件下合成了一系列苯并[b]氮杂卓及其衍生物的活性分子。这种方法是在单电子转移过程后紧跟氢原子转移步骤,以产生新的自由基中间体,并通过自由基在芳基到烷基到乙烯基的接力过程最终合成含有苯并[b]氮杂卓分子片段的结构,这种特定的反应模式是极其罕见的。
本发明采用的技术方案为:
一种抗肿瘤活性的苯并[b]氮杂卓衍生物活性分子的合成方法,具体步骤如下:将炔酰胺化合物(1),使用过渡金属Pd催化剂,在配体、碱性物质和溶剂存在下,在氮气气氛中,在波长为420~500nm的可见光照射下,生成苯并[b]氮杂卓衍生物活性分子,如通式(2)所示;
R
R
R
X为氧原子、氮原子或带取代基的碳原子;
Y为氧原子、氮原子或带取代基的碳原子;
进一步的,过渡金属Pd催化剂为醋酸钯、双(二亚芐基丙酮)钯、三氟乙酸钯、双(2-二苯基膦苯基)醚二氯化钯、双三苯基磷二氯化钯、双三苯基磷醋酸钯、四(三苯基膦)钯、双(1,2-双(二苯基膦)乙烷)钯中的一种或多种组合,其中优选四(三苯基膦)钯或/和双(1,2-双(二苯基膦)乙烷)钯。
进一步的,配体为1,1'-联萘-2,2'-双二苯膦,双(2-二苯基膦苯基)醚、4,5-双二苯基膦-9,9-二甲基氧杂蒽、1,3-双(二苯基膦)丙烷、1,2-双(二苯膦)乙烷、1,1'-双(二苯基膦)二茂铁、三苯基膦、2,2'-联吡啶,其中优选双(2-二苯基膦苯基)醚。
进一步的,碱性物质为磷酸钾、碳酸钾、碳酸铯、氧化银、乙酸钠、磷酸钾、叔丁醇钠、乙醇钠、甲醇钠、N,N-二己基甲胺,其中优选甲醇钠。
进一步的,溶剂为苯、三氟甲苯、四氢呋喃、1,4-二氧六环,其中优选苯。
进一步的,反应温度为0℃~100℃,其中优选为25℃。
进一步的,反应时间为2~48h,其中优选为12h。
进一步的,所述炔酰胺和金属钯催化剂的摩尔比例为1:0.01~1:0.2,其中优选为1:0.1。
进一步的,所述金属钯催化剂与配体的摩尔比例为1:0~1:4,其中优选为1:1。
进一步的,所述炔酰胺和碱的摩尔比例为1:0~1:5,其中优选为1:2。
进一步的,所述化合物1在溶剂中的浓度为0.01~0.2M,其中优选为0.05M。
进一步的,还包括纯化步骤:反应结束后,将混合物用水洗涤并有机相萃取,合并有机相,无水硫酸钠干燥并减压浓缩,用柱层析纯化。
优选地,物料添加方法包括:先将通式(1)的炔酰胺、配体、碱性物质加入反应瓶;再在氮气环境中加入金属催化钯催化剂和溶剂。
优选地,反应过程中辅以剧烈搅拌。
通式(2)化合物,优选为以下具体通式(2)化合物:
制备的抗肿瘤活性分子苯并[b]氮杂卓衍生物在制备抗肿瘤药物中的应用。
与现有技术相比,本发明的有益效果为:(1)通过过渡金属Pd催化实现芳基到烷基到乙烯基的自由基接力串联环化反应合成含有苯并[b]氮杂卓分子片段的结构,这种特定的反应模式是极其罕见的。目前没有报导过使用该方法合成此类分子。
(2)本发明方法操作简单,具有优秀的的底物普适性,所合成的一系列苯并[b]氮杂卓分子及其衍生物活性分子经过测试具有抗肿瘤活性。
附图说明
图1为苯并[b]氮杂卓衍生物在不同肿瘤细胞系中的抗增殖表型筛选图。
图2为苯并[b]氮杂卓衍生物2u的线性回归分析图。
图3为苯并[b]氮杂卓衍生物2n的线性回归分析图。
具体实施方式
下面结合实施例,对本发明的具体实施方式作进一步详细描述。
实施例1
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1a 83.4mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2a(产物50.6mg,收率87%)。
对采用上述合成方法得到的目标产物2a的进行核磁共振氢谱和碳谱检测,测试结果如下:
研究改变反应条件(单一变量)对收率的影响,具体如下:
实施例2
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1b 89.5mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2b(产物42.3mg,收率66%)。
对采用上述合成方法得到的目标产物2b的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例3
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1c 86.2mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2c(产物37.6mg,收率62%)。
对采用上述合成方法得到的目标产物2c的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例4
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1d 87.1mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2d(产物46.0mg,收率75%)。
对采用上述合成方法得到的目标产物2d的进行核磁共振氢谱、碳谱和氟谱检测,测试结果如下:
实施例5
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1e 90.3mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2e(产物46.9mg,收率73%)。
对采用上述合成方法得到的目标产物2e的进行核磁共振氢谱、碳谱和氟谱检测,测试结果如下:
实施例6
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1f97.1 mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2f(产物42.0mg,收率55%)。
对采用上述合成方法得到的目标产物2f的进行核磁共振氢谱、碳谱和氟谱检测,测试结果如下:
实施例7
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1g 95.0mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2g(产物34.0mg,收率49%)。
对采用上述合成方法得到的目标产物2g的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例8
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1h 90.3mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2h(产物34.0mg,收率53%)。
对采用上述合成方法得到的目标产物2h的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例9
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1i 94.7mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2i(产物60.0mg,收率88%)。
对采用上述合成方法得到的目标产物2i的进行核磁共振氢谱和碳谱检测,测试结果如下:δ7.34(d,J=8.0Hz,2H),7.29-7.23(m,2H),7.12(d,J=8.0Hz,2H),7.04-6.98(m,2H),3.44(s,3H),2.84(d,J=7.6Hz,1H),2.74-2.59(m,2H),2.46-2.40(m,1H),1.93-1.85(m,2H),1.82-1.72(m,1H),1.33(s,9H).
实施例10
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1j 89.5mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2j(产物39.0mg,收率61%)。
对采用上述合成方法得到的目标产物2j的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例11
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1h 87.1mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2h(产物54.5mg,收率89%)。
对采用上述合成方法得到的目标产物2h的进行核磁共振氢谱、碳谱和氟谱检测,测试结果如下:
实施例12
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1l 90.3mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2l(产物50.0mg,收率77%)。
对采用上述合成方法得到的目标产物2l的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例13
在手套箱中向8mL的西林瓶中,加入双(1,2-双(二苯基膦)乙烷)钯18.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1m 99.2mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2m(产物36.1mg,收率49%)。
对采用上述合成方法得到的目标产物2m的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例14
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1n 97.1mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2n(产物54.1mg,收率76%)。
对采用上述合成方法得到的目标产物2n的进行核磁共振氢谱、碳谱和氟谱检测,测试结果如下:
实施例15
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1o 88.5mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2o(产物45.0mg,收率72%)。
对采用上述合成方法得到的目标产物2o的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例16
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1p 95.1mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2p(产物45.2mg,收率65%)。
对采用上述合成方法得到的目标产物2p的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例17
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1q 89.1mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2q(产物41.0mg,收率65%)。
对采用上述合成方法得到的目标产物2q的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例18
在手套箱中向8mL的西林瓶中,加入双(1,2-双(二苯基膦)乙烷)钯18.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1r 89.5mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2r(产物33.2mg,收率52%)。
对采用上述合成方法得到的目标产物2r的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例19
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1s 95.5mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2s(产物52.2mg,收率75%)。
对采用上述合成方法得到的目标产物2s的进行核磁共振氢谱、碳谱和氟谱检测,测试结果如下:
实施例20
/>
在手套箱中向8mL的西林瓶中,加入双(1,2-双(二苯基膦)乙烷)钯18.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1t 95.3mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌24小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2t(产物43.2mg,收率62%)。
对采用上述合成方法得到的目标产物2t的进行核磁共振氢谱、碳谱和氟谱检测,测试结果如下:
实施例21
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1u 93.5mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2u(产物53.1mg,收率78%)。
对采用上述合成方法得到的目标产物2u的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例22
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1v 93.5mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2v(产物47.0mg,收率81%)。
对采用上述合成方法得到的目标产物2v的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例23
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1w 84.7mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2w(产物44.3mg,收率70%)。
对采用上述合成方法得到的目标产物2w的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例24
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1x 79.5mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2x(产物39.3mg,收率73%)。
对采用上述合成方法得到的目标产物2x的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例25
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1y 68.2mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2y(产物29.9mg,收率70%)。
对采用上述合成方法得到的目标产物2y的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例26
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1z 98.7mg(0.20mmol),最后加入4mL的1,4-二氧六环。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2z(产物40.2mg,收率55%)。
对采用上述合成方法得到的目标产物2z的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例27
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1aa 112.3mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2aa(产物46.0mg,收率53%)。
对采用上述合成方法得到的目标产物2aa的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例28
在手套箱中向8mL的西林瓶中,加入双(1,2-双(二苯基膦)乙烷)钯18.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1ab 84.0mg(0.20mmol),最后加入4mL的苯。混合物在24W的蓝色LED灯的照射下室温搅拌12小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2ab(产物47.2mg,收率81%)。
对采用上述合成方法得到的目标产物2ab的进行核磁共振氢谱和碳谱检测,测试结果如下:
实施例29
在手套箱中向8mL的西林瓶中,加入四(三苯基膦)钯23.1mg(0.02mmol),双(2-二苯基膦苯基)醚10.8mg(0.02mmol),甲醇钠21.6mg(0.40mmol)、化合物1ac 99.3mg(0.20mmol),最后加入4mL的1,4-二氧六环。混合物在24W的蓝色LED灯的照射下室温搅拌36小时。反应结束后,将混合物用水洗涤并用乙酸乙酯萃取,合并有机相,无水硫酸钠干燥并减压浓缩。残渣用柱层析分离得到目标化合物2ac(产物62.2mg,收率70%)。
对采用上述合成方法得到的目标产物2ac的进行核磁共振氢谱和碳谱检测,测试结果如下:
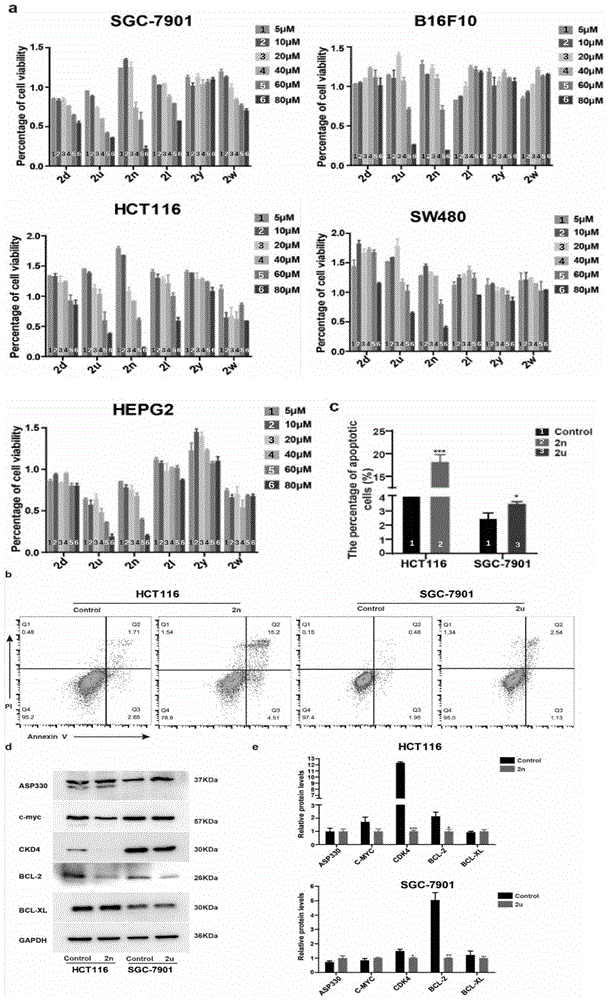
苯并[b]氮杂卓衍生物对多种癌细胞系的抗肿瘤活性测试实验:
1.细胞培养
胃癌细胞株SGC-7901、黑色素瘤细胞株B16F10、肝癌细胞株HEPG2、结直肠癌细胞株HCT116和SW480分别培养于含10%胎牛血清(FBS)的RPMI-1640(SGC-7901,B16F10)、DMEM(HEPG2、HCT116)和L15(SW480)培养基中,置于37℃、5%CO2培养箱中培养。
2.细胞毒性分析
用3-(4,5-二甲基噻唑-2-基)-2,四甲基偶氮唑盐法(MTT)法检测细胞毒性。将5000个SGC-7901细胞,B16F10细胞,HEPG2细胞,HCT116细胞和SW480细胞接种于含有100μL培养基的96孔板中。当细胞密度达到70%左右时,在细胞中加入不同浓度的化合物(5、10、20、40、60、80μM)。二甲基亚砜(DMSO)作为对照组。24h后,用MTT(每孔20μL)分析细胞活力,浓度为5mg/mL,用iMark
根据线性回归分析得到的最佳直线拟合和回归线,利用各自的回归方程计算IC
表1.苯并[b]氮杂卓衍生物的IC
3.蛋白印迹实验
从HCT116和SGC-7901中提取蛋白质样品,经10%SDS-聚丙烯酰胺凝胶电泳分离,转移到聚偏氟乙烯膜上。用抗ASP330、抗c-myc、抗CDK4、抗BCL-2和抗BCL-XL兔多克隆抗体(1:1000)检测ASP330、c-myc、CDK4、BCL-2和BCL-XL蛋白。小鼠GAPDH单克隆抗体(1:2000)作为内参。HRP标记的二抗(1:10000)。使用持久性化学发光底物对结合蛋白进行可视化。
4.细胞凋亡实验
采用流式细胞仪检测HCT116和SGC-7901细胞在不同浓度下按各自IC
活性数据分析:
使用溴化噻唑蓝四氮唑(MTT)法在体外进行抗增殖活性的表型筛选。结果显示,化合物2n和2u在5种不同的癌细胞系(SGC-7901胃癌细胞株、B16F10黑色素瘤细胞株、HEPG2肝癌细胞株、HCT116和SW480结直肠癌细胞株)中均表现出显著的抗癌活性,且具有剂量依赖性。其中,化合物2n和2u对HCT116和SGC-7901细胞的IC
- 由杂环衍生物取代的抗真菌5,6-二氢-4H-吡咯并[1,2-a][1,4]苯并-二氮杂卓类及6H-吡咯并[1,2-a][1,4]苯并二氮杂卓类
- 新颖的由杂环衍生物取代的抗真菌5,6-二氢-4H-吡咯并1,2-a1,4苯并-二氮杂卓类及6H-吡咯并1,2-a1,4苯并二氮杂卓类