喹啉胺类化合物、其制备方法及其在医药上的应用
文献发布时间:2024-04-18 19:58:30
本公开属于医药领域,涉及一种喹啉胺类化合物、其制备方法及其在医药上的应用。特别地,本公开涉及通式(I)所示的喹啉胺类化合物、其制备方法、含有该类化合物的药物组合物以及其作为治疗剂的用途,特别是作为miRNA调节剂的用途和在制备用于治疗通过调节miRNA水平而改善的疾病或病况的药物中的用途。
MicroRNA(miRNA)是一类由内源基因编码的长度约为22个核苷酸的非编码单链RNA分子,它们在动植物中参与转录后基因表达调控。每个miRNA可以有多个靶基因,而几个miRNA也可以调节同一个基因。利用这种复杂的网络可以对靶基因进行精准的调控。miR-124在全身各组织有广泛表达,尤其在脑部组织中高表达。研究表明,过表达miR-124可促进激活的巨噬细胞-小胶质细胞向静态转变,从而抑制自身免疫疾病脑脊髓炎(Ponomarev ED等人,Nat Med,2011;17:67-70)。另外,miR-124可促进巨噬细胞向M2型转化,从而发挥抗炎作用(Veremeyko T,et.al,Plos One,2013;8:e81774)。miR-124也影响T细胞分化,miR-124处理的T细胞IFN-γ和TNFα水平都有所下降。过表达miR-124通过下调STAT3蛋白,进而减少炎症细胞因子IL-17的表达,抑制Th17细胞的分化发挥抗炎作用(Wei J et.al,Cancer Res,2013;73:3913-3926)。有统计研究报道,在小儿溃疡性结肠炎中miR-124的水平远低于健康人,暗示上调miR-124或可抑制肠道炎症反应(Koukos G,et.al,Gastroenterology,2013;145:842-852)。另外,Nakamachi团队发现,相比于骨关节炎病人,类风湿关节炎病人的滑膜细胞中miR-124显著下调(Nakamachi,Y,et.al,Arthritis Rheum,2009;60:1294-1304)。以上研究表明,发展一种新型的小分子药物,通过上调miR-124,可以用于有效地治疗相关炎症疾病。
炎症是免疫系统对局部感染或者组织损伤的一种保护性反应,严重的炎症反应可以损伤肌体。炎症反应的一般表现为疼痛、发热、发红、肿胀和功能丧失。炎性疾病包含多种情形,它包括与自身免疫有关的炎性疾病、中枢神经系统(CNS)中的炎性疾病、关节中的炎性疾病、消化道中的炎性疾病和皮肤中的炎性疾病等。炎性肠病(IBD)和类风湿性关节炎(RA)是两种最常见的炎性疾病,受到了人们广泛地关注。
炎性肠病是一种特发性肠道炎症性疾病,临床表现腹泻、腹痛,甚至可有血便。目前IBD病因和发病机制尚未完全明确,已知肠道黏膜免疫系统异常反应所导致的炎症反应在IBD发病中起重要作用,环境、遗传、感染和免疫因素等多因 素都可能导致该疾病。IBD通常是指溃疡性结肠炎(UC)和克罗恩病(CD),溃疡性结肠炎是结肠黏膜层和黏膜下层连续性炎症,疾病通常先累及直肠,逐渐向全结肠蔓延,克罗恩病可累及全消化道,为非连续性全层炎症,最常累及部位为末端回肠、结肠和肛周。IBD通常表现为过多的免疫细胞侵染肠道粘膜,T细胞亚群包括Th17、Th1和Treg的失衡,过度激活的巨噬细胞和树突细胞。目前已上市或正在临床实验的药物包含减弱炎症反应的JAK抑制剂、TNFα抗体,抑制Th1和Th17分化的IL-12、IL-23的抗体,阻断炎症细胞浸润的整合素α4β7抗体等。
类风湿关节炎是一种影响关节内膜组织(被称为滑膜)的系统性炎性疾病,其特征是手、足小关节的多关节、对称性、侵袭性关节炎症,经常伴有关节外器官受累,可以导致关节畸形及功能丧失。炎症细胞因子(像肿瘤坏死因子TNFα、白细胞介素IL-1和IL-6)在类风湿关节炎(RA)的发病机理中起重要作用。通常使用缓解疾病的抗风湿类小分子药品(DMARD)或者TNFα抑制剂之类的生物药来治疗。然而,这些药物的应答患者通常在使用几年后变成无应答的。因此,需要发展新型作用机理的疗效好并可以长期安全使用的治疗方法。
公开的相关专利申请包括WO2010143169A2、WO2015001518A1、WO2016009065A2、WO2017158201A1和WO2020127843A1等。
发明内容
本公开的目的在于提供一种通式(I)所示的化合物或其可药用的盐:
其中:
环A为环烷基或杂环基;
G为N原子或CR
各个R
各个R
各个R
或者其中的两个相邻的R
R
R
R
R
R
R
n为0、1、2、3或4;
m为0、1或2;
p为1、2、3或4;
q为0、1、2或3。
在本公开的一些实施方案中,所述的通式(I)所示的化合物或其可药用的盐,其中:
环A为环烷基或杂环基;
G为N原子或CR
各个R
各个R
各个R
或者其中的两个相邻的R
R
R
R
R
R
R
n为0、1、2、3或4;
m为0、1或2;
p为1、2、3或4;且
q为0、1、2或3。
在本公开的一些实施方案中,所述的通式(I)所示的化合物或其可药用的盐,其为通式(IC)所示的化合物或其可药用的盐:
其中:
环A、G、R
在本公开的一些实施方案中,所述的通式(I)、通式(IC)所示的化合物或其可药用的盐,其中
在本公开的一些实施方案中,所述的通式(I)所示的化合物或其可药用的盐,其为通式(I-1)或通式(I-2)所示的化合物或其可药用的盐:
其中:
环A、G、R
在本公开的一些实施方案中,所述的通式(I)所示的化合物或其可药用的盐,其为通式(I-3)或通式(I-4)所示的化合物或其可药用的盐:
其中:
环B为环烷基或杂环基;其中所述的环烷基或杂环基各自独立地任选被选自卤素、羟基、羧基、烷基、卤代烷基、烷氧基、卤代烷氧基、羟烷基和氰基中的一个或多个相同或不同的取代基取代;
各个R
r为0、1或2;
环A、G、R
在本公开的一些实施方案中,所述的通式(I)所示的化合物或其可药用的盐,其为通式(I-3C)或通式(I-4C)所示的化合物或其可药用的盐:
其中:
环A、环B、G、R
在本公开的一些实施方案中,所述的通式(I-3)、通式(I-3C)、通式(I-4)或通式(I-4C)所示的化合物或其可药用的盐,其中环B为3至8元环烷基或3至8元杂环基;优选地,环B为5或6元环烷基或5或6元杂环基;更优选地,环B为环戊基。
在本公开的一些实施方案中,所述的通式(I)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)或通式(IC)所示的化合物或其可药用的盐,其中环A为3至8元环烷基或3至8元杂环基;优选地,环A为5或6元环烷基或5或6元杂环基。
在本公开的一些实施方案中,所述的通式(I)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)或通式(IC)所示的化合物或其可药用的盐,其中:各个R
在本公开的一些实施方案中,所述的通式(I)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)或通式(IC)所示的化合物或其可药用的盐,其中各个R
在本公开的一些实施方案中,所述的通式(I)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)或通式(IC)所示的化合物或其可药用的盐,其中:各个R
在本公开的一些实施方案中,所述的通式(I)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)或通式(IC)所示的化合物或其可药用的盐,其中:R
在本公开的一些实施方案中,所述的通式(I)所示的化合物或其可药用的盐,其为通式(II)所示的化合物或其可药用的盐:
其中:
G
R
R
G、R
在本公开的一些实施方案中,所述的通式(II)所示的化合物或其可药用的盐,其中:
G
R
R
G、R
在本公开的一些实施方案中,所述的通式(I)或通式(II)所示的化合物或其可药用的盐,其为通式(IIC)所示的化合物或其可药用的盐:
其中:
G、G
在本公开的一些实施方案中,所述的通式(I)或通式(II)所示的化合物或其可药用的盐,其为通式(IID-1)或(IID-2)所示的化合物或其可药用的盐:
其中:
G、G
在本公开的一些实施方案中,所述的通式(II)、通式(IIC)、通式(IID-1)或通式(IID-2)所示的化合物或其可药用的盐,其中G
在本公开的一些实施方案中,所述的通式(II)、通式(IIC)、通式(IID-1)或通式(IID-2)所示的化合物或其可药用的盐,其中G
在本公开的一些实施方案中,所述的通式(II)、通式(IIC)、通式(IID-1)或通式(IID-2)所示的化合物或其可药用的盐,其中G
在本公开的一些实施方案中,所述的通式(II)、通式(IIC)、通式(IID-1)或通式(IID-2)所示的化合物或其可药用的盐,其中G
在本公开的一些实施方案中,所述的通式(II)、通式(IIC)、通式(IID-1)或通式(IID-2)所示的化合物或其可药用的盐,其中G
在本公开的一些实施方案中,所述的通式(II)、通式(IIC)、通式(IID-1)或通式 (IID-2)所示的化合物或其可药用的盐,其中
在本公开的一些实施方案中,所述的通式(I)所示的化合物或其可药用的盐,其为通式(III)所示的化合物或其可药用的盐:
其中:
L
R
R
G、R
在本公开的一些实施方案中,所述的通式(III)所示的化合物或其可药用的盐,其中:
L
R
R
G、R
在本公开的一些实施方案中,所述的通式(I)或通式(III)所示的化合物或其可药用的盐,其为通式(IIIC)所示的化合物或其可药用的盐:
其中:
G、L
在本公开的一些实施方案中,所述的通式(I)或通式(III)所示的化合物或其可药用的盐,其为通式(IIID-1)或通式(IIID-2)所示的化合物或其可药用的盐:
其中:
G、L
在本公开的一些实施方案中,所述的通式(III)、通式(IIIC)、通式(IIID-1)或通式(IIID-2)所示的化合物或其可药用的盐,其中L
在本公开的一些实施方案中,所述的通式(III)、通式(IIIC)、通式(IIID-1)或通式(IIID-2)所示的化合物或其可药用的盐,其中L
在本公开的一些实施方案中,所述的通式(III)、通式(IIIC)、通式(IIID-1)或通式(IIID-2)所示的化合物或其可药用的盐,其中L
在本公开的一些实施方案中,所述的通式(III)、通式(IIIC)、通式(IIID-1)或通式(IIID-2)所示的化合物或其可药用的盐,其中L
在本公开的一些实施方案中,所述的通式(III)、通式(IIIC)、通式(IIID-1)或通式(IIID-2)所示的化合物或其可药用的盐,其中L
在本公开的一些实施方案中,所述的通式(III)、通式(IIIC)、通式(IIID-1)或通式(IIID-2)所示的化合物或其可药用的盐,其中L
在本公开的一些实施方案中,所述的通式(III)、通式(IIIC)、通式(IIID-1)或通式(IIID-2)所示的化合物或其可药用的盐,其中L
在本公开的一些实施方案中,所述的通式(III)、通式(IIIC)、通式(IIID-1)或通 式(IIID-2)所示的化合物或其可药用的盐,其中
在本公开的一些实施方案中,所述的通式(I)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-4)、通式(IC)、通式(I-3C)、通式(I-4C)、通式(IIC)、通式(IIIC)、通式(II)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)和通式(III)所示的化合物或其可药用的盐,其中各个R
在本公开的一些实施方案中,所述的通式(I)、通式(I-1)、通式(I-2)、通式(IC)、通式(IIC)、通式(IIIC)、通式(II)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)或通式(III)所示的化合物或其可药用的盐,其中各个R
在本公开的一些实施方案中,所述的通式(I-3)、通式(I-4)、通式(I-3C)或通式(I-4C)所示的化合物或其可药用的盐,其中各个R
在本公开的一些实施方案中,所述的通式(I)、通式(I-1)、通式(I-2)、通式(IC)、通式(IIC)、通式(IIIC)、通式(II)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)或通式(III)所示的化合物或其可药用的盐,其中各个R
在本公开的一些实施方案中,所述的通式(I)、通式(I-1)、通式(I-2)、通式(IC)、通式(IIC)、通式(IIIC)、通式(II)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)或通式(III)所示的化合物或其可药用的盐,其中各个R
在本公开的一些实施方案中,所述的通式(I-3)、通式(I-4)、通式(I-3C)或通式(I-4C)所示的化合物或其可药用的盐,其中各个R
在本公开的一些实施方案中,所述的通式(I)、通式(I-1)、通式(I-2)、通式(IC)、通式(IIC)、通式(IIIC)、通式(II)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)或通式(III)所示的化合物或其可药用的盐,其中p为2、3或4;且其中两个相邻的R
在本公开的一些实施方案中,所述的通式(I)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-4)、通式(IC)、通式(I-3C)、通式(I-4C)、通式(IIC)、通式(IIIC)、通式(II)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)或通式(III)所示的化合物或其可药用的盐,其中m为0。
在本公开的一些实施方案中,所述的通式(I)、通式(I-1)、通式(I-2)、通式(IC)、通式(IIC)、通式(IIIC)、通式(II)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)或通式(III)所示的化合物或其可药用的盐,其中p为1或2。
在本公开的一些实施方案中,所述的通式(I)、通式(I-1)、通式(I-2)、通式(IC)、通式(IIC)、通式(IIIC)、通式(II)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)或通式(III)所示的化合物或其可药用的盐,其中p为3。
在本公开的一些实施方案中,所述的通式(I-3)、通式(I-3C)、通式(I-4C)、或通式(I-4)所示的化合物或其可药用的盐,其中r为1或0。
在本公开的一些实施方案中,所述的通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(II)、通式(IIC)、通式(IID-1)、通式(IID-2)、通式(III)、通式(IIIC)、通式(IIID-1)、通式(IIID-2)所示的化合物或其可药用的盐,其中
在本公开的一些实施方案中,所述的通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(II)、通式(IIC)、通式(IID-1)、通式(IID-2)、通式(III)、通式(IIIC)、通式(IIID-1)、通式(IIID-2)所示的化合物或其可药用的盐,当
在本公开的一些实施方案中,所述的通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(II)、通式(IIC)、通式(IID-1)、通式(IID-2)、通式(III)、通式(IIIC)、通式(IIID-1)、通式(IIID-2)所示的化合物或其可药用的盐,当
在本公开的一些实施方案中,所述的通式(I)或通式(II)所示的化合物或其可药用的盐,其为通式(II-1)所示的化合物或其可药用的盐:
其中:
G、R
在本公开的一些实施方案中,所述的通式(I)、通式(II)或通式(II-1)所示的化合物或其可药用的盐,其为通式(II-1C)所示的化合物或其可药用的盐:
其中:
G、G
在本公开的一些实施方案中,所述的通式(II-1)或(II-1C)所示的化合物或其可药用的盐,其中G
在本公开的一些实施方案中,所述的通式(II-1)或(II-1C)所示的化合物或其可药用的盐,其中G
在本公开的一些实施方案中,所述的通式(II-1)或(II-1C)所示的化合物或其可药用的盐,其中G
在本公开的一些实施方案中,所述的通式(I)或通式(III)所示的化合物或其可药用的盐,其为通式(III-1)所示的化合物或其可药用的盐:
其中:
G、R
在本公开的一些实施方案中,所述的通式(I)、通式(III)或通式(III-1)所示的化合物或其可药用的盐,其为通式(III-1C)所示的化合物或其可药用的盐:
其中:
G、L
在本公开的一些实施方案中,所述的通式(III-1)或通式(III-1C)所示的化合物或其可药用的盐,其中L
在本公开的一些实施方案中,所述的通式(III-1)或通式(III-1C)所示的化合物或其可药用的盐,其中L
在本公开的一些实施方案中,所述的通式(III-1)或通式(III-1C)所示的化合物或其可药用的盐,其中L
在本公开的一些实施方案中,所述的通式(II-1)、通式(II-1C)、通式(III-1)或通式(III-1C)所示的化合物或其可药用的盐,其中R
在本公开的一些实施方案中,所述的通式(II-1)、通式(II-1C)、通式(III-1)或通式(III-1C)所示的化合物或其可药用的盐,其中R
在本公开的一些实施方案中,所述的通式(I)、通式(I-3)、通式(I-4)、通式(II)、通式(II-1)、通式(III)或通式(III-1)所示的化合物或其可药用的盐,其中R
在本公开的一些实施方案中,所述的通式(I)、通式(I-3)、通式(I-4)、通式(II)、通式(II-1)、通式(III)或通式(III-1)所示的化合物或其可药用的盐,其中R
在本公开的一些实施方案中,所述的通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、 通式(III-1)或通式(III-1C)所示的化合物或其可药用的盐,其中R
在本公开的一些实施方案中,所述的通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1)或通式(III-1C)所示的化合物或其可药用的盐,其中R
在本公开的一些实施方案中,所述的通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1)或通式(III-1C)所示的化合物或其可药用的盐,其中R
在本公开的一些实施方案中,所述的通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1)或通式(III-1C)所示的化合物或其可药用的盐,其中G为CR
在本公开的一些实施方案中,所述的通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1)或通式(III-1C)所示的化合物或其可药用的盐,其中R
在本公开的一些实施方案中,所述的通式(I)所示的化合物或其可药用的盐,其中环A为3至8元环烷基或3至8元杂环基;G为N原子或CR
在本公开的一些实施方案中,所述的通式(I)所示的化合物或其可药用的盐,其中环A为5或6元环烷基或5或6元杂环基;G为CR
在本公开的一些实施方案中,所述的通式(II)所示的化合物或其可药用的盐,其中
在本公开的一些实施方案中,所述的通式(III)所示的化合物或其可药用的盐,其中
在本公开的一些实施方案中,所述的通式(I-3)或通式(I-4)所示的化合物或其可 药用的盐,其中环B为3至8元环烷基或3至8元杂环基;环A为5或6元环烷基或5或6元杂环基;G为CR
表A 本公开的典型化合物包括但不限于:
本公开的另一方面涉及通式(I-1C)或(I-2C)所示的化合物或其盐:
其中:
R和R
环A、G、R
本公开的另一方面涉及通式(IID-1A)或(IID-2A)所示的化合物或其盐:
其中:
R和R
环A、G、G
本公开的另一方面涉及通式(IIID-1A)或(IIID-2A)所示的化合物或其盐:
其中:
R和R
环A、G、L
在本公开的一些实施方案中,所述的通式(I-1C)、(I-2C)、(IID-1A)、(IID-2A)、(IIID-1A)或(IIID-2A)所示的化合物或其可药用的盐,其中R为C
在本公开的一些实施方案中,所述的通式(I-1C)、(I-2C)、(IID-1A)、(IID-2A)、(IIID-1A)或(IIID-2A)所示的化合物或其可药用的盐,其中R
表B 本公开的典型中间体化合物包括但不限于:
本公开的另一方面涉及一种制备通式(I)所示的化合物或其可药用的盐的方法,该方法包括:
通式(IC)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
环A、G、R
本公开的另一方面涉及一种制备通式(IC)所示的化合物或其可药用的盐的方法,该方法包括:
通式(IA)化合物或其盐与通式(IB)化合物或其盐反应,得到通式(IC)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
环A、G、R
本公开的另一方面涉及一种制备通式(I-1)所示的化合物或其可药用的盐的方法,该方法包括:
通式(I-1C)化合物或其盐发生酯水解反应,得到通式(I-1)的化合物或其可药用的盐;
其中:
R和R
环A、G、R
本公开的另一方面涉及一种制备通式(I-2)所示的化合物或其可药用的盐的方法,该方法包括:
通式(I-2C)化合物或其盐发生酯水解反应,得到通式(I-2)的化合物或其可药用的盐;
其中:
R和R
环A、G、R
本公开的另一方面涉及一种制备通式(I-3)所示的化合物或其可药用的盐的方法,该方法包括:
通式(I-3C)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
环A、环B、G、R
本公开的另一方面涉及一种制备通式(I-3C)所示的化合物或其可药用的盐的方法,该方法包括:
通式(I-3A)化合物或其盐与通式(IB)化合物或其盐反应,得到通式(I-3C)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
环A、环B、G、R
本公开的另一方面涉及一种制备通式(I-4)所示的化合物或其可药用的盐的方法,该方法包括:
通式(I-4C)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
环A、环B、G、R
本公开的另一方面涉及一种制备通式(I-4C)所示的化合物或其可药用的盐的方法,该方法包括:
通式(I-4A)化合物或其盐与通式(IB)化合物或其盐反应,得到通式(I-4C)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
环A、环B、G、R
本公开的另一方面涉及一种制备通式(II)所示的化合物或其可药用的盐的方法,该方法包括:
通式(IIC)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
G、G
本公开的另一方面涉及一种制备通式(IIC)所示的化合物或其可药用的盐的方法,该方法包括:
通式(IA)化合物或其盐与通式(IIB)化合物或其盐反应,得到通式(IIC)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
G、G
本公开的另一方面涉及一种制备通式(IID-1)所示的化合物或其可药用的盐的方法,该方法包括:
通式(IID-1A)化合物或其盐发生酯水解反应,得到通式(IID-1)的化合物或其可药用的盐;
其中:
R和R
G、G
本公开的另一方面涉及一种制备通式(IID-2)所示的化合物或其可药用的盐的方法,该方法包括:
通式(IID-2A)化合物或其盐发生酯水解反应,得到通式(IID-2)的化合物或其可药用的盐;
其中:
R和R
G、G
本公开的另一方面涉及一种制备通式(II-1)所示的化合物或其可药用的盐的方法,该方法包括:
通式(II-1C)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
G、G
本公开的另一方面涉及一种制备通式(II-1C)所示的化合物或其可药用的盐的方法,该方法包括:
通式(II-1A)化合物或其盐与通式(II-1B)化合物或其盐反应,得到通式(II-1C)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
G、G
本公开的另一方面涉及一种制备通式(III)所示的化合物或其可药用的盐的方法,该方法包括:
通式(IIIC)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
G、L
本公开的另一方面涉及一种制备通式(IIIC)所示的化合物或其可药用的盐的方法,该方法包括:
通式(IA)化合物或其盐与通式(IIIB)化合物或其盐反应,得到通式(IIIC)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
G、L
本公开的另一方面涉及一种制备通式(IIID-1)所示的化合物或其可药用的盐的方法,该方法包括:
通式(IIID-1A)化合物或其盐发生酯水解反应,得到通式(IIID-1)的化合物或其可药用的盐;
其中:
R和R
G、L
本公开的另一方面涉及一种制备通式(IIID-2)所示的化合物或其可药用的盐的方法,该方法包括:
通式(IIID-2A)化合物或其盐发生酯水解反应,得到通式(IIID-2)的化合物或其可药用的盐;
其中:
R和R
G、L
本公开的另一方面涉及一种制备通式(III-1)所示的化合物或其可药用的盐的方法,该方法包括:
通式(III-1C)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
G、L
本公开的另一方面涉及一种制备通式(III-1C)所示的化合物或其可药用的盐的方法,该方法包括:
通式(II-1A)化合物或其盐与通式(III-1B)化合物或其盐反应,得到通式(III-1C)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
G、L
本公开的另一方面涉及一种药物组合物,所述药物组合物含有治疗有效量的本公开通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐,以及一种或多种药学上可接受的载体、稀释剂或赋形剂。
本公开进一步涉及通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐,或包含其的药物组合物在制备用于调节miRNA水平的药物中的用途;优选地,所述miRNA为miR-124。
本公开进一步涉及通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐,或包含其的药物组合物在制备用于治疗通过调节miRNA水平而改善的疾病或病况的药物中的用途。
本公开进一步涉及通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式 (III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐,或包含其的药物组合物在制备用于治疗和/或预防疾病或病况的药物中的用途,所述的疾病或病况选自病毒感染、炎症和癌症。
本公开进一步涉及通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐,或包含其的药物组合物在制备用于治疗和/或预防AIDS或AIDS相关的病况或人类免疫缺陷病毒(HIV)药物中的用途。
本公开进一步涉及通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐,或包含其的药物组合物在制备用于治疗和/或预防疾病或病况的药物中的用途,所述的疾病或病况为炎症,所述的炎症选自自身免疫有关的炎性疾病、中枢神经系统(CNS)中的炎性疾病、关节中的炎性疾病、消化道中的炎性疾病、皮肤中的炎性疾病、上皮细胞有关的其他炎性疾病、与癌症有关的炎症、与刺激有关的炎症和与损伤有关的炎症。
本公开进一步涉及一种通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐、或包括其的药物组合物,其用作治疗和/或预防疾病或病况的药物,其中所述的疾病或病况选自病毒感染、炎症和癌症;其中所述的炎症选自炎性肠病、类风湿性关节炎、多发性硬化、阿尔茨海默病、帕金森病、骨关节炎、动脉粥样硬化、强直性脊柱炎、银屑癣、皮炎、系统性红斑狼疮、斯耶格伦(Sjogren)综合征、支气管炎、哮喘和与结肠癌有关的炎症;优选地,所述炎症为炎性肠病。
本公开进一步涉及通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用盐,或包含其的药物组合物在制备用于治疗和/或预防疾病或病况的药物中的用途,所述的疾病或病况为癌症,所述的癌症选自白血病、淋巴瘤、巨球蛋白血症、重链病、肉瘤、癌瘤、胰腺癌、乳腺癌、卵巢癌、前列腺癌、鳞癌、汗腺癌、皮脂腺癌、乳头状癌、囊腺癌、髓样癌、支气管癌、肝癌、胆管癌、绒毛膜癌、精原细胞瘤、胚胎癌、威尔姆氏肿瘤、宫颈癌、子宫癌、睾丸癌、肺癌、膀胱癌、神经胶质瘤、髓母细胞瘤、颅咽管瘤、室管膜瘤、松果体瘤、血管母细胞瘤、听神经瘤、神经鞘瘤、神经纤维瘤、 视网膜母细胞瘤、黑色素瘤、皮肤癌、肾癌、鼻咽癌、胃癌、食道癌、头颈癌、结肠直肠癌、小肠癌、胆囊癌、儿科肿瘤、尿路上皮癌、输尿管肿瘤、甲状腺癌、骨瘤、成神经细胞瘤、脑瘤和骨髓瘤。
本公开进一步涉及一种调节miRNA水平的方法,其包括给予所需患者治疗有效量的通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐、或包括其的药物组合物;优选地,所述miRNA为miR-124。
本公开进一步涉及一种治疗和/或预防疾病或病况的方法,其包括给予所需患者治疗有效量的通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐、或包括其的药物组合物,其中所述的疾病或病况选自病毒感染、炎症和癌症;所述的炎症优选选自自身免疫有关的炎性疾病、中枢神经系统(CNS)中的炎性疾病、关节中的炎性疾病、消化道中的炎性疾病、皮肤中的炎性疾病、上皮细胞有关的其他炎性疾病、与癌症有关的炎症、与刺激有关的炎症和与损伤有关的炎症。
本公开进一步涉及一种通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐、或包括其的药物组合物,其用作治疗和/或预防疾病或病况的药物,其中所述的疾病或病况选自病毒感染、炎症和癌症;其中所述的炎症选自炎性肠病、类风湿性关节炎、多发性硬化、阿尔茨海默病、帕金森病、骨关节炎、动脉粥样硬化、强直性脊柱炎、银屑癣、皮炎、系统性红斑狼疮、斯耶格伦(Sjogren)综合征、支气管炎、哮喘和与结肠癌有关的炎症;优选地,所述炎症为炎性肠病。
本公开进一步涉及一种治疗和/或预防疾病或病况的方法,其包括给予所需患者治疗有效量的通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐、或包括其的药物组合物,其中所述的疾病或病况选自病毒感染、炎症和癌症;所述的癌症选自白血病、淋巴瘤、巨球蛋白血症、重链病、肉瘤、癌瘤、胰腺癌、乳腺癌、卵巢癌、前列腺癌、鳞癌、汗腺癌、皮脂腺癌、乳头状癌、囊腺癌、髓样癌、支气管癌、肝癌、胆管癌、绒毛膜癌、精原细胞瘤、胚胎癌、威尔姆氏肿瘤、宫颈癌、子宫癌、睾丸癌、肺 癌、膀胱癌、神经胶质瘤、髓母细胞瘤、颅咽管瘤、室管膜瘤、松果体瘤、血管母细胞瘤、听神经瘤、神经鞘瘤、神经纤维瘤、视网膜母细胞瘤、黑色素瘤、皮肤癌、肾癌、鼻咽癌、胃癌、食道癌、头颈癌、结肠直肠癌、直肠癌、小肠癌、胆囊癌、儿科肿瘤、尿路上皮癌、输尿管肿瘤、甲状腺癌、骨瘤、成神经细胞瘤、脑瘤和骨髓瘤。
本公开进一步涉及一种治疗和/或预防疾病或病况的方法,其包括给予所需患者治疗有效量的通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐、或包括其的药物组合物,其中所述的疾病或病况选自AIDS或AIDS相关的病况和人类免疫缺陷病毒(HIV)。
本公开进一步涉及一种通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐、或包括其的药物组合物,其用作药物。
本公开进一步涉及一种通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐、或包括其的药物组合物,其用于调节miRNA;优选地,所述miRNA为miR-124。
本公开进一步涉及一种通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐、或包括其的药物组合物,其用于治疗和/或预防疾病或病况的用途,其中所述的疾病或病况选自病毒感染、炎症和癌症;所述的炎症优选选自自身免疫有关的炎性疾病、中枢神经系统(CNS)中的炎性疾病、关节中的炎性疾病、消化道中的炎性疾病、皮肤中的炎性疾病、上皮细胞有关的其他炎性疾病、与癌症有关的炎症、与刺激有关的炎症和与损伤有关的炎症。
本公开进一步涉及一种通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐、或包括其的药物组合物,其用于治疗和/或预防疾病或病况的用途,其中所述的疾病或病况选自病毒感染、炎症和癌症;其中所述的炎症选自炎性肠病、类风湿性关节炎、多发性硬 化、阿尔茨海默病、帕金森病、骨关节炎、动脉粥样硬化、强直性脊柱炎、银屑癣、皮炎、系统性红斑狼疮、斯耶格伦(Sjogren)综合征、支气管炎、哮喘和与结肠癌有关的炎症;优选地,所述炎症为炎性肠病。
本公开进一步涉及一种通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐、或包括其的药物组合物,其用于治疗和/或预防疾病或病况的用途,其中所述的疾病或病况选自病毒感染、炎症和癌症;其中所述的炎症为炎性肠病;其中所述的炎性肠病为溃疡性结肠炎(UC)或克罗恩病(CD)。
本公开进一步涉及一种通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐、或包括其的药物组合物,其用于治疗和/或预防疾病或病况的用途,其中所述的疾病或病况选自病毒感染、炎症和癌症;所述的癌症选自白血病、淋巴瘤、巨球蛋白血症、重链病、肉瘤、癌瘤、胰腺癌、乳腺癌、卵巢癌、前列腺癌、鳞癌、汗腺癌、皮脂腺癌、乳头状癌、囊腺癌、髓样癌、支气管癌、肝癌、胆管癌、绒毛膜癌、精原细胞瘤、胚胎癌、威尔姆氏肿瘤、宫颈癌、子宫癌、睾丸癌、肺癌、膀胱癌、神经胶质瘤、星形细胞瘤、髓母细胞瘤、颅咽管瘤、室管膜瘤、松果体瘤、血管母细胞瘤、听神经瘤、神经鞘瘤、神经纤维瘤、视网膜母细胞瘤、黑色素瘤、皮肤癌、肾癌、鼻咽癌、胃癌、食道癌、头颈癌、结肠直肠癌、小肠癌、胆囊癌、儿科肿瘤、尿路上皮癌、输尿管肿瘤、甲状腺癌、骨瘤、成神经细胞瘤、脑瘤和骨髓瘤。
本公开进一步涉及一种通式(I)、通式(IC)、通式(I-1)、通式(I-2)、通式(I-3)、通式(I-3C)、通式(I-4)、通式(I-4C)、通式(II)、通式(IIC)、通式(II-1)、通式(II-1C)、通式(III)、通式(IID-1)、通式(IID-2)、通式(IIID-1)、通式(IIID-2)、通式(IIIC)、通式(III-1C)、通式(III-1)以及表A所示的化合物或其可药用的盐,或包含其的药物组合物,其用于治疗和/或预防AIDS或AIDS相关的病况或人类免疫缺陷病毒(HIV)的用途。
本公开所述的疾病或病况是通过调节miRNA水平来治疗和/或预防的疾病或病况;优选地,所述miRNA为miR-124。
优选地,本公开所述的病毒感染为逆转录病毒感染。
优选地,本公开所述的炎性肠病为溃疡性结肠炎(UC)或克罗恩病(CD)。
优选地,本公开所述的淋巴瘤为霍奇金氏疾病或非霍奇金淋巴瘤(例如套细胞淋巴瘤、弥漫性大B细胞淋巴瘤、滤泡中心淋巴瘤、边缘区B细胞淋巴瘤、淋巴浆细胞淋巴瘤和外周T细胞淋巴瘤);肝癌优选为肝细胞癌;肺癌(又称支气管肺 癌)选自非小细胞肺癌(NSCLC)(例如鳞状细胞癌)和小细胞肺癌(SCLC);肾癌选自肾细胞癌、透明细胞和肾嗜酸细胞瘤;白血病选自慢性淋巴细胞性白血病(CLL)、慢性粒细胞性白血病、急性成淋巴细胞性白血病(ALL)、T-细胞急性成淋巴细胞性白血病(T-ALL)、慢性髓细胞性白血病(CML)和急性骨髓性白血病(AML);皮肤癌选自恶性黑色素瘤、鳞状细胞癌、基底细胞癌和血管肉瘤;骨髓瘤优选为多发性骨髓瘤;结直肠癌优选为结肠癌或直肠癌;神经胶质瘤(即胶质瘤或胶质细胞瘤)优选选自胶质母细胞瘤、星形细胞瘤和少突胶质细胞瘤。
可将活性化合物制成适合于通过任何适当途径给药的形式,通过常规方法使用一种或多种药学上可接受的载体来配制本公开的组合物。因此,本公开的活性化合物可以配制成用于口服给药,注射(例如静脉内、肌肉内或皮下)给药,吸入或吹入给药的各种剂型。本公开的化合物也可以配制成例如片剂、硬或软胶囊、水性或油性混悬液、乳剂、注射液、可分散性粉末或颗粒、栓剂、锭剂或糖浆等剂型。
作为一般性指导,活性化合物优选是以单位剂量的方式,或者是以患者可以以单剂自我给药的方式。本公开化合物或组合物的单位剂量的表达方式可以是片剂、胶囊、扁囊剂、瓶装药水、药粉、颗粒剂、锭剂、栓剂、再生药粉或液体制剂。合适的单位剂量可以是0.1~1000mg。
本公开的药物组合物除活性化合物外,可含有一种或多种辅料,所述辅料选自以下成分:填充剂(稀释剂)、粘合剂、润湿剂、崩解剂或赋形剂等。根据给药方法的不同,组合物可含有0.1至99重量%的活性化合物。
片剂含有活性成分和用于混合的适宜制备片剂的无毒的可药用的赋形剂。这些赋形剂可以是惰性赋形剂、造粒剂、崩解剂、粘合剂和润滑剂。这些片剂可以不包衣或可通过掩盖药物的味道或在胃肠道中延迟崩解和吸收,因而在较长时间内提供缓释作用的已知技术将其包衣。
也可用其中活性成分与惰性固体稀释剂或其中活性成分与水溶性载体或油溶媒混合的软明胶胶囊提供口服制剂。
水混悬液含有活性物质和用于混合的适宜制备水混悬液的赋形剂。此类赋形剂是悬浮剂、分散剂或湿润剂。水混悬液也可以含有一种或多种防腐剂、一种或多种着色剂、一种或多种矫味剂和一种或多种甜味剂。
油混悬液可通过使活性成分悬浮于植物油或矿物油配制而成。油混悬液可含有增稠剂。可加入上述的甜味剂和矫味剂,以提供可口的制剂。可通过加入抗氧化剂保存这些组合物。
本公开的药物组合物也可以是水包油乳剂的形式。油相可以是植物油、或矿物油或其混合物。适宜的乳化剂可以是天然产生的磷脂,乳剂也可以含有甜味剂、矫味剂、防腐剂和抗氧剂。此类制剂也可含有缓和剂、防腐剂、着色剂和抗氧剂。
本公开的药物组合物可以是无菌注射水溶液形式。可以使用的可接受的溶媒或溶剂有水、林格氏液和等渗氯化钠溶液。无菌注射制剂可以是其中活性成分溶于油相的无菌注射水包油微乳,可通过局部大量注射将注射液或微乳注入患者的血流中。或者,最好按可保持本公开化合物恒定循环浓度的方式给予溶液和微乳。为保持这种恒定浓度,可使用连续静脉内递药装置。这种装置的实例是Deltec CADD-PLUS.TM.5400型静脉注射泵。
本公开的药物组合物可以是用于肌内和皮下给药的无菌注射水或油混悬液的形式。可按已知技术,用上述那些适宜的分散剂或湿润剂和悬浮剂配制该混悬液。无菌注射制剂也可以是在肠胃外可接受的无毒稀释剂或溶剂中制备的无菌注射溶液或混悬液。此外,可方便地用无菌固定油作为溶剂或悬浮介质。为此目的,可使用任何调和固定油。此外,脂肪酸也可以制备注射剂。
可按用于直肠给药的栓剂形式给予本公开化合物。可通过将药物与在普通温度下为固体但在直肠中为液体,因而在直肠中会溶化而释放药物的适宜的无刺激性赋形剂混合来制备这些药物组合物。
可通过加入水来制备水混悬的可分散粉末和颗粒给予本公开化合物。可通过将活性成分与分散剂或湿润剂、悬浮剂或一种或多种防腐剂混合来制备这些药物组合物。
如本领域技术人员所熟知的,药物的给药剂量依赖于多种因素,包括但并非限定于以下因素:所用具体化合物的活性、患者的年龄、患者的体重、患者的健康状况、患者的行为、患者的饮食、给药时间、给药方式、排泄的速率、药物的组合、疾病的严重性等;另外,最佳的治疗方式如治疗的模式、化合物的日用量或可药用的盐的种类可以根据传统的治疗方案来验证。
术语说明
除非有相反陈述,在说明书和权利要求书中使用的术语具有下述含义。
术语“烷基”指饱和的直链或支链脂肪族烃基,其为包含1至20个碳原子的直链或支链基团,优选含有1至12个(例如1、2、3、4、5、6、7、8、9、10、11和12个)碳原子的烷基(即C
术语“亚烷基”指饱和的直链或支链脂肪族烃基,其为从母体烷的相同碳原子或两个不同的碳原子上除去两个氢原子所衍生的残基,其为包含1至20个碳原子的直链或支链基团,优选含有1至12个(例如1、2、3、4、5、6、7、8、9、10、11和12个)碳原子(即C
术语“烯基”指分子中含有至少一个碳碳双键的烷基化合物,其中烷基的定义如上所述。优选含有2至12个(例如2、3、4、5、6、7、8、9、10、11和12个)碳原子的烯基,更优选含有2至6个碳原子的烯基(即C
术语“炔基”指分子中含有至少一个碳碳三键的烷基化合物,其中烷基的定义如上所述。优选含有2至12个(例如2、3、4、5、6、7、8、9、10、11和12个)碳原子的炔基(即C
术语“环烷基”指饱和或部分不饱和单环或多环环状烃取代基,环烷基环包含3至20个碳原子,优选包含3至12个(例如3、4、5、6、7、8、9、10、11和12个)碳原子(即3至12元环烷基),优选包含3至8个碳原子(即3至8元环烷基),更优选包含3至6个碳原子(即3至6元环烷基),最优选包含5或6个碳原子(即5或6元环烷基)。单环环烷基的非限制性实例包括环丙基、环丁基、 环戊基、环戊烯基、环己基、环己烯基、环己二烯基、环庚基、环庚三烯基和环辛基等;多环环烷基包括螺环烷基、稠环烷基和桥环烷基。
术语“螺环烷基”指5至20元,单环之间共用一个碳原子(称螺原子)的多环基团,其可以含有一个或多个双键。优选6至14元,更优选7至10元(例如7、8、9或10元)。根据环与环之间共用螺原子的数目将螺环烷基分为单螺环烷基或多螺环烷基(如双螺环烷基),优选单螺环烷基和双螺环烷基。更优选为3元/5元、3元/6元、4元/4元、4元/5元、4元/6元、5元/5元、5元/6元、6元/4元、6元/5元或6元/6元单螺环烷基。螺环烷基的非限制性实例包括:
术语“稠环烷基”指5至20元,系统中的每个环与体系中的其他环共享毗邻的一对碳原子的全碳多环基团,其中一个或多个环可以含有一个或多个双键。优选6至14元,更优选7至10元(例如7、8、9或10元)。根据组成环的数目可以分为双环、三环、四环等多环稠环烷基,优选双环或三环,更优选3元/4元、3元/5元、3元/6元、4元/4元、4元/5元、4元/6元、5元/4元、5元/5元、5元/6元、6元/3元、6元/4元、6元/5元、6元/6元、6元/7元、7元/5元或7元/6元的双环烷基。稠环烷基的非限制性实例包括:
术语“桥环烷基”指5至20元,任意两个环共用两个不直接连接的碳原子的全碳多环基团,其可以含有一个或多个双键。优选6至14元,更优选7至10元(例如7、8、9或10元)。根据组成环的数目可以分为双环、三环、四环等多环桥环烷基,优选双环、三环或四环桥环烷基,更优选双环或三环桥环烷基。桥环烷基的非限制性实例包括:
所述环烷基环包括如上所述的环烷基(包括单环、螺环、稠环和桥环)稠合于芳基、杂芳基或杂环烷基环上,其中与母体结构连接在一起的环为环烷基,非 限制性实例包括
环烷基可以是取代的或非取代的,当被取代时,其可以在任何可使用的连接点上被取代,取代基优选选自卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、环烷基氧基、杂环基氧基、羟基、羟烷基、氧代基、氰基、氨基、硝基、环烷基、杂环基、芳基和杂芳基中的一个或多个。
术语“烷氧基”指-O-(烷基),其中烷基的定义如上所述。烷氧基的非限制性实例包括:甲氧基、乙氧基、丙氧基和丁氧基。烷氧基可以是任选取代的或非取代的,当被取代时,其取代基优选选自氘原子、卤素、烷氧基、卤代烷基、卤代烷氧基、环烷基氧基、杂环基氧基、羟基、羟烷基、氰基、氨基、硝基、环烷基、杂环基、芳基和杂芳基中的一个或多个。
术语“杂环基”指饱和或部分不饱和单环或多环环状取代基,其包含3至20个环原子,其中一个或多个环原子为选自氮、氧和硫的杂原子,所述的硫可任选被氧代(即形成亚砜或砜),但不包括-O-O-、-O-S-或-S-S-的环部分,其余环原子为碳。优选包含3至12个(例如3、4、5、6、7、8、9、10、11和12个)环原子,其中1~4个(例如1、2、3和4个)是杂原子(即3至12元杂环基);更优选包含3至8个环原子(例如3、4、5、6、7和8个),其中1-3是杂原子(例如1、2和3个)(即3至8元杂环基);更优选包含3至6个环原子,其中1-3个是杂原子(即3至6元杂环基);最优选包含5或6个环原子,其中1-3个是杂原子(即5或6元杂环基)。单环杂环基的非限制性实例包括吡咯烷基、四氢吡喃基、1,2,3,6-四氢吡啶基、哌啶基、哌嗪基、吗啉基、硫代吗啉基、高哌嗪基等。多环杂环基包括螺杂环基、稠杂环基和桥杂环基。
术语“螺杂环基”指5至20元,单环之间共用一个原子(称螺原子)的多环杂环基团,其中一个或多个环原子为选自氮、氧和硫的杂原子,所述的硫可任选被氧代(即形成亚砜或砜),其余环原子为碳。其可以含有一个或多个双键。优选为6至14元,更优选为7至10元(例如7、8、9或10元)。根据环与环之间共用螺原子的数目将螺杂环基分单螺杂环基或多螺杂环基(如双螺杂环基),优选为单螺杂环基和双螺杂环基。更优选为3元/5元、3元/6元、4元/4元、4元/5元、4元/6元、5元/5元、5元/6元或6元/6元单螺杂环基。螺杂环基的非限制性实例包括:
术语“稠杂环基”指5至20元,系统中的每个环与体系中的其他环共享毗邻的一对原子的多环杂环基团,一个或多个环可以含有一个或多个双键,其中一个或多个环原子为选自氮、氧和硫的杂原子,所述的硫可任选被氧代(即形成亚砜或砜),其余环原子为碳。优选6至14元,更优选7至10元(例如7、8、9或10元)。根据组成环的数目可以分双环、三环、四环等多环稠杂环基,优选双环或三环,更优选为3元/4元、3元/5元、3元/6元、4元/4元、4元/5元、4元/6元、5元/3元、5元/4元、5元/5元、5元/6元、5元/7元、6元/3元、6元/4元、6元/5元、6元/6元、6元/7元、7元/5元或7元/6元双环稠杂环基。稠杂环基的非限制性实例包括:
术语“桥杂环基”指5至14元,任意两个环共用两个不直接连接的原子的多环杂环基团,其可以含有一个或多个双键,其中一个或多个环原子为选自氮、氧和硫的杂原子,所述的硫可任选被氧代(即形成亚砜或砜),其余环原子为碳。优选6至14元,更优选7至10元(例如7、8、9或10元)。根据组成环的数目可以分双环、三环、四环等多环桥杂环基,优选双环、三环或四环桥杂环基,更优选双环或三环桥杂环基。桥杂环基的非限制性实例包括:
所述杂环基环包括如上所述的杂环基(包括单环、螺杂环、稠杂环和桥杂环)稠合于芳基、杂芳基或环烷基环上,其中与母体结构连接在一起的环为杂环基,其非限制性实例包括:
等。
杂环基可以是取代的或非取代的,当被取代时,其可以在任何可使用的连接点上被取代,取代基优选选自卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、环烷基氧基、杂环基氧基、羟基、羟烷基、氰基、氨基、硝基、环烷基、杂环基、芳基和杂芳基中的一个或多个。
术语“芳基”指具有共轭的π电子体系的6至14元全碳单环或稠合多环(稠合多环是共享毗邻碳原子对的环)基团,优选6至10元,例如苯基和萘基。所述芳基环包括如上所述的芳基环稠合于杂芳基、杂环基或环烷基环上,其中与母体结构连接在一起的环为芳基环,其非限制性实例包括:
芳基可以是取代的或非取代的,当被取代时,其可以在任何可使用的连接点上被取代,取代基优选选自卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、环烷基氧基、杂环基氧基、羟基、羟烷基、氰基、氨基、硝基、环烷基、杂环基、芳基和杂芳基中的一个或多个。
术语“杂芳基”指包含1至4个杂原子(例如1、2、3和4个)、5至14个环原子的杂芳族体系,其中杂原子选自氧、硫和氮。杂芳基优选5至10元(例如5、6、7、8、9或10元),更优选5元或6元,例如呋喃基、噻吩基、吡啶基、吡咯基、N-烷基吡咯基、嘧啶基、吡嗪基、哒嗪基、咪唑基、吡唑基、三唑基、四唑基等。所述杂芳基环包括如上述的杂芳基稠合于芳基、杂环基或环烷基环上,其中与母体结构连接在一起的环为杂芳基环,其非限制性实例包括:
杂芳基可以是取代的或非取代的,当被取代时,其可以在任何可使用的连接点上被取代,取代基优选选自卤素、烷基、烷氧基、卤代烷基、卤代烷氧基、环烷基氧基、杂环基氧基、羟基、羟烷基、氰基、氨基、硝基、环烷基、杂环基、芳基和杂芳基中的一个或多个。
上述环烷基、杂环基、芳基和杂芳基包括从母体环原子上除去一个氢原子所衍生的残基,或从母体的相同环原子或两个不同的环原子上除去两个氢原子所衍生的残基即“二价环烷基”、“二价杂环基”、“亚芳基”、“亚杂芳基”。
术语“氨基保护基”是为了使分子其它部位进行反应时氨基保持不变,用易于脱去的基团对氨基进行保护。非限制性实例包括:(三甲基硅)乙氧基甲基、四氢吡喃基、叔丁氧羰基、乙酰基、苄基、烯丙基和对甲氧苄基等。这些基团可任选地被选自卤素、烷氧基和硝基中的1-3个取代基所取代。
术语“羟基保护基”是指通常用于阻断或保护羟基而反应在化合物的其它官能团上进行的羟基衍生物。作为示例,优选地,所述的羟基保护基可以是例如:三乙基硅基、三异丙基硅基、叔丁基二甲基硅烷基(TBS)、叔丁基二苯基硅基、甲基、叔丁基、烯丙基、苄基、甲氧基甲基(MOM)、乙氧基乙基、甲酰基、乙酰基、苯甲酰基、对硝基苯甲酰基等。
术语“环烷基氧基”指环烷基-O-,其中环烷基如上所定义。
术语“杂环基氧基”指杂环基-O-,其中杂环基如上所定义。
术语“芳基氧基”指芳基-O-,其中芳基如上所定义。
术语“杂芳基氧基”指杂芳基-O-,其中杂芳基如上所定义。
术语“烷硫基”指烷基-S-,其中烷基如上所定义。
术语“卤代烷基”指烷基被一个或多个卤素取代,其中烷基如上所定义。
术语“卤代烷氧基”指烷氧基被一个或多个卤素取代,其中烷氧基如上所定义。
术语“氘代烷基”指烷基被一个或多个氘原子取代,其中烷基如上所定义。
术语“羟烷基”指烷基被一个或多个羟基取代,其中烷基如上所定义。
术语“卤素”指氟、氯、溴或碘。
术语“羟基”指-OH。
术语“巯基”指-SH。
术语“氨基”指-NH
术语“氰基”指-CN。
术语“硝基”指-NO
术语“氧代基”或“氧代”指“=O”。
术语“羰基”指C=O。
术语“羧基”指-C(O)OH。
术语“羧酸酯基”指-C(O)O(烷基)、-C(O)O(环烷基)、(烷基)C(O)O-或(环烷基)C(O)O-,其中烷基和环烷基如上所定义。
本公开的化合物包括其同位素衍生物。术语“同位素衍生物”指结构不同仅在于存在一种或多种同位素富集原子的化合物。例如,具有本公开的结构,用“氘”或“氚”代替氢,或者用
本公开化合物可以存在特定的立体异构体形式。术语“立体异构体”是指结构相同但原子在空间中的排列不同的异构体。其包括顺式和反式(或Z和E)异构体、(-)-和(+)-异构体、(R)-和(S)-对映异构体、非对映异构体、(D)-和(L)-异构体、互变异构体、阻转异构体、构象异构体及其混合物(如外消旋体、非对映异构体的混合物)。本公开化合物中的取代基可以存在另外的不对称原子。所有这些立体异构体以及它们的混合物,均包括在本公开的范围内。对于所有的碳-碳双键,即使仅命名了一个构型,Z型和E型均包括在内。可以通过手性合成、手性试剂或者其他常规技术制备光学活性的(-)-和(+)-异构体、(R)-和(S)-对映异构体以及(D)-和(L)-异构体。本公开某化合物的一种异构体,可以通过不对称合成或者具有手性助剂的衍生作用来制备,或者,当分子中含有碱性官能团(如氨基)或酸性官能团(如羧基)时,与适当的光学活性的酸或碱形成非对映异构体的盐,然后通过本领域所公知的常规方法进行非对映异构体拆分,得到纯的异构体。此外,对映异构体和非对映异构体的分离通常是通过色谱法完成。
本公开的化合物可以以不同的互变异构体形式存在,并且所有这样的形式包含在本公开的范围内。术语“互变异构体”或“互变异构体形式”是指平衡存在并且容易从一种异构形式转化为另一种异构形式的结构异构体。其包括所有可能的互变异构体,即以单一异构体的形式或以所述互变异构体的任意比例的混合物的形式存在。非限制性的实例包括:酮-烯醇、亚胺-烯胺、内酰胺-内酰亚胺等。内酰胺-内酰亚胺的平衡实例如下所示:
如当提及吡唑基时,应理解为包括如下两种结构中的任何一种或两种互变异构体的混合物:
所有的互变异构形式在本公开的范围内,且化合物的命名不排除任何互变异构体。
本公开所述化合物的化学结构中,键
“任选地”或“任选”是指意味着随后所描述的事件或环境可以但不必发生,该说明包括该事件或环境发生或不发生的场合。例如“任选的被卤素或者氰基取 代的C
“取代的”指基团中的一个或多个氢原子,优选为1~5个,更优选为1~3个氢原子彼此独立地被相应数目的取代基取代。本领域技术人员能够在不付出过多努力的情况下(通过实验或理论)确定可能或不可能的取代。例如,具有游离氢的氨基或羟基与具有不饱和(如烯属)键的碳原子结合时可能是不稳定的。
“药物组合物”表示含有一种或多种本文所述化合物或其可药用的盐或前体药物与其他化学组分的混合物,以及其他组分例如药学上可接受的载体和赋形剂。药物组合物的目的是促进对生物体的给药,利于活性成分的吸收进而发挥生物活性。
“可药用的盐”是指本公开化合物的盐,可选自无机盐或有机盐。这类盐用于哺乳动物体内时具有安全性和有效性,且具有应有的生物活性。可以在化合物的最终分离和纯化过程中,或通过使合适的基团与合适的碱或酸反应来单独制备盐。通常用于形成药学上可接受的盐的碱包括无机碱,例如氢氧化钠和氢氧化钾,以及有机碱,例如氨。通常用于形成药学上可接受的盐的酸包括无机酸以及有机酸。
针对药物或药理学活性剂而言,术语“治疗有效量”是指足以能达到或至少部分达到预期效果的药物或药剂的用量。有效量的确定因人而异,取决于受体的年龄和一般情况,也取决于具体的活性物质,个案中合适的有效量可以由本领域技术人员根据常规试验确定。
本文所用的术语“药学上可接受的”是指这些化合物、材料、组合物和/或剂型,在合理的医学判断范围内,适用于与患者组织接触而没有过度毒性、刺激性、过敏反应或其他问题或并发症,具有合理的获益/风险比,并且对预期的用途是有效。
本文所使用的,单数形式的“一个”、“一种”和“该”包括复数引用,反之亦然,除非上下文另外明确指出。
当将术语“约”应用于诸如pH、浓度、温度等的参数时,表明该参数可以变化±10%,并且有时更优选地在±5%之内。如本领域技术人员将理解的,当参数不是关键时,通常仅出于说明目的给出数字,而不是限制。
本公开化合物的合成方法
为了完成本公开的目的,本公开采用如下技术方案:
方案一
本公开通式(I)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(IC)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
环A、G、R
方案二
本公开通式(IC)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(IA)化合物或其盐与通式(IB)化合物或其盐任选地在碱性条件或酸性条件下发生芳香亲核取代反应;或者任选地在碱性条件和催化剂存在下发生偶联反应,得到通式(IC)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
环A、G、R
方案三
本公开通式(I-1)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(I-1C)化合物或其盐在碱性条件下发生酯水解反应,得到通式(I-1)的化合物或其可药用的盐;
其中:
R和R
环A、G、R
方案四
本公开通式(I-2)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(I-2C)化合物或其盐在碱性条件下发生酯水解反应,得到通式(I-2)的化合物或其可药用的盐;
其中:
R和R
环A、G、R
方案五
本公开通式(I-3)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(I-3C)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
环A、环B、G、R
方案六
本公开通式(I-3C)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(I-3A)化合物或其盐与通式(IB)化合物或其盐任选地在碱性条件或酸性条件下发生亲核取代反应;或者任选地在碱性条件和催化剂存在下发生偶联反应,得到通式(I-3C)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
环A、环B、G、R
方案七
本公开通式(I-4)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(I-4C)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
环A、环B、G、R
方案八
本公开通式(I-4C)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(I-4A)化合物或其盐与通式(IB)化合物或其盐任选地在碱性条件或酸性条件下发生亲核取代反应;或者任选地在碱性条件和催化剂存在下发生偶联反应,得到通式(I-4C)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
环A、环B、G、R
方案九
本公开通式(II)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(IIC)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
G、G
方案十
本公开通式(IIC)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(IA)化合物或其盐与通式(IIB)化合物或其盐任选地在碱性条件或酸性条件下发生亲核取代反应;或者任选地在碱性条件和催化剂存在下发生偶联反应,得到通式(IIC)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
G、G
方案十一
本公开通式(IID-1)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(IID-1A)化合物或其盐在碱性条件下发生酯水解反应,得到通式(IID-1)的化合物或其可药用的盐;
其中:
R和R
G、G
方案十二
本公开通式(IID-2)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(IID-2A)化合物或其盐在碱性条件下发生酯水解反应,得到通式(IID-2)的化合物或其可药用的盐;
其中:
R和R
G、G
方案十三
本公开通式(II-1)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(II-1C)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
G、G
方案十四
本公开通式(II-1C)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(II-1A)化合物或其盐与通式(II-1B)化合物或其盐任选地在碱性条件或酸性条件下发生亲核取代反应;或者任选地在碱性条件和催化剂存在下发生偶联反应,得到通式(II-1C)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
G、G
方案十五
本公开通式(III)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(IIIC)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
G、L
方案十六
本公开通式(IIIC)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(IA)化合物或其盐与通式(IIIB)化合物或其盐任选地在碱性条件或酸性条件下发生亲核取代反应;或者任选地在碱性条件和催化剂存在下发生偶联反应,得到通式(IIIC)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
G、L
方案十七
本公开通式(IIID-1)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(IIID-1A)化合物或其盐在碱性条件下发生酯水解反应,得到通式(IIID-1) 的化合物或其可药用的盐;
其中:
R和R
G、L
方案十八
通式(IIID-2A)化合物或其盐在碱性条件下发生酯水解反应,得到通式(IIID-2)的化合物或其可药用的盐;
其中:
R和R
G、L
方案十九
本公开通式(III-1)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(III-1C)的化合物或其可药用的盐与R
其中:
Y为卤素;优选为Br原子;
R
R和R
R
G、L
方案二十
本公开通式(III-1C)所示的化合物或其可药用的盐的制备方法,包括以下步骤:
通式(II-1A)化合物或其盐与通式(III-1B)化合物或其盐任选地在碱性条件或酸性条件下发生亲核取代反应;或者任选地在碱性条件和催化剂存在下发生偶联反应,得到通式(III-1C)的化合物或其可药用的盐;
其中:
X为卤素;优选为Cl原子;
G、L
以上合成方案中提供所述碱性条件的试剂包括有机碱和无机碱类,所述的有机碱类包括但不限于三乙胺、吡啶、N,N-二异丙基乙胺、正丁基锂、二异丙基氨基锂、醋酸钠、醋酸钾、叔丁醇钠、叔丁醇钾或1,8-二氮杂二环十一碳-7-烯,所述的无机碱类包括但不限于氢化钠、磷酸钾、碳酸钠、碳酸钾、碳酸铯、碳酸镉、氢氧化钠、一水合氢氧化锂、氢氧化锂和氢氧化钾;优选地,所述提供碱性条件的试剂选自一水合氢氧化锂、碳酸钾、碳酸铯和碳酸镉;方案二、六、八、十、十四、十六和二十中所述提供碱性条件的试剂更优选为碳酸钾或碳酸铯;方案一、五、七、九、十三、十五和十九中所述亲核取代反应中提供碱性条件的试剂更优选为碳酸镉。
方案三、四、十一、十二、十七和十八中所述碱性条件的试剂优选为一水合氢氧化锂,更优选为一水合氢氧化锂和过氧化氢。
方案一、五、七、九、十三、十五和十九中所述脱去R
以上合成方案中提供所述酸性条件的试剂包括但不限于苯六甲酸、氮硫方酸、三氯乙酸、三硝基苯磺酸、三氟甲磺酸和三氟乙酸;优选地,所述提供酸性条件的试剂为三氟乙酸。
以上合成方案中所涉及的酯水解反应优选在一水合氢氧化锂和过氧化氢条件下进行。
以上合成方案中所述的催化剂包括但不限于四(三苯基膦)钯、二氯化钯、醋酸钯、甲烷磺酸(2-二环己基膦)-3,6-二甲氧基-2',4',6'-三异丙基-1,1'-联苯)(2'-氨基-1,1'-联苯-2-基)钯(II)、1,1’-双(二苄基膦)二氯二茂铁钯、[1,1'-双(二苯基膦基)二茂铁]二氯化钯、[1,1'-双(二苯基膦基)二茂铁]二氯化钯二氯甲烷络合物、三(二亚苄基丙酮)二钯和4,5-双(二苯基膦)-9,9-二甲基氧杂蒽;优选地,催化剂选自甲烷磺酸(2-二环己基膦)-3,6-二甲氧基-2',4',6'-三异丙基-1,1'-联苯)(2'-氨基-1,1'-联苯-2-基)钯(II)、三(二亚苄基丙酮)二钯和4,5-双(二苯基膦)-9,9-二甲基氧杂蒽。
上述步骤的反应优选在溶剂中进行,所用的溶剂包括但不限于:吡啶、乙二醇二甲醚、醋酸、甲醇、乙醇、乙腈、正丁醇、甲苯、四氢呋喃、二氯甲烷、石油醚、乙酸乙酯、正己烷、二甲基亚砜、1,4-二氧六环、水、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、1,2-二溴乙烷及其混合物。
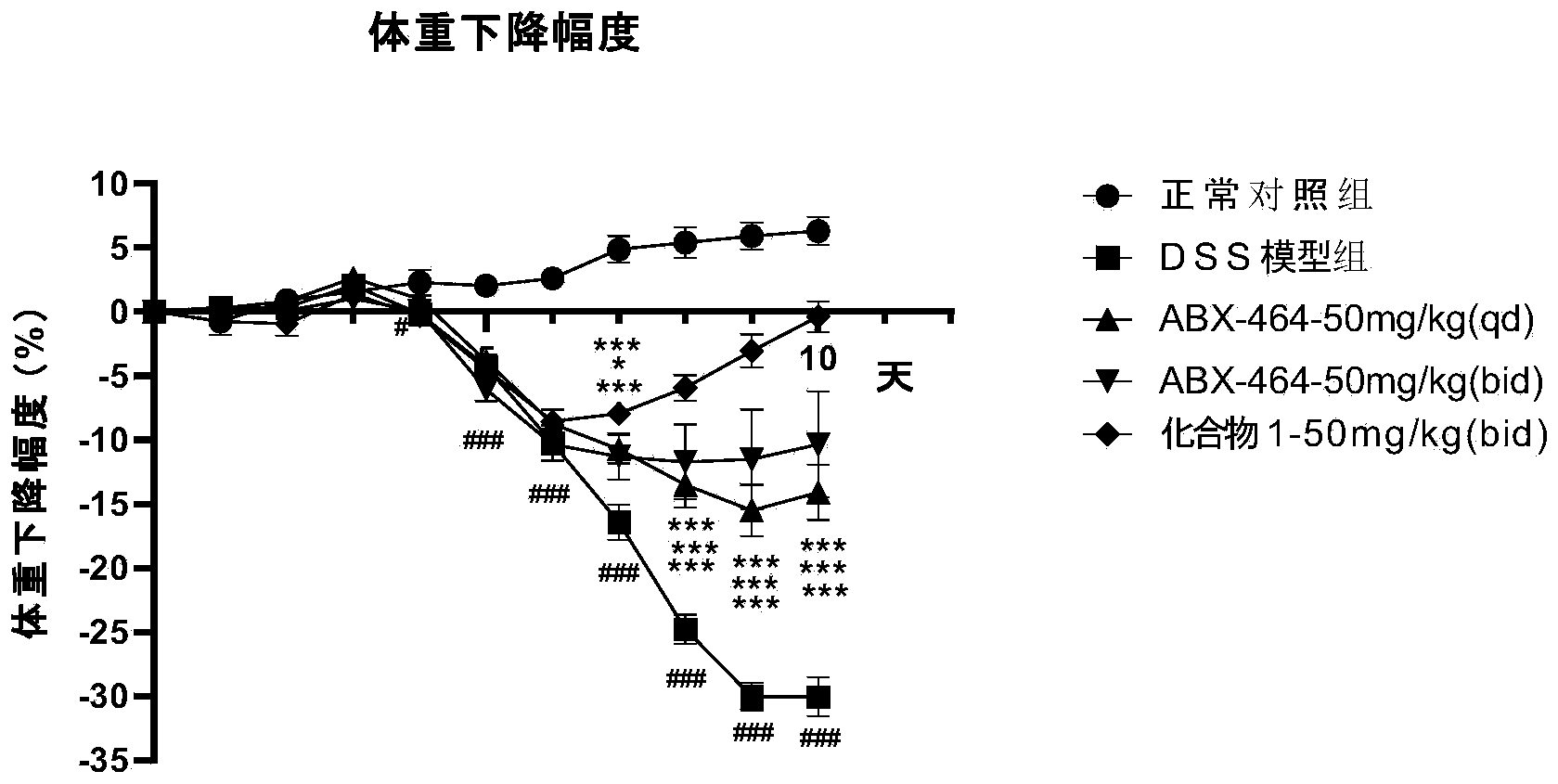
图1:本公开化合物1对硫酸钠葡聚糖(DSS)诱导的UC小鼠体重的影响。其中,#P<0.05表示模型组与正常对照组比较具有显著性差异,##P<0.01表示模型组与正常对照组比较具有高度显著性差异,###P<0.001表示模型组与正常对照组比较具有极高度显著性差异;*P<0.05表示给药组与模型组比较具有显著性差异,**P<0.01表示给药组与模型组比较具有高度显著性差异,***P<0.001表示给药组与模型组比较具有极高度显著性差异。
图2:本公开化合物1对硫酸钠葡聚糖(DSS)诱导的UC小鼠结肠长度的影响。其中,#P<0.05表示模型组与正常对照组比较具有显著性差异,##P<0.01表示模型组与正常对照组比较具有高度显著性差异,###P<0.001表示模型组与正常对照组比较具有极高度显著性差异;*P<0.05表示给药组与模型组比较具有显著性差异,**P<0.01表示给药组与模型组比较具有高度显著性差异,***P<0.001表示给药组与模型组比较具有极高度显著性差异。
以下结合实施例进一步描述本公开,但这些实施例并非限制着本公开的范围。
实施例
化合物的结构是通过核磁共振(NMR)或/和质谱(MS)来确定的。NMR位移(δ)以10
MS的测定用Agilent 1200/1290 DAD-6110/6120 Quadrupole MS液质联用仪(生产商:Agilent,MS型号:6110/6120 Quadrupole MS)、waters ACQuity UPLC-QD/SQD(生产商:waters,MS型号:waters ACQuity Qda Detector/waters SQ Detector)、THERMO Ultimate 3000-Q Exactive(生产商:THERMO,MS型号:THERMO Q Exactive)。
高效液相色谱法(HPLC)分析使用Agilent HPLC 1200DAD、Agilent HPLC 1200VWD和Waters HPLC e2695-2489高效液相色谱仪。
手性HPLC分析测定使用Agilent 1260 DAD高效液相色谱仪。
高效液相制备使用Waters 2545-2767、Waters 2767-SQ Detecor2、Shimadzu LC-20AP和Gilson GX-281制备型色谱仪。
手性制备使用Shimadzu LC-20AP制备型色谱仪。
CombiFlash快速制备仪使用Combiflash Rf200(TELEDYNE ISCO)。
薄层层析硅胶板使用烟台黄海HSGF254或青岛GF254硅胶板,薄层色谱法(TLC)使用的硅胶板采用的规格是0.15mm~0.2mm,薄层层析分离纯化产品采用的规格是0.4mm~0.5mm。
硅胶柱色谱法一般使用烟台黄海硅胶200~300目硅胶为载体。
激酶平均抑制率及IC
本公开的已知的起始原料可以采用或按照本领域已知的方法来合成,或可购买自ABCR GmbH & Co.KG、Acros Organics、Aldrich Chemical Company、韶远化学科技(Accela ChemBio Inc)、达瑞化学品等公司。
实施例中无特殊说明,反应均能够在氩气氛或氮气氛下进行。
氩气氛或氮气氛是指反应瓶连接一个约1L容积的氩气或氮气气球。
氢气氛是指反应瓶连接一个约1L容积的氢气气球。
加压氢化反应使用Parr 3916EKX型氢化仪和清蓝QL-500型氢气发生器或HC2-SS型氢化仪。
氢化反应通常抽真空,充入氢气,反复操作3次。
微波反应使用CEM Discover-S 908860型微波反应器。
实施例中无特殊说明,溶液是指水溶液。
实施例中无特殊说明,反应的温度为室温,为20℃~30℃。
实施例中的反应进程的监测采用薄层色谱法(TLC),反应所使用的展开剂,纯化化合物采用的柱层析的洗脱剂的体系和薄层色谱法的展开剂体系包括:A:二氯 甲烷/甲醇体系,B:正己烷/乙酸乙酯体系,溶剂的体积比根据化合物的极性不同而进行调节,也可以加入少量的三乙胺和醋酸等碱性或酸性试剂进行调节。
实施例1
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)喹啉-2-胺1
将2,8-二氯喹啉1a(100mg,0.51mmol,毕得医药)、5-氨基-2,2-二氟-1,3-苯并[1,3]二氧杂环戊烷1b(105mg,0.61mmol,上海皓鸿)溶解在异丙醇(1mL)中,加热升温至90℃反应12小时。冷却至室温,反应液过滤后经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:0.1%的甲酸水溶液和乙腈,乙腈的梯度:65%-85%,流速:30mL/min)分离纯化,得到标题化合物1(150mg,产率89%)。
MS m/z(ESI):335.0[M+1]。
1
实施例2
(2S,3S,4S,5R,6R)-6-((8-氯喹啉-2-基)(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)氨基)-3,4,5-三羟基四氢-2H-吡喃-2-羧酸2
第一步
(2R,3R,4S,5S,6S)-2-((8-氯喹啉-2-基)(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)氨基)-6-(甲氧基羰基)四氢-2H-吡喃-3,4,5-三醋酸酯2b
将化合物1(500mg,1.49mmol)溶解在甲苯(30mL)中,加入碳酸镉(155mg,0.90mmol),加热至140℃分水反应12小时,再加入1-溴-1-脱氧-2,3,4-三-O-乙酰基-α-D-葡萄糖醛酸甲酯2a(1.05g,2.64mmol,韶远化学科技),140℃分水反应24小时。冷却至室温,减压浓缩除去甲苯,用柱层析色谱法以洗脱剂体系B纯化得到标题化合物2b(340mg,产率35%)。
MS m/z(ESI):651.0[M+1]。
第二步
(2S,3S,4S,5R,6R)-6-((8-氯喹啉-2-基)(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)氨基)-3,4,5-三羟基四氢-2H-吡喃-2-羧酸2
将一水合氢氧化锂(450mg,10.74mmol)溶解在水(5mL)中,加入过氧化氢(1mL),室温搅拌10分钟。将该溶液加入到化合物2b(340mg,0.52mmol)的四氢呋喃溶液(15mL)中,室温搅拌2小时。用饱和硫代硫酸钠溶液(20mL)淬灭后,用1N盐酸溶液调节至pH为3,用乙酸乙酯(50mL×3)萃取,饱和氯化钠溶液(100mL)洗涤,有机相减压浓缩后经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L的碳酸氢铵水溶液和乙腈,乙腈的梯度:25%-45%,流速:30mL/min)分离纯化,得到标题化合物2(95mg,产率36%)。
MS m/z(ESI):511.0[M+1]。
1
实施例3
8-氯-N-(2,3-二氢苯并呋喃-5-基)喹啉-2-胺3
将化合物1a(100mg,0.51mmol,毕得医药)、5-氨基-2,3-二氢苯并呋喃3a(105mg,0.61mmol,毕得医药)、三氟乙酸(0.15mL,2.0mmol)加入到异丙醇(1mL)中,加热升温至90℃反应12小时。冷却至室温,反应液过滤后经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:0.1%的甲酸水溶液和乙腈,乙腈的梯度:45%-65%,流速:30mL/min)分离纯化,得到标题化合物3(115mg,产率77%)。
MS m/z(ESI):297.0[M+1]。
1
实施例4
8-氯-N-(2,3-二氢-1H-茚-5-基)喹啉-2-胺
将5-氨基茚满4a(168mg,1.26mmol,TCI)、化合物1a(100mg,0.51mmol,上海皓鸿)和三氟乙酸(173mg,1.51mmol)溶于异丙醇(3mL)中。将反应加热至100℃反应12小时。冷却至室温,反应液过滤经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:65%-85%,流速:30mL/min)分离纯化,得到标题化合物4(129mg,产率:87%)。
MS m/z(ESI):295.1[M+1]。
1
实施例5
1-(5-((8-氯喹啉-2-基)氨基)吲哚啉-1-基)乙烷-1-酮5
第一步
1-(5-硝基吲哚啉-1-基)乙烷-1-酮5b
将5-硝基二氢吲哚5a(2.0g,12.19mmol,毕得医药)、三乙胺(1.6g,15.85mmol)加入到40mL二氯甲烷,0℃缓慢加入乙酰氯(1.24g,15.85mmol)。然后自然升至室温,继续反应2小时。向体系内加入30mL水,用二氯甲烷萃取(50mL×3),合并有机相,用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物5b(1.8g,产率:72%)。
MS m/z(ESI):207.1[M+1]。
第二步
1-(5-氨基吲哚啉-1-基)乙烷-1-酮5c
将化合物5b(0.8g,3.88mmol)加入到10mL甲醇中,加入钯碳(10%,0.2g),氢气置换三次排出空气,在氢气氛下反应2小时。过滤除去钯碳,减压浓缩滤液,得到粗品标题化合物5c(0.5g),粗产品直接用于下一步反应。
MS m/z(ESI):177.1[M+1]。
第三步
1-(5-((8-氯喹啉-2-基)氨基)吲哚啉-1-基)乙烷-1-酮5
将化合物5c(100mg,0.57mmol)、2,8-二氯喹啉1a(112mg,0.57mmol,毕得医药)、三氟乙酸(64.72mg,0.57mmol)溶于异丙醇(1mL)中,然后加热至90℃反应12小时。冷却至室温,反应液减压浓缩除去溶剂,残余物用薄层色谱法(TLC),以展开剂体系B纯化得到标题产物5(48mg,产率:25%)。
MS m/z(ESI):338.1[M+1]。
1
实施例6
8-氯-N-(2,3-二氢苯并[b][1,4]二氧杂环己烷-6-基)喹啉-2-胺6
将6-氨基-1,4-苯并二氧杂环己烷6a(95mg,0.63mmol,TCI)、2,8-二氯喹啉1a(50mg,0.25mmol,毕得医药)溶于异丙醇(3mL)中。将反应液加热至100℃反应12小时。冷却至室温,反应液过滤后经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:58%-78%,流速:30mL/min)分离纯化,得到标题化合物6(63mg,产率:80%)。
MS m/z(ESI):313.1[M+1]。
1
实施例7
8-氯-N-(5,6,7,8-四氢萘-2-基)喹啉-2-胺7
将5,6,7,8-四氢-2-萘胺7a(147mg,0.51mmol,TCI)、2,8-二氯喹啉1a(100mg,0.51mmol,毕得医药)和三氟乙酸(173mg,1.51mmol)溶于异丙醇(3mL)中。将反应加热至100℃反应12小时。冷却至室温,反应液过滤,经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:60%-80%,流速:30mL/min)分离纯化,得到标题化合物7(136mg,产率:87%)。
MS m/z(ESI):309.1[M+1]。
1
实施例8
8-氯-N-(2,2,3,3-四氟-2,3-二氢苯并[b][1,4]二氧杂环己烷-6-基)喹啉-2-胺8
第一步
1-(2-溴-1,1,2,2-四氟乙氧基)-2-甲氧基-4-硝基苯8c
将2-甲氧基-4-硝基苯酚8a(2.0g,11.82mmol,毕得医药)、碳酸铯(5.8g,17.74mmol)、1,2-二溴四氟乙烷8b(6.2g,23.65mmol,上海泰坦科技)和1-丙硫醇(0.45g,5.91mmol,TCI)溶于30mL二甲亚砜,升至100℃反应12小时。冷却至室温,加入50mL的2N氢氧化钠溶液,用乙醚萃取(50mL×3),合并有机相,随 后用2N的氢氧化钠溶液洗涤(50mL×3),饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物8c(2.6g,产率:64%)。
第二步
2-(2-溴-1,1,2,2-四氟乙氧基)-5-硝基苯酚8d
将化合物8c(2.6g,7.56mmol)加入到46mL醋酸中,加入氢溴酸溶液(40%质量分数,19mL),升温至120℃,反应30小时。冷却至室温,反应加入50mL水,使用二氯甲烷萃取(50mL×3)。有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱层析色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物8d(2.0g,产率:79%)。
MS m/z(ESI):331.9[M-1]。
第三步
2,2,3,3-四氟-6-硝基-2,3-二氢苯并[b][1,4]二氧杂环己烷8e
将化合物8d(200mg,0.60mmol)溶于甲醇中,随后加入氢氧化钾(37mg,0.66mmol),反应1小时。减压除去溶剂,加入4mL环丁砜,加热至140℃反应8小时。冷却至室温,反应加入20mL水,用乙酸乙酯萃取(50mL×3),饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥。过滤,滤液减压浓缩,用硅胶柱层析色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物8e(98mg,产率:65%)。
第四步
2,2,3,3-四氟-2,3-二氢苯并[b][1,4]二氧杂环己烷-6-胺8f
将化合物8e(95mg,0.38mmol)溶于3mL甲醇中,随后加入二氧化铂,氢气置换三次排出空气,在氢气氛下反应4小时。反应液过滤,滤液减压浓缩,用硅胶柱层析色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物8f(60mg,产率:72%)。
MS m/z(ESI):224.0[M+1]。
第五步
8-氯-N-(2,2,3,3-四氟-2,3-二氢苯并[b][1,4]二氧杂环己烷-6-基)喹啉-2-胺8
将化合物8f(60mg,0.27mmol)、2,8-二氯喹啉1a(50mg,0.25mmol,毕得医药)、碳酸钾(32mg,0.30mmol)、4,5-双(二苯基膦)-9,9-二甲基氧杂蒽(52mg,0.1mmol)和三(二亚苄基丙酮)二钯(28mg,0.03mmol)溶于3mL 1,4-二氧六环中,将反应液加热至100℃反应12小时。反应冷却至室温,加入30mL水,用乙酸乙酯萃取(50mL×3),饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩,残余物经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:80%-95%,流速:30mL/min)纯化,得到标题化合物8(23mg,产率:24%)。
MS m/z(ESI):385.0[M+1]。
1
实施例9
N-(8-氯喹啉-2-基)-[1,3]二氧杂戊环并[4,5-b]吡啶-6-胺9
第一步
6-溴-[1,3]二氧杂戊环并[4,5-b]吡啶9b
将2,3-二羟基-5-溴吡啶9a(2.0g,10.53mmol,毕得医药)、无水碳酸钾(4.36g,31.55mmol)、二溴甲烷(2.2g,12.65mmol)溶于30mL N-甲基吡咯烷酮,升温至100℃反应12小时。冷至室温,倒入30mL水中,用乙酸乙酯萃取(50mL×3),合并有机相,用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱层析色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物9b(300mg,产率:14%)。
MS m/z(ESI):201.9[M+1],203.9[M+3]。
第二步
[1,3]二氧杂戊环并[4,5-b]吡啶-6-基氨基甲酸叔丁酯9c
将化合物9b(100mg,0.49mmol)、氨基甲酸叔丁酯(104mg,0.89mmol)溶于8mL 1,4-二氧六环中,氮气氛下加入三(二亚苄基丙酮)二钯(45mg,0.049mmol)、2-二环己基膦-2',4',6'-三异丙基联苯(47mg,0.098mmol)和叔丁醇钠(85mg,0.89mmol),升温至105℃,反应20小时。反应液冷却至室温,加入20mL水,用乙酸乙酯萃取(20mL×3),饱和氯化钠溶液洗涤(30mL),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱层析色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物9c(50mg,产率:42%)。
MS m/z(ESI):239.1[M+1]。
第三步
[1,3]二氧杂戊环并[4,5-b]吡啶-6-胺盐酸盐9d
将化合物9c(50mg,0.21mmol)溶于5mL二氯甲烷中,冰浴下滴加4N氯化氢的二氧六环溶液(425μL,1.7mmol),滴完后自然升至室温,室温反应3小时。反应液减压浓缩,得到粗产品标题化合物9d(36mg),粗产品直接用于下一步反应。
MS m/z(ESI):139.0[M+1]。
第四步
N-(8-氯喹啉-2-基)-[1,3]二氧杂戊环并[4,5-b]吡啶-6-胺9
将化合物9d(36mg,0.21mmol)、2,8-二氯喹啉1a(30mg,0.15mmol,毕得医药)、碳酸铯(148mg,0.45mmol)、甲烷磺酸(2-二环己基膦)-3,6-二甲氧基-2',4',6'-三异丙基-1,1'-联苯)(2'-氨基-1,1'-联苯-2-基)钯(II)(20mg,0.02mmol)溶于5mL 1,4-二氧六环中,将反应液加热至100℃反应12小时。冷却至室温,加入30mL水,用乙酸乙酯萃取(50mL×3),饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩,残余物经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:千分之一的三氟乙酸水溶液和乙腈,乙腈的梯度:50%-95%,流速:30mL/min)分离纯化,得到标题化合物9(12mg,产率:26%)。
MS m/z(ESI):300.0[M+1]。
1
实施例10
N-(苯并[d][1,3]二氧杂环戊烷-5-基)-8-氯喹啉-2-胺10
将化合物苯并[d][1,3]二氧杂环戊烷-5-胺10a(693mg,5.05mmol,毕得医药)、化合物1a(1.0g,5.05mmol)、碳酸铯(2.64g,8.10mmol)、4,5-双二苯基膦-9,9-二甲基氧杂蒽(584mg,1.01mmol)、三(二亚苄基丙酮)二钯(462mg,0.50mol)溶于30mL 1,4-二氧六环中,将反应液加热至105℃反应12小时。反应液冷却至室温,加入25mL水,用乙酸乙酯萃取(25mL×3),饱和氯化钠溶液洗涤(25mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩,残余物经高效液相制备色谱法(Waters-2545,洗脱体系:10mmol/L的碳酸氢铵水溶液和乙腈,乙腈的梯度:10%-26%,流速:30mL/min)纯化,得到目标化合物10(750mg,产率:50%)。
MS m/z(ESI):299.0[M+1]。
1
实施例11
N-(苯并[d][1,3]二氧杂环戊烷-5-基2,2-二氘)-8-氯喹啉-2-胺11
将化合物苯并[d][1,3]二氧杂环戊烷-2,2-二氘-5-胺11a(170mg,1.21mmol,采用专利申请“WO2012037351A1中说明书第41页的中间体合成method 2”公开的方法制备而得)、化合物1a(240mg,1.21mmol)、碳酸铯(636mg,1.95mmol)、4,5-双二苯基膦-9,9-二甲基氧杂蒽(141mg,0.24mmol)、三(二亚苄基丙酮)二钯(111mg,0.12mol)溶于10mL 1,4-二氧六环中,将反应加热至105℃反应12小时。反应冷却至室温,加入25mL水,用乙酸乙酯萃取(25mL×3),饱和氯化钠溶液洗涤(25mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩,残余物经高效液相制备色谱法(Waters-2767 Autopurification,洗脱体系:千分之一的甲酸水溶液和乙腈,乙腈的梯度:45%-95%,流速:30mL/min)分离纯化,得到目标化合物11(100mg,产率:27%)。
MS m/z(ESI):300.9[M+1]。
1
实施例12
8-氯-N-(2,2-二甲基苯并[d][1,3]二氧杂环戊烷-5-基)喹啉-2-胺12
将化合物2,2-二甲基苯并[d][1,3]二氧杂环戊烷-5-胺12a(141mg,0.85mmol,上海皓鸿生物医药)、化合物1a(146mg,0.74mmol)、碳酸铯(362mg,1.11mmol)、三(二亚苄基丙酮)二钯(68mg,0.074mmol)、4,5-双二苯基膦-9,9-二甲基氧杂蒽(86mg,0.15mmol)溶于5mL 1,4-二氧六环中,将反应加热至105℃反应12小时。反应冷却至室温,加入30mL水,用乙酸乙酯萃取(50mL×3),饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩,经高效液相制备色谱法(Waters-2545,洗脱体系:10mmol/L的碳酸氢铵水溶液和乙腈,乙腈的梯度:60%-95%,流速:30mL/min)分离纯化,得到目标化合物12(132mg,产率:55%)。
MS m/z(ESI):327.1[M+1]。
1
实施例13
8-氯-N-(2,2-二甲基-2,3-二氢苯并呋喃-5-基)喹啉-2-胺13
将化合物2,2-二甲基-2,3-二氢苯并呋喃-5-胺13a(59mg,0.36mmol,采用专利申请“WO2014169845中说明书第72页的实施例13”公开的方法制备而得)、化合物1a(60mg,0.30mmol)、三氟乙酸(62mg,0.54mmol)和2mL异丙醇置于25mL封管中,将反应加热至90℃反应12小时。反应液冷却至室温,加入15mL水,用饱和碳酸氢钠溶液调节至pH为8左右,用乙酸乙酯萃取(25mL×3),饱和氯化钠溶液洗涤(25mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩,残余物经高效液相制备色谱法(Waters-2767 Autopurification,洗脱体系:千分之一的甲酸水溶液和甲醇,甲醇的梯度:60%-95%,流速:30mL/min)分离纯化,得到目标化合物13(55mg,产率:56%)。
MS m/z(ESI):325.1[M+1]。
1
实施例14
5-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-8,9-二氢-7H-环戊烷并[f]喹啉-3-胺14
第一步
6-氯-2,3-二氢-1H-茚-5-胺14b
向100mL单口瓶中依次加入50mL二氯甲烷、(6-氯-2,3-二氢-1H-茚-5-基)氨基甲酸叔丁酯14a(3.4g,12.73mmol,采用公知的方法“ACS Catalysis,2018,8,4783–4788”制备而得)、三氟乙酸10mL,室温反应3小时。减压浓缩除去溶剂,用100mL二氯甲烷稀释,用饱和碳酸氢钠溶液洗涤(50mL×2),饱和氯化钠溶液洗涤(50mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩得到粗产品标题化合物14b(1.55g,产率:73%),粗产品直接用于下一步反应。
MS m/z(ESI):168.1[M+1]。
第二步
N-(6-氯-2,3-二氢-1H-茚-5-基)-3,3-二甲氧基丙酰胺14d
向100mL三口瓶中依次加入16mL四氢呋喃、化合物14b(1.55g,9.28mmol)、3,3-二甲氧基丙酸甲酯14c(1.65g,11.14mmol,百灵威),然后0℃缓慢滴加双(三甲基硅基)氨基钠(2M四氢呋喃溶液,6.96mL,13.92mmol)。反应自然升至室温,反应12小时。冰浴下向体系内加入50mL饱和碳酸氢铵溶液淬灭反应,用乙酸乙酯萃取(50mL×3)。合并有机相,用饱和氯化钠溶液洗涤(50mL×3),有机 相干燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物14d(950mg,产率:36%)。
第三步
5-氯-4,7,8,9-四氢-3H-环戊烷并[f]喹啉-3-酮14e
向50mL单口瓶中依次加入10mL二氯甲烷、化合物14d(800mg,2.83mmol),0℃向体系内缓慢滴加浓硫酸(2.27mL,42.40mmol),加完后室温反应30分钟。减压浓缩除去溶剂,将残余物滴入到冰水中析出固体,过滤,固体干燥得到标题化合物14e(500mg,产率:81%)。
MS m/z(ESI):220.0[M+1]。
第四步
3,5-二氯-8,9-二氢-7H-环戊烷并[f]喹啉14f
向50mL单口瓶中依次加入2mL N,N-二甲基甲酰胺、化合物14e(300mg,1.37mmol),将体系温度升至95℃后缓慢滴加三氯氧磷(0.1mL,1.10mmol),继续反应30分钟。减压浓缩除去三氯氧磷,加入20mL乙酸乙酯稀释,用0.2N氢氧化钠溶液洗涤(30mL×2),饱和氯化钠溶液洗涤(30mL×3)。有机相用无水硫酸钠干燥,过滤,滤液减压浓缩得到粗品标题化合物14f(0.25g,产率:78%),粗品直接用于下一步反应。
MS m/z(ESI):238.0[M+1]。
第五步
5-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-8,9-二氢-7H-环戊烷并[f]喹啉-3-胺14
向25mL单口瓶中依次加入2mL异丙醇、化合物14f(100mg,0.42mmol)、三氟乙酸(47.89mg,0.42mmol)、化合物1b(73mg,0.42mmol),然后将温度升至85℃反应16小时。减压除去溶剂,用1mL甲醇稀释,采用薄层色谱法(TLC),展开剂体系B纯化得标题化合物14(78mg,产率:49%)。
MS m/z(ESI):375.0[M+1]。
1
实施例15
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-7-甲基喹啉-2-胺15
第一步
N-(2-氯-3-甲基苯基)-3,3-二甲氧基丙酰胺15b
向100mL三口瓶中依次加入25mL四氢呋喃、2-氯-3-甲基苯胺15a(1.5g,10.59mmol,毕得医药)、化合物14c(1.88g,11.69mmol),将体系温度降至0℃后缓慢滴加双(三甲基硅基)氨基钠(2M四氢呋喃溶液,10.57mL,21.14mmol),然后体系升至室温,反应24小时。向体系内加入50mL饱和碳酸氢钠水溶液淬灭反应,反应液减压浓缩除去大部分有机相,用乙酸乙酯萃取(50mL×3),合并有机相并用饱和氯化钠溶液洗涤(50mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,得到粗品标题化合物15b(2.70g,产率:98%),粗产品直接用于下一步反应。
第二步
8-氯-7-甲基喹啉-2(1H)-酮15c
向100mL单口瓶中依次加入15mL二氯甲烷、化合物15b(2.70g,10.48mmol),0℃缓慢滴加浓硫酸(8.37mL,157.11mmol),加完后撤去冰浴,继续反应16小时。减压除去溶剂,将残余物滴入到冰水中析出固体,过滤,固体干燥得到标题化合物15c(1.8g,产率:89%)。
MS m/z(ESI):194.0[M+1]。
第三步
2,8-二氯-7-甲基喹啉15d
向50mL单口瓶中依次加入化合物15c(800mg,4.67mmol)、三氯氧磷(3.16g,20.66mmol),将体系温度升至95℃反应90分钟。减压除去三氯氧磷,加入冰水50mL,用饱和碳酸氢钠溶液调节至pH为8左右,用乙酸乙酯萃取(50mL×3),有机相用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物15d(620mg,产率:71%)。
MS m/z(ESI):211.9[M+1]。
第四步
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-7-甲基喹啉-2-胺15
向25mL封管中依次加入8mL异丙醇、化合物15d(300mg,1.29mmol)、化合物1b(268mg,1.54mmol)、三氟乙酸(260mg,2.28mmol)。然后将反应升温至95℃反应16小时。减压除去溶剂,加入乙酸乙酯100mL稀释,用饱和碳酸氢钠溶液洗涤(50mL×2),饱和氯化钠溶液洗涤(50mL×2)。有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,残余物经高效液相制备色谱法(Waters-2767 Autopurification,洗脱体系:千分之一的甲酸水溶液和甲醇,甲醇的梯度:65%-95%,流速:30mL/min)纯化,得到标题化合物15(300mg,产率:73%)。
MS m/z(ESI):349.1[M+1]。
1
实施例16
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-7-甲氧基喹啉-2-胺16
第一步
N-(2-氯-3-甲氧基苯基)-3,3-二甲氧基丙酰胺16b
向100mL三口瓶中依次加入25mL四氢呋喃、2-氯-3-甲氧基苯胺16a(1.5g,9.51mmol,毕得医药)、化合物14c(1.69g,11.40mmol),0℃缓慢滴加双(三甲基硅基)氨基钠(2M四氢呋喃溶液,9.51mL,19.03mmol),自然升至室温,反应24小时。向体系内加入50mL饱和碳酸氢钠溶液淬灭反应,反应液减压浓缩除去大部分有机相,用乙酸乙酯萃取(50mL×3),合并有机相,用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩,得到粗产品标题化合物16b(2.50g,产率:96%),粗产品直接用于下一步反应。
第二步
8-氯-7-甲氧基喹啉-2(1H)-酮16c
向100mL单口瓶中依次加入10mL二氯甲烷、化合物16b(2.50g,9.13mmol),0℃缓慢滴加浓硫酸(7.29mL,136.93mmol),撤去冰浴,继续反应16小时。减压除去溶剂,将残余物滴入到冰水中析出固体,过滤,固体干燥得到标题化合物16c(1.6g,产率:84%)。
MS m/z(ESI):210.0[M+1]。
第三步
2,8-二氯-7-甲氧基喹啉16d
向50mL单口瓶中依次加入甲苯8mL、化合物16c(800mg,3.81mmol)、三氯氧磷(1.17g,7.63mmol),将体系温度升至100℃反应3小时。反应冷至室温,加入20mL冰水,用饱和碳酸氢钠调节至pH为8左右,用乙酸乙酯萃取(50mL×3),有机相用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物16d(680mg,产率:78%)。
MS m/z(ESI):227.9[M+1]。
第四步
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-7-甲氧基喹啉-2-胺16
向25mL封管中依次加入4mL异丙醇、化合物16d(250mg,1.09mmol)、化合物1b(227mg,1.31mmol)、三氟乙酸(224mg,1.96mmol),然后将温度升至95℃后反应16小时。减压浓缩除去溶剂,加入乙酸乙酯100mL稀释,用饱和碳酸氢钠溶液洗涤(50mL×2),饱和氯化钠溶液洗涤(50mL×2)。有机相用无水硫酸钠干燥,过滤,浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得目标产物16(280mg,产率:70%)。
MS m/z(ESI):365.1[M+1]。
1
实施例17
7,8-二氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-喹啉-2-胺17
第一步
N-((2,3-二氯苯基)-3,3-二甲氧基丙酰胺17b
向100mL三口瓶中依次加入25mL四氢呋喃、2,3-二氯苯胺17a(1.5g,9.26mmol,百灵威)、化合物14c(1.65g,11.11mmol),0℃缓慢滴加双(三甲基硅基)氨基钠(2M四氢呋喃溶液,9.30mL,18.60mmol),然后体系升至室温,反应24小时。向体系内加入50mL饱和碳酸氢钠水溶液淬灭反应,反应液减压浓缩除去大部分有机相,用乙酸乙酯萃取(50mL×3),合并有机相,用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩,得到粗品标题化合物17b(2.50g,产率:97%),粗产品直接用于下一步反应。
第二步
7,8-二氯喹啉-2(1H)-酮17c
向100mL单口瓶中依次加入15mL二氯甲烷、化合物17b(2.50g,8.99mmol),0℃缓慢滴加浓硫酸(7.18mL,134.79mmol),然后撤去冰浴,继续反应16小时。减压除去溶剂,将残余物滴入到冰水中析出固体,过滤,固体干燥得到标题化合物17c(1.70g,产率:89%)。
MS m/z(ESI):214.0[M+1]。
第三步
2,7,8-三氯喹啉17d
向50mL单口瓶中依次加入化合物17c(1.0g,4.67mmol)、三氯氧磷(3.58g,23.36mmol),将体系温度升至95℃反应90分钟。减压除去三氯氧磷,加入50mL冰水,用饱和碳酸氢钠调节至pH为8左右,用乙酸乙酯萃取(50mL×3),有机相用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得标题化合物17d(950mg,产率:87%)。
MS m/z(ESI):231.9[M+1]。
第四步
7,8-二氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-喹啉-2-胺17
向25mL封管反应瓶中依次加入8mL异丙醇、化合物17d(300mg,1.29 mmol)、化合物1b(268mg,1.54mmol)、三氟乙酸(260mg,2.28mmol),然后将温度升至95℃反应16小时。减压除去溶剂,加入100mL乙酸乙酯稀释,用饱和碳酸氢钠溶液洗涤(50mL×2),饱和氯化钠溶液洗涤(50mL×2)。有机相用无水硫酸钠干燥,过滤,浓缩,用硅胶柱色谱法以洗脱剂体系A纯化所得残余物,得目标产物17(300mg,产率:63%)。
MS m/z(ESI):369.0[M+1]。
1
实施例18
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-6-甲基喹啉-2-胺18
第一步
N-(2-氯-4-甲基苯基)-3,3-二甲氧基丙酰胺18b
向100mL三口瓶中依次加入25mL四氢呋喃、2-氯-4-甲基苯胺18a(1g,7.06mmol,毕得医药)、化合物14c(1.26g,8.47mmol),0℃缓慢滴加双(三甲基硅基)氨基钠(2M四氢呋喃溶液,5.30mL,10.60mmol),然后体系升至室温,反应24小时。向体系内加入50mL饱和碳酸氢钠水溶液淬灭反应,反应液减压浓缩除去大部分有机相,用乙酸乙酯萃取(50mL×3),合并有机相,用饱和氯化钠溶液洗涤(50mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物18b(1.50g,产率:82%)。
第二步
8-氯-6-甲基喹啉-2(1H)-酮18c
向100mL单口瓶中依次加入2mL二氯甲烷、化合物18b(1.50g,5.82mmol), 0℃缓慢滴加浓硫酸(4.7mL,87.31mmol),然后撤去冰浴,继续反应4小时。减压浓缩除去溶剂,将残余物滴入到冰水中析出固体,过滤,固体干燥得到标题化合物18c(0.8g,产率:71%)。
MS m/z(ESI):194.0[M+1]。
第三步
2,8-二氯-6-甲基喹啉18d
向50mL单口瓶中依次加入化合物18c(0.5g,2.58mmol)、三氯氧磷2mL,将体系温度升至95℃反应90分钟。减压除去三氯氧磷,加入冰水50mL,用乙酸乙酯萃取(50mL×3),有机相用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物18d(0.3g,产率:55%)。
MS m/z(ESI):212.0[M+1]。
第四步
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-6-甲基喹啉-2-胺18
向25mL封管中依次加入3mL异丙醇、化合物18d(50mg,0.24mmol)、化合物1b(102mg,0.59mmol)、三氟乙酸(67mg,0.59mmol),然后将温度升至95℃反应16小时。反应冷却至室温,加入25mL水,用乙酸乙酯萃取(25mL×3),饱和氯化钠溶液洗涤(25mL×2),无水硫酸钠干燥。减压浓缩除去溶剂,残余物经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:70%-90%,流速:30mL/min)纯化,得到标题化合物18(55mg,产率:67%)。
MS m/z(ESI):349.1[M+1]。
1
实施例19
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-6-甲氧基喹啉-2-胺19
第一步
N-(2-氯-4-甲氧基苯基)-3,3-二甲氧基丙酰胺19b
向100mL三口瓶中依次加入25mL四氢呋喃、2-氯-4-甲氧基苯胺19a(1g,6.35mmol,毕得医药)、化合物14c(1.13g,7.61mmol),0℃缓慢滴加双(三甲基硅基)氨基钠(2M四氢呋喃溶液,4.76mL,9.52mmol),然后体系升至室温,反应24小时。向体系内加入50mL饱和碳酸氢钠水溶液淬灭反应,反应液减压浓缩除去大部分有机相,用乙酸乙酯萃取(50mL×3),合并有机相并用饱和氯化钠溶液洗涤(50mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物19b(1.20g,产率:69%)。
第二步
8-氯-6-甲氧基喹啉-2(1H)-酮19c
向100mL单口瓶中依次加入2mL二氯甲烷、化合物19b(1.20g,4.38mmol),0℃缓慢滴加浓硫酸(3.52mL,65.76mmol),然后撤去冰浴,继续反应4小时。减压除去溶剂,将残余物滴入到冰水中析出固体,过滤,固体干燥得到标题化合物19c(0.6g,产率:65%)。
MS m/z(ESI):210.0[M+1]。
第三步
2,8-二氯-6-甲氧基喹啉19d
向50mL单口瓶中依次加入化合物19c(500mg,2.39mmol)、三氯氧磷2.5mL,将体系温度升至95℃反应90分钟。减压除去三氯氧磷后,加入50mL冰水,用乙酸乙酯萃取(50mL×3),有机层用饱和氯化钠溶液洗(50mL×2),无水硫酸钠干燥,过滤,浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物19d(75mg,产率:14%)。
MS m/z(ESI):228.0[M+1]。
第四步
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-6-甲氧基喹啉-2-胺19
向25mL封管反应瓶中依次加入3mL异丙醇、化合物19d(75mg,0.33 mmol)、化合物1b(142mg,0.82mmol)、三氟乙酸(94mg,0.82mmol),然后将温度升至95℃后反应16小时。反应冷却至室温,加入25mL水,用乙酸乙酯萃取(25mL×3),饱和氯化钠溶液洗涤(25mL×2),无水硫酸钠干燥。减压除去溶剂,残余物经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:65%-85%,流速:30mL/min)纯化,得到标题化合物19(55mg,产率:67%)。
MS m/z(ESI):365.1[M+1]。
1
实施例20
6,8-二氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)喹啉-2-胺20
第一步
N-(2,4-二氯苯基)-3,3-二甲氧基丙酰胺20b
向100mL三口瓶中依次加入25mL四氢呋喃、2,4-二氯苯胺20a(1.0g,6.17mmol,百灵威)、化合物14c(1.10g,7.41mmol),0℃缓慢滴加双(三甲基硅基)氨基钠(2M四氢呋喃溶液,4.63mL,9.26mmol),然后体系升至室温,反应24小时。向体系内加入50mL饱和碳酸氢钠水溶液淬灭反应,反应液减压浓缩除去大部分有机相,用乙酸乙酯萃取(50mL×3),合并有机相用饱和氯化钠溶液洗涤(50mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物20b(1.40g,产率:82%)。
第二步
6,8-二氯喹啉-2(1H)-酮20c
向100mL单口瓶中依次加入2mL二氯甲烷、化合物20b(1.20g,4.31mmol),0℃缓慢滴加浓硫酸(3.52mL,65.76mmol),然后撤去冰浴,加热至90℃继续反应4小时。减压除去溶剂,将残余物滴入到冰水中析出固体,过滤,固体干燥得到标题化合物20c(0.5g,产率:54%)。
MS m/z(ESI):213.9[M+1]。
第三步
2,6,8-三氯喹啉20d
向50mL单口瓶中依次加入化合物20c(300mg,1.40mmol)、三氯氧磷2mL,将体系温度升至95℃反应90分钟。减压除去三氯氧磷,加入冰水50mL,用乙酸乙酯萃取(50mL×3),有机相用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物20d(240mg,产率:74%)。
MS m/z(ESI):231.9[M+1]。
第四步
6,8-二氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)喹啉-2-胺20
向25mL封管中依次加入3mL异丙醇、化合物20d(100mg,0.43mmol)、化合物1b(82mg,0.47mmol)、三氟乙酸(123mg,1.08mmol),然后将温度升至95℃反应16小时。反应冷却至室温,加入25mL水,用乙酸乙酯萃取(25mL×3),饱和氯化钠溶液洗涤(25mL×2),无水硫酸钠干燥。减压浓缩除去溶剂,残余物经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:80%-95%,流速:30mL/min)纯化,得到标题化合物20(55mg,产率:63%)。
MS m/z(ESI):369.0[M+1]。
1
实施例21
(2S,3S,4S,5R,6R)-6-(苯并[d][1,3]二氧杂环戊烷-5-基(8-氯喹啉-2-基)氨基)-3,4,5-三羟基四氢-2H-吡喃-2-羧酸21
第一步
(2R,3R,4S,5S,6S)-2-(苯并[d][1,3]二氧杂环戊烷-5-基)(8-氯喹啉-2-基)氨基)-6-(甲氧基羰基)四氢-2H-吡喃-3,4,5-三醋酸酯21a
将化合物10(380mg,1.27mmol)溶解在甲苯(15mL)中,加入碳酸镉(131mg,0.76mmol),加热至145℃分水反应12小时,再加入化合物2a(606mg,1.53mmol),145℃分水反应24小时。冷却至室温,减压浓缩除去甲苯,残余物用柱层析色谱法以洗脱剂体系B纯化得到标题化合物21a(450mg,产率58%)。
MS m/z(ESI):615.0[M+1]。
第二步
(2S,3S,4S,5R,6R)-6-(苯并[d][1,3]二氧杂环戊烷-5-基(8-氯喹啉-2-基)氨基)-3,4,5-三羟基四氢-2H-吡喃-2-羧酸21
将一水合氢氧化锂(645mg,15.35mmol)溶解在水(4mL)中,加入30%过氧化氢溶液(1.83mL),室温搅拌10分钟。将该溶液加入到化合物21a(450mg,0.51mmol)的四氢呋喃溶液(16mL)中,室温搅拌16小时。用饱和硫代硫酸钠溶液(20mL)淬灭后,用1N盐酸溶液调节至pH为4,用乙酸乙酯(50mL×3)萃取,饱和氯化钠溶液(100mL)洗涤,有机相减压浓缩后经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L的碳酸氢铵水溶液和乙腈,乙腈的梯度:15%-95%,流速:30mL/min)纯化,得到标题化合物21(85mg,产率35%)。
MS m/z(ESI):475.1[M+1]。
1
实施例22
(2S,3S,4S,5R,6R)-6-((8-氯喹啉-2-基)(2,3-二氢苯并[b][1,4]二氧杂环己烷-6-基)氨基)-3,4,5-三羟基四氢-2H-吡喃-2-羧酸22
第一步
(2R,3R,4S,5S,6S)-2-((8-氯喹啉-2-基)(2,3-二氢苯并[b][1,4]二氧杂环己烷-6-基)氨基)-6-(甲氧基羰基)四氢-2H-吡喃-3,4,5-三醋酸酯22a
将化合物6(500mg,1.60mmol)溶解在甲苯(30mL)中,加入碳酸镉(165mg,0.96mmol),加热至140℃分水反应12小时,再加入化合物2a(1.90g,4.80mmol),140℃分水反应24小时。冷却至室温,减压浓缩除去甲苯,残余物用柱层析色谱法以洗脱剂体系B纯化得到标题化合物22a(400mg,产率40%)。
MS m/z(ESI):629.0[M+1]。
第二步
(2S,3S,4S,5R,6R)-6-((8-氯喹啉-2-基)(2,3-二氢苯并[b][1,4]二氧杂环己烷-6-基)氨基)-3,4,5-三羟基四氢-2H-吡喃-2-羧酸22
将一水合氢氧化锂(534mg,12.72mmol)溶解在水(5mL)中,加入30%过氧化氢溶液(1.1mL),室温搅拌10分钟。将该溶液加入到化合物22a(400mg,0.64mmol)的四氢呋喃溶液(15mL)中,室温搅拌2小时。用饱和硫代硫酸钠溶液(20mL)淬灭后,用1N盐酸溶液调节至pH为4,用乙酸乙酯(50mL×3)萃取,饱和氯化钠溶液(100mL)洗涤,有机相减压浓缩后经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L的碳酸氢铵水溶液和乙腈,乙腈的梯度:20%-65%, 流速:30mL/min)纯化,得到标题化合物22(95mg,产率36%)。
MS m/z(ESI):489.1[M+1]。
1
实施例23
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-5-甲基喹啉-2-胺23
第一步
N-(2-氯-5-甲基苯基)-3,3-二乙氧基丙酰胺23c
向100mL三口瓶中依次加入5mL N,N-二甲基甲酰胺、2-氯-5-甲基苯胺23a(0.5g,3.53mmol,毕得医药)、3,3-二乙氧基丙酸23b(0.63g,3.88mmol,百灵威),0℃缓慢加入2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(2.01g,5.29mmol)、N,N-二异丙基乙胺(0.91g,7.06mmol),然后体系升至室温,反应24小时。向体系内加入50mL饱和碳酸氢钠溶液淬灭反应,用乙酸乙酯萃取(50mL×3),合并有机相,用饱和氯化钠溶液洗涤(50mL×3),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,得到粗产品标题化合物23c(1.0g,产率:99%),粗产品直接用于下一步反应。
第二步
8-氯-5-甲基喹啉-2(1H)-酮23d
向100mL单口瓶中依次加入2mL二氯甲烷、化合物23c(1.00g,3.5mmol),0℃缓慢滴加浓硫酸(3.35mL,62.98mmol),然后撤去冰浴,继续反应4小时。减 压除去溶剂,将残余物滴入到50mL冰水中,用乙酸乙酯萃取(50mL×5),合并有机相,用饱和氯化钠溶液洗(50mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物23d(0.26g,产率:38%)。
MS m/z(ESI):194.0[M+1]。
第三步
2,8-二氯-5-甲基喹啉23e
向50mL单口瓶中依次加入化合物23d(0.26g,1.34mmol)、三氯氧磷(2mL),将体系温度升至95℃反应90分钟。减压除去三氯氧磷,加入冰水(50mL),用乙酸乙酯萃取(50mL×3),有机相用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,浓缩,得到粗产品标题化合物23e(0.28g,产率:99%),粗产品直接用于下一步反应。
MS m/z(ESI):212.0[M+1]。
第四步
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-5-甲基喹啉-2-胺23
向25mL封管中依次加入3mL异丙醇、化合物23e(100mg,0.47mmol)、化合物1b(97mg,0.56mmol)、三氟乙酸(161mg,1.41mmol),然后将温度升至95℃反应16小时。反应冷却至室温,加入25mL水,用乙酸乙酯萃取(25mL×3),饱和氯化钠溶液洗涤(25mL×2),无水硫酸钠干燥。减压除去溶剂,残余物经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:70%-90%,流速:30mL/min)纯化,得到标题化合物23(80mg,产率:48%)。
MS m/z(ESI):349.1[M+1]。
1
实施例24
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-5-甲氧基喹啉-2-胺24
第一步
N-(2-氯-5-甲氧基苯基)-3,3-二乙氧基丙酰胺24b
向100mL三口瓶中依次加入5mL N,N-二甲基甲酰胺、2-氯-5-甲氧基苯胺24a(0.5g,3.17mmol,毕得医药)、化合物23b(0.63g,3.88mmol),0℃缓慢加入2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(2.01g,5.29mmol)、N,N-二异丙基乙胺(0.91g,7.06mmol),然后体系升至室温,反应24小时。向体系内加入50mL饱和碳酸氢钠溶液淬灭反应,用乙酸乙酯萃取(50mL×3),合并有机相,用饱和氯化钠溶液洗涤(50mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,得到粗产品标题化合物24b(0.95g,产率:99%),粗产品直接用于下一步反应。
第二步
8-氯-5-甲氧基喹啉-2(1H)-酮24c
向100mL单口瓶中依次加入2mL二氯甲烷、化合物24b(0.95g,3.14mmol),0℃缓慢滴加浓硫酸(2.52mL,47.22mmol),然后撤去冰浴,继续反应4小时。减压除去溶剂,将残余物滴入到50mL冰水中,用乙酸乙酯萃取(50mL×5),合并有机相,用饱和氯化钠溶液洗涤(50mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物24c(0.26g,产率:40%)。
MS m/z(ESI):210.0[M+1]。
第三步
2,8-二氯-5-甲氧基喹啉24d
向50mL单口瓶中依次加入化合物24c(365mg,1.26mmol)、三氯氧磷2.5mL,将体系温度升至95℃反应90分钟。减压除去三氯氧磷,加入冰水50mL,用乙酸乙酯萃取(50mL×3),有机层用饱和氯化钠溶液洗(50mL×2),无水硫酸钠干燥,过滤,浓缩,得到粗产品标题化合物24d(288mg,产率:99%),粗产品直接用于下一步反应。
MS m/z(ESI):228.0[M+1]。
第四步
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-5-甲氧基喹啉-2-胺24
向25mL封管中依次加入3mL异丙醇、化合物24d(288mg,1.26mmol)、化合物1b(218mg,1.26mmol)、三氟乙酸(431mg,3.78mmol),然后将温度升至95℃反应16小时。反应冷却至室温,加入25mL水,用乙酸乙酯萃取(25mL×3),饱和氯化钠溶液洗涤(25mL×2),无水硫酸钠干燥。减压除去溶剂,残余物经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:65%-85%,流速:30mL/min)纯化,得到标题化合物24(230mg,产率:49%)。
MS m/z(ESI):365.1[M+1]。
1
实施例25
5,8-二氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)喹啉-2-胺25
第一步
N-(2,5-二氯苯基)-3,3-二乙氧基丙酰胺25b
向100mL三口瓶中依次加入5mL N,N-二甲基甲酰胺、2,5-二氯苯胺25a(0.5g,3.08mmol,毕得医药)、化合物23b(0.50g,3.39mmol),0℃缓慢加入2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(2.01g,5.29mmol)、N,N-二异丙基乙胺(0.91g,7.06mmol),然后体系升至室温,反应24小时。向体系内加入50mL饱和碳酸氢钠水溶液淬灭反应,用乙酸乙酯萃取(50mL×3),合并有机相,用饱和氯化钠溶液洗涤(50mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,得到粗产品标题化合物25b(0.94g,产率:99%),粗产品直接用于下一步反应。
第二步
5,8-二氯喹啉-2(1H)-酮25c
向100mL单口瓶中依次加入2mL二氯甲烷、化合物25b(0.51g,1.67mmol),0℃缓慢滴加浓硫酸(1.33mL,25.05mmol),然后撤去冰浴,继续反应4小时。减压除去溶剂,将反应液滴入到50mL冰水中,用乙酸乙酯萃取(50mL×3),合并有机相,用饱和氯化钠溶液洗涤(50mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物25c(0.21g,产率:56%)。
MS m/z(ESI):214.1[M+1]。
第三步
2,5,8-三氯喹啉25d
向50mL单口瓶中依次加入化合物25c(213mg,0.99mmol)、三氯氧磷2mL,将体系温度升至95℃反应90分钟。减压除去三氯氧磷,加入冰水50mL,用乙酸乙酯萃取(50mL×3),有机相用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,浓缩,得到粗产品标题化合物25d(230mg,产率:99%),粗产品直接用于下一步反应。
MS m/z(ESI):231.9[M+1]。
第四步
5,8-二氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)喹啉-2-胺25
向25mL封管中依次加入3mL异丙醇、化合物25d(234mg,1.01mmol)、化合物1b(174mg,1.01mmol)、三氟乙酸(344mg,3.02mmol),然后将温度升至95℃反应16小时。反应冷却至室温,加入25mL水,用乙酸乙酯萃取(25mL×3),饱和氯化钠溶液洗涤(25mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩除去溶剂,残余物经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:80%-95%,流速:30mL/min)纯化,得到标题化合物25(230mg,产率:59%)。
MS m/z(ESI):369.0[M+1]。
1
实施例26
8-氯-2-((2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)氨基)喹啉-6-腈26
第一步
N-(4-溴-2-氯苯基)-3,3-二甲氧基丙酰胺26b
向100mL三口瓶中依次加入25mL四氢呋喃、4-溴-2-氯苯胺26a(3.0g,14.53mmol,百灵威)、化合物14c(2.36g,15.98mmol),0℃缓慢滴加双(三甲基硅基)氨基钠(2M四氢呋喃溶液,10.89mL,21.79mmol),然后体系升至室温,反应24小时。向体系内加入50mL饱和氯化铵溶液淬灭反应,减压除去溶剂,用乙酸乙酯萃取(50mL×3),合并有机相用饱和氯化钠溶液洗涤(50mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,得到粗产品标题化合物26b(4.60g,产率:98%),粗产品直接用于下一步反应。
第二步
6-溴-8-氯喹啉-2(1H)-酮26c
向100mL单口瓶中依次加入5mL二氯甲烷、化合物26b(4.60g,14.25mmol),0℃缓慢滴加浓硫酸(11.40mL,213.89mmol),然后撤去冰浴,加热至90℃继续反应2小时。减压除去溶剂,将残余物滴入到50mL冰水中,用乙酸乙酯萃取(50mL×5),合并有机相用饱和氯化钠溶液洗涤(50mL×2),有机相用无水硫酸钠干燥,过滤,滤液减压浓缩,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物26c(0.46g,产率:12%)。
MS m/z(ESI):257.9[M+1]。
第三步
6-溴-2,8-二氯喹啉26d
向50mL单口瓶中依次加入化合物26c(460mg,1.78mmol)、三氯氧磷2mL,将体系温度升至95℃反应90分钟。减压除去三氯氧磷,加入冰水50mL,用乙酸乙酯萃取(50mL×3),有机相用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,浓缩,得到粗产品标题化合物26d(440mg,产率:89%),粗产品直接用于下一步反应。
第四步
6-溴-8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)喹啉-2-胺26e
向25mL封管中依次加入3mL异丙醇、化合物26d(280mg,1.01mmol)、化 合物1b(175mg,1.01mmol)、三氟乙酸(345mg,3.03mmol),然后将温度升至95℃反应16小时。反应冷却至室温,加入25mL饱和碳酸氢钠溶液,用乙酸乙酯萃取(25mL×3)。有机相用饱和氯化钠溶液洗涤(25mL×2),无水硫酸钠干燥。减压除去溶剂,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物26e(260mg,产率:62%)。
MS m/z(ESI):412.9[M+1]。
第五步
8-氯-2-((2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)氨基)喹啉-6-腈26
向25mL三口瓶中依次加入3mL N,N-二甲基乙酰胺、化合物26e(150mg,0.36mmol)、氰化锌(126mg,1.01mmol)、锌粉(3.50mg,0.05mmol)、三(二亚苄基丙酮)二钯(36mg,0.04mmol)、1,1'-双(二苯基膦)二茂铁(46mg,0.08mmol),加热至135℃反应3小时。反应冷却至室温,加入25mL水,用乙酸乙酯萃取(25mL×3),饱和氯化钠溶液洗涤(25mL×2),无水硫酸钠干燥。减压除去溶剂,残余物经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:80%-95%,流速:30mL/min)纯化,得到标题化合物26(55mg,产率:63%)。
MS m/z(ESI):360.1[M+1]。
1
实施例27
8-氯-6-环丙基-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)喹啉-2-胺27
将化合物26e(165mg,0.40mmol)溶于7.5mL 1,4-二氧六环和水(V/V=4:1)中,加入环丙基硼酸27a(41mg,0.48mmol,韶远化学科技(上海)有限公司)、碳酸钠(127mg,1.20mmol)、二氯[1,1’-双(二叔丁基膦)二茂铁钯(II)(39mg,0.06mmol,毕得医药)。氮气置换三次后,升温到100℃反应3小时。反应冷却至室温,向反应液中加入饱和碳酸氢钠溶液(25mL),用乙酸乙酯萃取(50mL×2),合并有机相,用无水硫酸钠干燥。减压除去溶剂,残余物经高效液相制备色谱法(Waters2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:75%-95%,流速:30mL/min)纯化,得到标题化合物27(20mg,产率:13%)。
MS m/z(ESI):375.1[M+1]。
1
实施例28
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-6-(四氢-2H-吡喃-4-基)喹啉-2-胺28
第一步
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-6-(3,6-二氢-2H-吡喃-4-基)喹啉-2-胺28b
将化合物26e(181mg,0.44mmol)溶于7.5mL 1,4-二氧六环和水(V/V=4:1)中,加入3,6-二氢-2H-吡喃-4-硼酸频那醇酯28a(92mg,0.44mmol,韶远化学科技(上海)有限公司)、碳酸钠(140mg,1.31mmol)、二氯[1,1’-双(二叔丁基膦)二茂铁钯(II)(29mg,0.044mmol)。氮气置换三次后,升温到100℃反应3小时。反应冷却至室温,向反应液中加入饱和碳酸氢钠溶液(25mL),用乙酸乙酯萃取(50mL×2),合并有机相,无水硫酸钠干燥。减压除去溶剂,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物28b(140mg,产率:77%)。
MS m/z(ESI):417.0[M+1]。
第二步
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-6-(四氢-2H-吡喃-4-基)喹啉-2-胺 28
将化合物28b(140mg,0.34mmol)溶于10mL乙酸乙酯中,加入铂碳(70mg,5%Wt.,50-70%含水量,韶远化学科技(上海)有限公司),氢气置换三次,氢气氛下反应24小时。用硅藻土过滤反应液,减压除去溶剂,残余物经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:70%-95%,流速:30mL/min)纯化,得到标题化合物28(70mg,产率:50%)。
MS m/z(ESI):419.0[M+1]。
1
实施例29
8-氯-7-环丙基-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)喹啉-2-胺29
第一步
N-(3-溴-2-氯苯基)-3,3-二甲氧基丙酰胺29b
将3-溴-2-氯-苯胺29a(2.0g,9.72mmol,毕得医药)溶于15mL四氢呋喃中,加入化合物14c(1.58g,10.68mmol),反应降温到0℃,滴加双(三甲基硅基)氨基钠(2M四氢呋喃溶液,5.4mL,10.7mmol),然后体系升至室温,反应16小时。向反应液中加入饱和氯化铵溶液(100mL),用乙酸乙酯萃取(100mL×3),饱和氯化钠溶液洗涤(100mL×2),合并有机相,用无水硫酸钠干燥,过滤,滤液减压浓缩,真空干燥,得到粗产品标题化合物29b(3.1g,产率:99%),粗产品不经纯化直接用于下一步反应。
MS m/z(ESI):321.9[M+1]。
第二步
7-溴-8-氯喹啉-2(1H)-酮29c
将化合物29b(3.1g,9.61mmol)溶于3mL二氯甲烷中,降温到0℃,加入浓硫酸(7.7mL,144mmol)。然后撤去冰浴,加热到90℃继续反应2小时。反应液冷却至室温,减压除去溶剂,将残余物滴入到50mL冰水中,用乙酸乙酯萃取(50mL×5),合并有机相用饱和氯化钠溶液洗涤(50mL×2),有机相用无水硫酸钠干燥,过滤,减压除去溶剂,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物29c(0.56g,产率:23%)。
MS m/z(ESI):257.9[M+1]。
第三步
7-溴-2,8-二氯喹啉29d
向50mL单口瓶中依次加入化合物29c(560mg,2.17mmol)、三氯氧磷3mL,将体系温度升至95℃反应90分钟。减压除去三氯氧磷,加入冰水50mL,用乙酸乙酯萃取(50mL×3),有机相用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,浓缩,得到粗产品标题化合物29d(599mg,产率:99%),粗产品不经纯化直接用于下一步反应。
第四步
7-溴-8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)喹啉-2-胺29e
向25mL封管中依次加入3mL异丙醇、化合物29d(600mg,2.17mmol)、化合物1b(319mg,1.84mmol)、三氟乙酸(741mg,6.50mmol),然后将温度升至95℃反应16小时。反应冷却至室温,加入25mL饱和碳酸氢钠溶液,用乙酸乙酯萃取(25mL×3)。有机相用饱和氯化钠溶液洗涤(25mL×2),无水硫酸钠干燥。过滤,滤液减压除去溶剂,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物29e(554mg,产率:62%)。
MS m/z(ESI):412.9[M+1]。
第五步
8-氯-7-环丙基-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)喹啉-2-胺29
将化合物29e(140mg,0.34mmol)溶于7.5mL 1,4-二氧六环和水(V/V=4:1)中,加入化合物27a(37.8mg,0.44mmol)、碳酸钠(107.6mg,1.02mmol)、二氯[1,1’-双(二叔丁基膦)二茂铁钯(II)(33.0mg,0.051mmol)。氮气置换三次后,升温至100℃反应3小时。反应冷却至室温,向反应液中加入饱和碳酸氢钠溶液(25mL),用乙酸乙酯萃取(50mL×2),合并有机相,有机相用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥。减压除去溶剂,残余物经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:55%-95%,流速:30mL/min)纯化,得到标题化合物29(30mg,产率:23%)。
MS m/z(ESI):375.4[M+1]。
1
实施例30
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-7-(四氢-2H-吡喃-4-基)喹啉-2-胺30
第一步
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-7-(3,6-二氢-2H-吡喃-4-基)喹啉-2-胺30a
将化合物29e(170mg,0.41mmol)溶于7.5mL 1,4-二氧六环和水(V/V=4:1)中,加入化合物28a(104mg,0.49mmol)、碳酸钠(131mg,1.23mmol)、二氯[1,1’-双(二叔丁基膦)二茂铁钯(II)(16mg,0.025mmol)。氮气置换三次后,升温到100℃反应3小时。反应冷却至室温,向反应液中加入饱和碳酸氢钠溶液(25mL),用乙酸乙酯萃取(50mL×2),合并有机相,有机相用饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥。过滤,滤液减压除去溶剂,用硅胶柱色谱法以洗脱剂体系B纯化所得残余物,得到标题化合物30a(70mg,产率:41%)。
MS m/z(ESI):417.0[M+1]。
第二步
8-氯-N-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)-7-(四氢-2H-吡喃-4-基)喹啉-2-胺30
将化合物30a(70mg,0.17mmol)溶于5mL乙酸乙酯中,加入铂碳(35mg, 5%Wt.,50-70%含水量,韶远化学科技(上海)有限公司),氢气置换三次,氢气氛下反应24小时。用硅藻土过滤反应液,减压除去溶剂,残余物经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和甲醇,甲醇的梯度:75%-95%,流速:30mL/min)纯化,得到标题化合物30(10mg,产率:14%)。
MS m/z(ESI):419.1[M+1]。
1
实施例31
5-((8-氯喹啉-2-基)氨基)-3-甲基苯并[d]噁唑-2(3H)-酮31
将5-氨基-3-甲基-1,3-苯并噁唑-2(3H)-酮31a(123mg,0.75mmol,毕得医药)、化合物1a(135mg,0.68mmol)、碳酸铯(333mg,1.02mmol)、4,5-双(二苯基膦)-9,9-二甲基氧杂蒽(78.9mg,0.14mmol)和三(二亚苄基丙酮)二钯(63mg,0.068mmol)溶于5mL 1,4-二氧六环中,将反应加热到100℃反应12小时。反应冷却至室温,加入30mL水,用乙酸乙酯萃取(50mL×3),饱和氯化钠溶液洗涤(50mL×2),无水硫酸钠干燥,过滤,滤液减压浓缩,残余物经高效液相制备色谱法(Waters 2767-SQ Detecor2,洗脱体系:10mmol/L碳酸氢铵的水溶液和乙腈,乙腈的梯度:45%-95%,流速:30mL/min)纯化,得到标题化合物31(74mg,产率:33%)。
MS m/z(ESI):324.1[M-1]。
1
生物学评价
以下内容中出现的简称说明:
p.o.:口服
bid:一日两次
qd:一日一次
MC:羧甲基纤维素钠
测试例1、本公开化合物对小鼠溃疡性结肠炎(UC)的预防治疗作用
1、摘要
本实验选用维通利华C57BL/6雌性小鼠建立由硫酸钠葡聚糖(DSS)诱导的溃疡性结肠炎(Ulcerative Colitis,UC)模型,评估阳性化合物ABX-464(见WO2015001518A1化合物90)与本公开化合物1对DSS诱导的溃疡性结肠炎的预防治疗作用。
2.实验方法和实验材料
2.1.实验动物和饲养条件
实验用C57BL/6雌性小鼠,维通利华实验动物有限公司(生产许可证编号:SCXK(浙)2019-0001,动物合格证编号:20210401Abzz0619000795,购入时体重20-22g,5只/笼饲养于独立SPF空间,12/12小时光/暗周期调节,温度23±1℃恒温,湿度50~60%,自由进食进水。动物购进后,进行适应性饲养至少1周后开始实验。
2.2.实验试剂和仪器
右旋糖酐硫酸酯钠盐(Dextrane sulfate sodium salt,DSS):MP Biomedicals,货号160110,批号S5036。无菌水配制,过滤,不能高压灭菌,每两天更换一次。
乙醇:上海百特医疗用品有限公司,批号S2001050。
橄榄油:国药集团化学试剂有限公司,货号30189828,批号20180104。
甲基纤维素M450:国药集团化学试剂有限公司,货号69016460,批号20170308。
酶标仪:厂家BMGlabtech,型号PHERAstar Fs。
台式低速离心机:厂家Eppendorf,型号5417R。
电子天平:梅特勒-托利多仪器有限公司,型号AL204。
2.3.实验设计和实验方法
2.3.1.动物分组:
小鼠适应性饲养后,分组如下:
溶剂:0.5%MC混悬液
2.3.2.药物配制:
DSS配制方法:25g DSS+1L超纯水,无菌过滤,4℃保存。
50mg/kg ABX-464配制方法:100mg ABX-464+20mL 0.5%MC,研磨,4℃保存。配制2次。
50mg/kg本公开化合物1配制方法:100mg本公开化合物1+20mL 0.5%MC,研磨,4℃保存。
2.3.3.实验方法:
小鼠按照体重随机分成5组:正常对照组(
2.4.数据表达和统计学处理
实验数据表示为平均数(Mean)±标准误(SEM)。采用Excel软件t检验进行统计比较。将模型组与正常对照组数据进行分析比较,#P<0.05表示模型组与正常对照组比较具有显著性差异,##P<0.01表示模型组与正常对照组比较具有高度显著性差异,###P<0.001表示模型组与正常对照组比较具有极高度显著性差异。*P<0.05表示给药组与模型组比较具有显著性差异,**P<0.01表示给药组与模型组比较具有高度显著性差异,***P<0.001表示给药组与模型组比较具有极高度显著性差异。
3.结果
3.1本公开化合物1对DSS诱导的UC小鼠体重的影响
体重实验结果显示(图1):与正常对照组相比,DSS模型组小鼠自第4天开始体重明显降低,体重下降幅度逐步增加,第10天体重下降幅度达到30.0%(P<0.001);与DSS模型组比较,所有给药组自第7天开始体重均明显回升,第10天50mg/kg的ABX-464(qd)、ABX-464(bid)、本公开化合物1(bid)其体重下降幅度分别降低到14.1%(P<0.001)、10.3%(P<0.001)和0.4%(P<0.001)。实验终点时体重恢复幅度从强到弱依次为:本公开化合物1 50mg/kg(bid)>ABX-464 50mg/kg(bid)>ABX-464 50mg/kg(qd)。
3.2本公开化合物1对DSS诱导的UC小鼠结肠长度的影响
结肠长度结果显示(图2):与正常对照组相比,DSS模型组结肠长度明显缩 短(P<0.001),仅为正常对照组的75.4%;与DSS模型组比较,所有给药组结肠长度明显增长,50mg/kg的ABX-464(qd)、ABX-464(bid)、本公开化合物1(bid)其结肠长度分别为正常对照组的85.5%(P<0.05)、89.3%(P<0.05)和91.9%(P<0.01)。结肠长度从长到短依次为:本公开化合物1 50mg/kg(bid)>ABX-464 50mg/kg(bid)>ABX-464 50mg/kg(qd)。
4.结论
DSS模型作为IBD疾病的UC模拟动物模型,DSS的分子量(36000-50000)、批号、保存配置和小鼠饲养环境、系别等均影响模型的效果,本次造模比较成功,小鼠体重和结肠长度均有明显的变化。结果显示:本公开化合物1在体重和结肠长度方面表现出强于阳性药ABX-464的良好药效。因此,50mg/kg ABX-464和本公开化合物1对DSS诱导的UC有一定的预防治疗效果,本公开化合物1药效最强,高于同剂量ABX-464。
测试例2.本公开化合物对miR-124的上调作用
一、摘要
本试验用于评估本公开化合物对miR-124的上调作用。
二、实验材料及仪器
1.人T细胞活化CD3/CD28磁珠(Dynabead Human T-Activator CD3/CD28 for T Cell Expansion and Activation)(Gibco,11131D)
2.人总T细胞分离试剂盒(Pan T Cell Isolation Kit,human)(Miltenyi,130-096-535)
3.人白介素2(Human IL-2)(Peprotech,200-02-100)
4.小RNA抽提试剂盒(microRNA抽提试剂盒)(Qiagen,217004)
5.小RNA反转录试剂盒(miScript II RT Kit)(Qiagen,218161)
6.小RNA SYBR Green PCR试剂盒(miScript SYBR Green PCR Kit)(Qiagen,218073)
7.磷酸缓冲液PBS,pH7.4(上海源培生物科技股份有限公司,B320)
8.牛血清白蛋白,BSA(碧云天,ST023)
9.EDTA(0.5M),pH 8.0(Invitrogen,AM9260G)
10.LS分离柱(LS Columns)(Miltenyi,130-042-401)
11. 24孔细胞培养板(Corning,3524)
12. 96孔板(Corning,3788)
13.细胞培养箱(Thermo,Steri cycle i160)
14.实时荧光定量PCR仪(Applied biosystem,QuantStudio6 Flex)
15.PCR仪(Applied biosystem,ProFlex)
16. 96孔透明PCR板,0.2mL(Applied biosystems,N8010560)
17.RPMI1640培养基(Gibco,11875119)
18.胎牛血清,FBS(Gibco,10099-141)
19.磁力架(Invitrogen,DynaMag
20.六孔细胞培养板(Thermo,150239)
21.分光光度计(IMPLEN,NP80)
22.磁珠分离铁架(QuadroMACS Separator)(美天旎,130-090-976)
23.miR124-3P-F引物(金唯智公司定制)
24.hsa-U6检测引物(天根,CD201-0145)
三、实验步骤
化合物对miR-124表达水平的影响在CD3/CD28抗体激活后的T细胞中检测。激活的T细胞经过化合物处理后,提取细胞的总RNA,反转录所得的cDNA作为模板,使用特异性miR-124引物用SYBR green荧光定量PCR法来定量。
T细胞的分离:购买所得的人外周血单核细胞(PBMC),计数离心后用分离缓冲液(PBS pH 7.4,含有0.5%BSA和2mM EDTA))洗一遍,弃去上清,按每1×10
T细胞的活化:按每1×10
化合物处理:化合物储存液为20mM,用DMSO稀释至200μM,再用完全培养基将化合物稀释4倍至50μM(50×),混匀待用。DMSO稀释4倍(25%DMSO)为阴性对照孔。活化两天的T细胞,将细胞吹打均匀,使用磁力架并安装上1.5mL离心管,去除活化磁珠,并收集细胞悬液。对细胞进行计数后,300xg,10min离心弃上清,重悬细胞至1.02×10
RNA抽提:将T细胞离心收集,1500rpm离心3分钟,PBS清洗1次,离心后弃 上清。使用小RNA抽提试剂盒,根据说明书抽提细胞总RNA。细胞沉淀加入700μL Trizol细胞裂解液,枪头吹打均匀,于室温放置5分钟。加入140μL氯仿,振荡混匀,于室温静置3分钟。将氯仿-细胞裂解液混合物于4℃以12000xg离心15分钟。将上层溶液转移至新的无RNA酶(RNase-free)的离心管中,加入1.5倍体积的无水乙醇,枪头吹打数次。将溶液转移至RNA吸附柱中,8000xg离心15s。将离心柱用700μL RWT溶液洗一遍,8000xg离心15s,加入500μL RPE溶液洗两遍,8000xg离心2分钟。将吸附柱放入新的2mL离心管中,12000xg离心1min去除残余洗液。将吸附柱放入新的1.5mL离心管中,加入30-50μL无RNA酶水(RNase-free water),12000xg离心2分钟,收集的溶液为RNA溶液,使用分光光度计测量RNA浓度。RNA溶液保存于-80度冰箱。
反转录:上述提取的RNA模板放置在冰上,取出小RNA反转录试剂盒,于室温解冻部分成分(包含5×miScript HiSpec Buffer,10×miScript nucleics Mix和无RNA酶水),于冰上解冻miScript Reverse Transcriptase mix成分。每个反应(10μL)成分为:5×miScript HiSpec Buffer(2μL),10×miScript nucleics Mix(1μL),miScript Reverse Transcriptase mix(1μL),无RNA酶水(2μL),RNA模板(4μL),在冰上配制上述反应。将样品放置于PCR仪中,设置程序如下:37℃,60分钟;95℃,5分钟;4℃保存。反应完成的样品为cDNA样品。
荧光定量PCR:使用SYBR green染色法检测miR-124的转录水平,同时检测管家基因U6的转录水平作为内参。解冻所有小RNA SYBR green PCR试剂盒所需试剂至常温,将每个cDNA样品模板用无RNA酶水稀释10倍,再稀释5倍。按照下表1配制反应混合物,并将反应混合物加入到96孔PCR板中,用封板膜封板,离心。将PCR反应在荧光定量PCR仪上按照表2步骤进行。
表1 荧光定量PCR反应成分表
表2 荧光定量PCR步骤
表3 荧光定量PCR检测引物表
数据分析:根据软件所算得的CT值,计算每个样品miR-124与内参U6表达水平的比值,即ΔCT(测试化合物)=CT
表4 本公开化合物对miR-124上调的活性情况
结论:本公开化合物具有良好的促进miR124上调的活性。
测试例3、本公开化合物的药代动力学测试
1、摘要
以大鼠为受试动物,应用LC/MS/MS法测定了大鼠灌胃和静脉注射给予实施例2化合物和对比化合物A(参见WO2016135052A1实施例3的化合物(1))后不同时刻血浆中的药物浓度。研究本公开化合物在大鼠体内的药代动力学行为,评价其药动学特征。
2、试验方案
2.1试验药品
实施例2化合物、对比化合物A。
2.2试验动物
健康成年SD大鼠16只,雌雄各半,平均分成4组,购自维通利华实验动物技术有限公司。
2.3药物配制
称取一定量药物,加入5%DMSO、5%吐温80和90%生理盐水配制成澄明溶液。
2.4给药
灌胃组:SD大鼠禁食过夜后灌胃给药,给药剂量为2mg/kg,给药体积均为10.0mL/kg。
静脉组:SD大鼠禁食过夜后静脉注射给药,给药剂量为1mg/kg,给药体积均为5.0mL/kg。
3、操作
灌胃组:大鼠灌胃给药实施例2化合物和对比化合物A,于给药前及给药后0.25小时、0.5小时、1.0小时、2.0小时、4.0小时、6.0小时、8.0小时、11.0小时、24.0小时由眼眶采血0.1mL,置于EDTA-K2抗凝试管中,4℃、10000转/分钟离心1分钟,1小时内分离血浆,-20℃保存待测。采血至离心过程在冰浴条件下操作。给药后2h进食。
静脉组:大鼠静脉注射实施例2化合物和对比化合物A,于给药前及给药后5分钟、15分钟、0.5小时、1.0小时、2.0小时、4.0小时、8.0小时、11.0小时、24.0 小时采血,处理同灌胃组。
测定不同浓度的药物灌胃、静脉注射给药后大鼠血浆中的待测化合物含量:取给药后各时刻的大鼠血浆20μL,加入内标溶液(喜树碱100ng/mL)50μL、乙腈200μL,涡旋混合5分钟,离心10分钟(3700-4000转/分钟),血浆样品取上清液1.0-2.0μL进行LC/MS/MS分析。
4、药代动力学参数结果
表5 本公开化合物的灌胃给药药代动力学参数
表6 本公开化合物的静脉注射给药药代动力学参数
结论:由表5和表6可知,相比于对比化合物A,本公开实施例2化合物的药代吸收良好,具有明显的药代动力学优势。
- 一种新型二硝基喹啉类化合物及其制备方法和应用
- 喹啉类化合物柠檬酸盐的晶型、制备方法、组合物与应用
- 喹啉类化合物硫酸盐的晶型、制备方法、组合物与应用
- 具有磷酸肌酸和银杏内酯B复合结构类化合物,其制备及其在医药上的应用
- 含吡啶并喹唑啉酮的三芳胺类化合物及其制备方法和应用
- 喹啉胺类化合物、其制备方法及其在医药上的应用
- 澳洲茄胺的制备方法及其澳洲茄胺在医药上的应用