一种艾拉莫德中间体的制备方法
文献发布时间:2024-04-18 20:01:23
技术领域
本发明属于药物合成领域,涉及一种化合物的制备方法,具体涉及一种艾拉莫德中间体的制备方法。
背景技术
艾拉莫德(Iguratimod),化学名为N-[3-(甲酰胺基)-4-氧-6-苯氧基-4H-1-苯并呋喃-7-基]-甲磺酰胺,是一种非甾体抗炎药(NSAIDs),用于治疗骨关节炎和类风湿性关节炎,其既能选择性抑制环氧化酶COX-II,还能调节T-细胞,具有自身免疫调节作用,起效迅速、副作用小。艾拉莫德的非临床研究表明其对动物中各种关节炎模型的改善作用主要是抑制免疫球蛋白和各种炎症因子的产生,对骨代谢产生合成代谢作用。
现有技术中,甲酰胺基甲基-2-甲氧基-4-甲磺酰胺基-5-苯氧基苯基酮是用于合成艾拉莫德的关键中间体,其可以通过化合物I(2-氨基-1-(2-甲氧基-4-甲磺酰胺基-5-苯氧基苯基)乙酮盐酸盐)制备得到,化合物I和艾拉莫德中间体的结构式如下:
现有报道的合成工艺中,制备上述中间体的过程中,反应速率慢、收率低、杂质大,产品外观发红。
现有的艾拉莫德中间体的合成路线主要如下:
路线一:TakihiroInaba在Chem.Pharm.bull.48(1)131-139(2000)文献中报道了特戊酰氯与甲酸钠反应得到酸酐,再与2-氨基-1-(2-甲氧基-4-甲磺酰胺基-5-苯氧基苯基)乙酮盐酸盐(化合物I)反应得到甲酰胺基甲基-2-甲氧基-4-甲磺酰胺基-5-苯氧基苯基酮(艾拉莫德中间体)。由于甲酸钠不溶于反应体系,甲酸钠与特戊酰氯进行固液反应,反应速率较低,且产生的氯化钠容易包裹在甲酸钠表面,降低甲酸钠与特戊酰氯的反应速率,进一步导致未能反应的特戊酰氯生成酰胺类副产物,降低了收率且增加了杂质。
酰胺类副产物结构如下:
路线二:中国专利CN108727232A对艾拉莫德中间体合成进行了改进,采用甲酸、甲酸钠与新戊酰氯反应制备混合酸酐,再与2-氨基-1-(2-甲氧基-4-甲磺酰胺基-5-苯氧基苯基)乙酮盐酸盐(化合物I)反应得到甲酰胺基甲基-2-甲氧基-4-甲磺酰胺基-5-苯氧基苯基酮(艾拉莫德中间体)。该路线虽然提升了反应速率,但仍加入的甲酸钠依旧不溶于反应体系,还是会与新戊酰氯进行固液反应,容易造成新戊酰氯继续与2-氨基-1-(2-甲氧基-4-甲磺酰胺基-5-苯氧基苯基)乙酮盐酸盐(化合物I)生成酰胺类副产物,从而影响艾拉莫德中间体的质量。另一方面,该方法依旧使用了强卤试剂,新戊酰氯腐蚀性强,易产生烟雾,有特殊臭味,对设备、环境不友好,也存在一定的安全风险。
路线三:中国专利CN112209859A报道了以甲酸为原料,与N,N-二羰基二咪唑进行活化,再与2-氨基-1-(2-甲氧基-4-甲磺酰胺基-5-苯氧基苯基)乙酮盐酸盐(化合物I)反应得到甲酰胺基甲基-2-甲氧基-4-甲磺酰胺基-5-苯氧基苯基酮(艾拉莫德中间体)。该路线易造成起始物料反应不完全,且引入N,N-二羰基二咪唑极易产生副产物,不易脱除。
因此,需要开发一种工艺操作简单、副反应少、杂质含量低、质量好、收率高、适宜于大规模生产的艾拉莫德中间体的制备方法。
发明内容
本发明的目的在于解决上述路线中存在的问题,提供一种更安全环保、收率高、质量好、适合工业化生产的艾拉莫德中间体的制备方法。
本发明中,化合物I可以采用现有技术制备得到,或者直接使用市售工业化生产的化合物I,含量符合化学纯(CP)要求(含量≥99.5%)。
本发明的发明人从现有技术中的缺陷出发,提出解决问题的多个技术路线猜想,开展了大量试剂筛选、加料顺序、物料配比、反应温度等不同条件的试验并进行验证和优化,最终得到了本发明的路线和方法。
现有技术中,已有各种高效液相法测试杂质含量或纯度的方法,可以直接或经适当的优化后应用于测试本发明实施例产物或杂质的含量。
一种艾拉莫德中间体的制备方法,包括以下步骤:
(1)将催化剂加至丙酮中,再加入酰化剂进行活化,得到活化溶液;
(2)所述活化溶液中加入甲酸和缚酸剂,保温反应得到混合酸酐溶液;
(3)所述混合酸酐溶液中加入化合物I,保温反应,加水析晶,过滤,干燥得所述艾拉莫德中间体;
所述化合物I、艾拉莫德中间体的结构式分别为:
进一步的,所述步骤(1)中,催化剂选自碘化钠、碘化钾、溴化钠、溴化钾中的一种或多种;酰化剂选自氯甲酸异丁酯、氯甲酸异丙酯、氯甲酸叔丁酯、氯甲酸苄酯中的一种或多种。
进一步的,所述步骤(1)中,活化条件为0-30℃搅拌1h。
进一步的,所述催化剂:所述酰化剂:所述化合物I的摩尔比为(0.05~0.3):(1.5~2.5):1,优选的摩尔比为(0.05-0.2):(1.5-2):1。
进一步的,所述步骤(2)中,缚酸剂为N,N-二异丙基乙胺、三乙胺、二乙胺、吡啶中的一种或多种。
进一步的,所述甲酸:所述缚酸剂:所述化合物I的摩尔比为(1~2):(2.5~4):1,优选的摩尔比为(1-1.5):(2.5-3):1。
进一步的,所述步骤(2)中,反应温度为0~30℃,反应时间为0.5~4h;优选地,反应温度为0~15℃,反应时间为0.5~2h。
进一步的,所述步骤(3)中,反应温度为0~30℃,反应时间为2~6h;优选地,反应温度为0~15℃,反应时间为2~4h。
进一步的,所述艾拉莫德中间体的制备方法进一步包括精制;精制包括打浆或重结晶。
更进一步的,所述重结晶的精制方法为:将所述艾拉莫德中间体用10倍体积(v/m)的乙腈加热溶解、冷却析晶、过滤、将固体干燥。
本发明提供了一种艾拉莫德的制备方法,与现有工艺相比,工艺操作简单,对环境友好,质量好,收率高,适宜于大规模生产。
具体而言,本发明具有以下优势:
(1)反应试剂易得,未采用强卤试剂,对设备、环境友好,生产成本较低。
(2)反应为均相反应,不易产生产物包裹,反应更完全,能有效避免非均相反应造成副产物偏大,有利于质量控制和生产放大。
(3)反应条件温和,操作简单,易于控制,安全性高,动力成本大大降低。并且反应体系为酸性,极大抑制了副产物的产生。
(4)产物可以不需要进行额外的打浆、重结晶等精制操作,避免收率损失,收率更高,单位生产成本进一步降低。
(5)制备得到的艾拉莫德中间体产品质量好,类白色固体,未经精制的纯度即达99.6%以上,可以进一步应用并提高艾拉莫德原料药的产量和质量。
附图说明
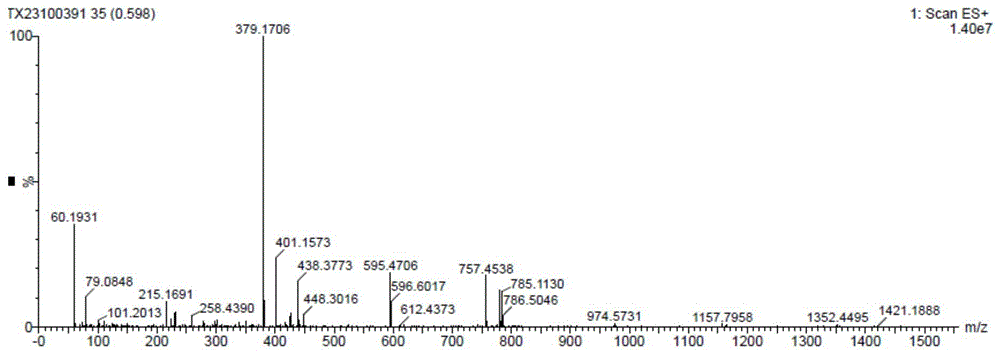
图1:实施例1中艾拉莫德中间体的MS图;
图2:实施例1中艾拉莫德中间体的
具体实施方式
以下通过具体实施例,进一步详细描述本发明。应理解的是,实施例仅为解释说明的目的,不是对本发明保护范围的限制。
下列实施例中未注明具体条件的实验方法,通常采用常规的实验仪器及按照常规条件下反应或操作,或采用制造厂商所建议的条件。
下列实施例中,化合物I可以采用现有技术制备得到,或者直接使用市售的工业化生产的化合物I,含量符合化学纯(CP)要求(含量≥99.5%),化合物I的结构如下所示:
实施例1:
0.387g碘化钠加至160ml丙酮中,再加入10.59g氯甲酸异丁酯,20-30℃搅拌1h进行活化,降温至0-5℃。
上述活化溶液中再加入2.37g甲酸和16.70gN,N-二异丙基乙胺,0-5℃搅拌0.5h得到混合酸酐溶液。
得到的混合酸酐溶液再加入20.0g化合物I,0-5℃反应2h。加入200ml水,0~15℃析晶2h,过滤,鼓风干燥得18.60g类白色固体(纯度99.65%,理论19.56g计)。
本实施例制备的艾拉莫德中间体的MS和
1
m/z:379.2[M+H]
实施例2:
2.575g碘化钾加至160ml丙酮中,再加入15.84g氯甲酸异丙酯,20-30℃搅拌1h进行活化,降温至10-15℃。
上述活化溶液中再加入4.76g甲酸和20.93g三乙胺,10-15℃搅拌2h得到混合酸酐溶液。
得到的混合酸酐溶液再加入20.0g化合物I,10-15℃反应6h。加入200ml水,0~15℃析晶2h,过滤,鼓风干燥得18.45g类白色固体(纯度99.72%,理论19.56g计)。
实施例3:
1.064g溴化钠加至160ml丙酮中,再加入14.12g氯甲酸叔丁酯,10-20℃搅拌1h进行活化,降温至5-10℃。
上述活化溶液中再加入3.57g甲酸和11.34g二乙胺,5-10℃搅拌2h得到混合酸酐溶液。
得到的混合酸酐溶液再加入20.0g化合物I,5-10℃反应4h。加入200ml水,0~15℃析晶2h,过滤,鼓风干燥得18.74g类白色固体(纯度99.69%,理论19.56g计)。
实施例4:
1.230g溴化钾加至160ml丙酮中,再加入17.64g氯甲酸苄酯,15-25℃搅拌1h进行活化,降温至5-10℃。
上述活化溶液中再加入3.57g甲酸和12.27g吡啶,5-10℃搅拌2h得到混合酸酐溶液。
得到的混合酸酐溶液再加入20.0g化合物I,5-10℃反应4h。加入200ml水,0~15℃析晶2h,过滤,鼓风干燥得18.56g类白色固体(纯度99.70%,理论19.56g计)。
实施例5:
1.22g溴化钾加至160ml丙酮中,再加入17.59g氯甲酸苄酯,0-10℃搅拌1h进行活化。
上述活化溶液中再加入3.52g甲酸和12.22g吡啶,25-30℃下搅拌4h得到混合酸酐溶液。
得到的混合酸酐溶液再慢慢加入20.0g化合物I,25-30℃反应6h。加入200ml水,0~15℃析晶2h,过滤,鼓风干燥得18.48g类白色固体(纯度99.62%,理论19.56g计)
实施例6:
将实施例1得到的15g类白色固体加入150ml乙腈中,加热回流10min,冷却至10-20℃析晶后,过滤收集固体,鼓风干燥得到14.25g白色固体,纯度99.93%,收率95.0%。
对比例1:
参照背景技术路线一提及的文献Chem.Pharm.bull.48(1)131-139(2000)中化合物24制备进行操作。
41.20g甲酸钠加至36.60g特戊酰氯和300ml丙酮混合液中,室温搅拌5h,加入100.00g2-氨基-1-(2-甲氧基-4-甲磺酰胺基-5-苯氧基苯基)乙酮盐酸盐,保温反应3h。加入900ml水析晶2h,过滤,异丙醇洗涤,干燥得80.25g固体(收率82.0%,纯度95.22%)。
对比例2:
参照背景技术路线二提及的CN108727232A实施例1进行操作。
14.30g甲酸和26.40g甲酸钠加至3000ml丙酮中,18~20℃搅拌,再加入37.40g新戊酰氯搅拌1h得到混合酸酐体系。向混合酸酐体系中加入120.20g2-氨基-1-(2-甲氧基-4-甲磺酰胺基-5-苯氧基苯基)乙酮盐酸盐和26.40g甲酸钠,10~20℃搅拌反应3h。降温至10±5℃,控温15±5℃以下加入1000ml水,搅拌1h,过滤,滤饼用2000ml乙腈重结晶,降温至10~25℃析晶,过滤,干燥得102.51g类白色固体(收率87.2%,纯度97.67%)。
对比例3:
参照背景技术路线三提及的CN112209859A实施例1进行操作。
10.20g甲酸溶于500ml二氯甲烷中,控温15~25℃搅拌,分批加入52.80gN,N-羰基二咪唑,加完搅拌1h。保温分批加入84.00g2-氨基-1-(2-甲氧基-4-甲磺酰胺基-5-苯氧基苯基)乙酮盐酸盐反应2h,再加入500ml水和500ml二氯甲烷搅拌0.5h,分层,有机相浓缩至干,加入500ml异丙醇打浆0.5h,抽滤,干燥得67.05g固体(收率81.6%,纯度88.41%)。
实施例1-5及对比例1-3的实验结果见下表:
以上实施例仅是解释说明的目的,以便于更好地理解本发明的内容。尽管上述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 一种泥皮画加工用加香辅助装置
- 一种成品油加工用计量调节装置
- 一种汽车配件加工用冲压装置及其冲压方法
- 一种新能源汽车车轴车削加工用测量装置
- 一种具有圆柱面汽车部件加工用输送装置
- 一种汽车传动轴加工用除油装置
- 一种等速传动轴移动端接头加工用加气清理辅助装置