酰胺类化合物作为Nav1.8抑制剂
文献发布时间:2024-05-31 01:29:11
本发明要求享有:
于2022年11月18日向中国国家知识产权局提交的,专利申请号为202211449362X,名称为“酰胺类化合物作为Nav1.8抑制剂”的在先申请的优先权;
于2023年2月23日向中国国家知识产权局提交的,专利申请号为2023101619060,名称为“酰胺类化合物作为Nav1.8抑制剂”的在先申请的优先权;
于2023年04月21日向中国国家知识产权局提交的,专利申请号为2023104370717,名称为“酰胺类化合物作为Nav1.8抑制剂”的在先申请的优先权;
于2023年11月14日向中国国家知识产权局提交的,专利申请号为2023115302997,名称为“酰胺类化合物作为Nav1.8抑制剂”的在先申请的优先权;
所述在先申请的全文通过引用的方式结合于本发明中。
技术领域
本发明属于医药领域,具体地,本发明涉及到一种Nav1.8抑制剂。
背景技术
疼痛是“一种令人不快的感觉和情绪上的感受,伴有实质上的或潜在的组织损伤,它是一种主观感受”。疼痛可以作为一种警戒信号,提醒机体注意潜在的危险,对机体正常的生命活动具有不可或缺的保护作用。同时,疼痛也是一种常见的临床症状,在引发疼痛的外界刺激消失后,强烈或持久的疼痛会造成生理功能的紊乱,严重影响生命体的生活质量。据统计,全世界约五分之一的人患有中度至重度慢性疼痛。2018年全球镇痛药市场约为360亿美元,预计2023年将达到560亿美元。其中急性中重度未来将以2.5%的年复合增长率稳定增长,慢性疼痛未来市场将18%左右的年复合增长率增长,慢性疼痛是驱动未来十年全球疼痛市场持续增长的主要推动力。
疼痛起源于周围神经系统的伤害感受器。这是一种游离的神经末梢,广泛分布于全身的皮肤、肌肉、关节和内脏组织中,它可以将感受到的热的、机械的或化学的刺激转化为神经冲动(动作电位)并经由传入神经纤维传递到其位于背根神经节(dorsalrootganglia,DRG)的胞体部分,最终传递到高级神经中枢,引起痛觉。而神经元中动作电位的产生和传导又依赖于细胞膜上的电压门控钠离子通道(voltage-gated sodiumchannels,NaV)。当细胞膜去极化时,钠离子通道激活,通道打开,引起钠离子内流,使细胞膜进一步去极化,导致动作电位的产生。因此,抑制异常的钠离子通道活动有助于疼痛的治疗、缓解。
人类钠离子是一类跨膜离子通道蛋白,由分子量260kD的α亚基和分子量为30-40kD的β亚基组成,根据α亚基的不同可以分为9种亚型,分别为Nav1.1~Nav1.9,Nav1.5、Nav1.8和Nav1.9是河豚毒素(tetrodotoxin,TTX)不敏感性钠通道,Nav1.5主要存在于心肌细胞中,Nav1.8、Navl.9存在于外周神经系统。其中Nav1.8是参与慢性疼痛、心房纤维性颤动、布加综合征的重要离子通道,是疼痛治疗的高选择性作用靶点。
Nav1.8编码基因为SCN10A,位于人类染色体3p21-22区域,主要编码α亚单位。研究发现人与大鼠Nav1.8基因的同源性高达93%。Nav1.8主要存在于三叉神经节神经元和DRG神经元中,具有慢速失活、迅速恢复的电生理特征。在表达Nav1.8的神经元内,动作电位的上升主要由Nav1.8电流构成。在神经性疼痛的模型中,神经损伤会使Nav1.8在轴突和神经元胞体中的表达水平上升。使用Nav1.8反义寡核苷酸在降低Nav1.8表达的同时可以明显地缓解疼痛。大鼠爪内注射角叉菜胶后,DRG神经元中Nav1.8的表达有所上升。Nav1.8敲除小鼠不能表现出正常的内脏炎症痛。人类的Nav1.8基因产生功能增益突变后,会导致外周神经痛。根据一系列动物实验以及人类基因证据,选择性抑制Nav1.8具有成为新型镇痛疗法的潜力,可以用于炎性疼痛、神经疼痛、手术后疼痛和癌痛等多种疼痛类型的治疗。
一些已知的Nav’s抑制剂的主要缺点是它们的治疗窗口差,这可能是它们缺乏同种型选择性的结果。由于Navl.8主要限于感知疼痛的神经元,因此选择性Nav1.8阻断剂不太可能诱导非选择性Nav’s阻断剂常见的不良反应。因此,本领域仍然需要开发新的Nav1.8选择性抑制剂,优选对Nav1.8选择性更好、更有效、代谢稳定性增加、溶解度增加和副作用更少的Nav通道抑制剂。
发明内容
本发明旨在提出一种Nav1.8抑制剂,可用于制备治疗、缓解或预防疼痛的药物,所述疼痛包括急性疼痛、慢性疼痛、炎性疼痛、癌症疼痛、神经性疼痛、肌肉骨骼痛、原发性疼痛、肠痛和特发性疼痛等。
本发明的第一方面,本发明提出了一种化合物,为式(I)所示的化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药:
其中,
环A、环B、环C各自独立地为苯环、C
R
R
或者,相邻的两个R
R
n、m、p各自独立地选自1、2、3、4、5、6。
在本发明一任选实施方案中,式(I)所示的化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药为:
其中,
环A、环B、环C各自独立地为苯环、C
R
R
或者,相邻的两个R
R
n、m、p各自独立地选自1、2、3、4、5、6。
在本发明一任选实施方案中,式(I)所示的化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药为:
其中,
环A、环B、环C各自独立地为苯环、C
R
R
而且,相邻的两个R
R
n、m、p各自独立地选自1、2、3、4、5、6。
在本发明一任选实施方案中,式(I)所示的化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药,其特征在于,其为式(II)所示的化合物,
R
在本发明一任选实施方案中,式(I)所示的化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药为:
其中,
R
R
而且,相邻的两个R
R
n、m、p各自独立地选自1、2、3、4、5、6。
在本发明一任选实施方案中,式(I)所示的化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药为式(I-A)所示的化合物,
其中,A
R
R
R
或者,R
R
较佳地,基团
在本发明一任选实施方案中,式(I)所示的化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药为式(I-A)所示的化合物,
A
较佳地,基团
在本发明一任选实施方案中,式(I)所示的化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药为式(I-A)所示的化合物,
其中,A
R
较佳地,基团
在本发明一任选实施方案中,基团
在本发明一任选实施方案中,式(I)所示的化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药为:
其中,所述
R
R
或者,R
R
在本发明一任选实施方案中,式(I)所示的化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药为:
其中,R
在本发明一任选实施方案中,式(I)所示的化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药为:
其中,
R
R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,式(I-A)所示的化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药为式(II-A)、式(II-B)、式(II-C)、式(II-D)、式(II-E)、式(II-F)所示的化合物,
其中,
R
R
或者,R
R
在本发明一任选实施方案中,式(I-A)所示的化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药为式(II-A)、式(II-B)或式(II-C)所示的化合物,
其中,
R
R
或者,相邻的两个R
R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,R
在本发明一任选实施方案中,所述
较佳地,
在本发明一任选实施方案中,所述
较佳地,所述
在本发明一任选实施方案中,所述
较佳地,所述
在本发明一任选实施方案中,所述化合物具有选自下列任一结构,或任一结构的互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药:
/>
本发明的第二方面,本发明提出了一种药物组合物,所述药物组合物包括治疗有效剂量的上述化合物,其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药以及药学上可接受的药用载体、稀释剂或赋形剂。
本发明的第三方面,本发明提出了上述化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药或上述药物组合物在制备用于治疗抑制电压门控钠离子通道相关药物中的用途,所述抑制电压门控钠离子通道包括Nav1.1~Nav1.9,优选为Nav1.8。
根据本发明的具体实施例,上述化合物或其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药或上述药物组合物在制备药物中的用途,所述药物可用于治疗、缓解或预防疼痛,所述疼痛包括急性疼痛、慢性疼痛、炎性疼痛、癌症疼痛、神经性疼痛、肌肉骨骼痛、原发性疼痛、肠痛和特发性疼痛。
本发明第四方面,提供一种抑制电压门控钠离子通道,或预防和/或治疗电压门控钠离子通道相关的疾病的方法,包括步骤:给需要的对象施用本发明第一方面所述的式I所示化合物、其互变异构体、立体异构体、水合物、溶剂化物、药学上可接受的盐或前药或本发明的第二方面所述的药物组合物。
所述电压门控钠离子通道包括Nav1.1~Nav1.9,Nav1.5、Nav1.8和Nav1.9,优选为Nav1.8。所述电压门控钠离子通道相关的疾病为疼痛,包括急性疼痛、慢性疼痛、炎性疼痛、癌症疼痛、神经性疼痛、肌肉骨骼痛、原发性疼痛、肠痛和特发性疼痛。
有益效果
根据本发明的实施例,本发明至少具有如下技术效果至少之一:
1)提供了结构新颖、药代动力学性质优良、药效或成药性好的Nav1.8抑制剂,可以用于有效治疗Nav1.8相关的疾病、病症;
2)本发明化合物具有较强的对Nav1.8离子通道抑制活性。
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
术语定义与说明
除非另有说明,本申请说明书和权利要求书中记载的基团和术语定义,包括其作为实例的定义、示例性的定义、优选的定义、表格中记载的定义、实施例中具体化合物的定义等,可以彼此之间任意组合和结合。这样的组合和结合后的基团定义及化合物结构,应当被理解为本申请说明书和/或权利要求书记载的范围内。
除非另有说明,用于本发明申请,包括本申请说明书和权利要求书中记载的术语和定义如下。
本领域技术人员可以理解,根据本领域中使用的惯例,在本申请的结构式中,
术语“药学上可接受的盐”是指药学上可接受的无毒酸或碱的盐,包括无机酸和碱、有机酸和碱的盐。
术语“药物组合物”表示一种或多种文本所述化合物或其生理学/药学上可接受的盐或前体药物与其它化学组分的混合物,其它组分例如生理学/药学上可接受的载体和赋形剂。药物组合物的目的是促进化合物对生物体的给药。
术语“辅料”是指可药用惰性成分。术语“赋形剂”的种类实例非限制性地包括粘合剂、崩解剂、润滑剂、助流剂、稳定剂、填充剂和稀释剂等。赋形剂能增强药物制剂的操作特性,即通过增加流动性和/或粘着性使制剂更适于直接压缩。
术语“前药”是指可以在生理条件下或者通过溶剂解转化为具有生物活性的本发明化合物。本发明的前药通过修饰在该化合物中的功能基团来制备,该修饰可以按常规的操作或者在体内被除去,而得到母体化合物。前药包括本发明化合物中的一个羟基或者氨基连接到任何基团上所形成的化合物,当本发明化合物的前药被施予哺乳动物个体时,前药被割裂而分别形成游离的羟基、游离的氨基。
术语“立体异构体”是指由分子中原子在空间上排列方式不同所产生的异构体,包括顺反异构体、对映异构体、非对应异构体和构象异构体。
术语“互变异构体”是指因分子中某一原子在两个位置迅速移动而产生的官能团异构体。本发明化合物可表现出互变异构现象。互变异构的化合物可以存在两种或多种可相互转化的种类。质子移变互变异构体来自两个原子之间共价键合的氢原子的迁移。互变异构体一般以平衡形式存在,尝试分离单一互变异构体时通常产生一种混合物,其理化性质与化合物的混合物是一致的。平衡的位置取决于分子内的化学特性。例如,在很多脂族醛和酮如乙醛中,酮型占优势;而在酚中,烯醇型占优势。本发明包含化合物的所有互变异构形式。
本发明的某些化合物可以具有不对称碳原子(光学中心)或双键。外消旋体、非对映异构体、几何异构体和单个的异构体都包括在本发明的范围之内。
本文中消旋体、ambiscalemic and scalemic或者对映体纯的化合物的图示法来自Maehr,J.Chem.Ed.1985,62:114-120。除非另有说明,用楔形键和虚线键表示一个立体中心的绝对构型。当本文所述化合物含有烯属双键或其它几何不对称中心,除非另有规定,它们包括E、Z几何异构体。同样地,所有的互变异构形式均包括在本发明的范围之内。
本发明的化合物可以存在特定的几何或立体异构体形式。本发明设想所有的这类化合物,包括顺式和反式异构体、(-)-和(+)-对对映体、(R)-和(S)-对映体、非对映异构体、D-异构体、L-异构体,及其外消旋混合物和其他混合物,例如对映异构体或非对映体富集的混合物,所有这些混合物都属于本发明的范围之内。烷基等取代基中可存在另外的不对称碳原子。所有这些异构体以及它们的混合物,均包括在本发明的范围之内。
可以通过的手性合成或手性试剂或者其他常规技术制备光学活性的(R)-和(S)-异构体以及D-和L-异构体。如果想得到本发明某化合物的一种对映体,可以通过不对称合成或者具有手性助剂的衍生作用来制备,其中将所得非对映体混合物分离,并且辅助基团裂开以提供纯的所需对映异构体。或者,当分子中含有碱性官能团(如氨基)或酸性官能团(如羧基)时,与适当的光学活性的酸或碱形成非对映异构体的盐,然后通过本领域所公知的分步结晶法或色谱法进行非对映异构体拆分,然后回收得到纯的对映体。此外,对映异构体和非对映异构体的分离通常是通过使用色谱法完成的,所述色谱法采用手性固定相,并任选地与化学衍生法相结合(例如由胺生成氨基甲酸盐)。
本发明的化合物可以在一个或多个构成该化合物的原子上包含非天然比例的原子同位素。例如,可用放射性同位素标记化合物,比如氚(
针对药物或药理学活性剂而言,术语“有效量”或“治疗有效量”是指无毒的但能达到预期效果的药物或药剂的足够用量。对于本发明中的口服剂型,组合物中一种活性物质的“有效量”是指与该组合物中另一种活性物质联用时为了达到预期效果所需要的用量。有效量的确定因人而异,取决于受体的年龄和一般情况,也取决于具体的活性物质,个案中合适的有效量可以由本领域技术人员根据常规试验确定。
术语“活性成分”、“治疗剂”,“活性物质”或“活性剂”是指一种化学实体,它可以有效地治疗目标紊乱、疾病或病症。
术语“被取代的”是指特定原子上的任意一个或多个氢原子被取代基取代,包括重氢和氢的变体,只要特定原子的价态是正常的并且取代后的化合物是稳定的。当取代基为酮基(即=O)时,意味着两个氢原子被取代。酮取代不会发生在芳香基上。术语“任选被取代的”是指可以被取代,也可以不被取代,除非另有规定,取代基的种类和数目在化学上可以实现的基础上可以是任意的。
前缀“C
术语“C
术语“-O-(C
术语“-S-(C
术语“C
术语“C
术语“C
术语“C
术语“4-8元杂环烷基”或“4-8元杂环基”或“4-8元杂环”是指总共具有4、5、6、7或8个环原子且包含一个或两个相同或不同的环杂原子或含杂原子的基团的单环饱和杂环,所述环杂原子或含杂原子的基团选自:N、NH、O、S、SO和SO
术语“4-8元不饱和杂环”是指含有4至8个环原子并且含有一个或多个C-C双键(优选地一个C-C双键)的环基团,所述环原子包含1、2或3个独立地选自N、NH、O、S、SO和SO
术语“6-10元芳基”或“6-10元芳环”应理解为具有6-10个碳原子的一价芳香性或部分芳香性的单环、双环或三环烃环,特别是具有6个碳原子的环(“C
术语“5-8元杂芳基”或“5-8元杂芳环”应理解为具有5-8个环原子,特别是5或6个碳原子,且包含1-5个独立选自N、O和S的杂原子的一价单环、双环或三环芳族环基团。优选1-3个且独立选自N、O和S的杂原子的一价单环、双环或三环芳族环基团,并且,另外在每一种情况下可为苯并稠合的。特别地,杂芳基选自噻吩基、呋喃基、吡咯基、噁唑基、噻唑基、咪唑基、吡唑基、异噁唑基、异噻唑基、噁二唑基、三唑基、噻二唑基等;或吡啶基、哒嗪基、嘧啶基、吡嗪基、三嗪基等;或噌啉基、酞嗪基、喹唑啉基、喹喔啉基、萘啶基、蝶啶基、咔唑基、吖啶基、吩嗪基、吩噻嗪基、吩噁嗪基等。
术语“卤代基”或“卤素”为氟、氯、溴和碘。
术语“任选”或“任选地”意思是随后所述的事件或情形可发生或可不发生,且所述描述包括其中所述事件或情形发生的情况以及其中所述事件或情形不发生的情况。
另外,需要说明的是,除非以其他方式明确指出,在本发明中所采用的描述方式“……独立地”应作广义理解,是指所描述的各个个体之间是相互独立的,可以独立地为相同或不同的具体基团。更详细地,描述方式“……独立地”既可以是指在不同基团中,相同符合之间所表达的具体选项之间互相不影响,也可以表示在相同的基团中,相同符号之间所表达的具体选项之间互相不影响。
附图说明
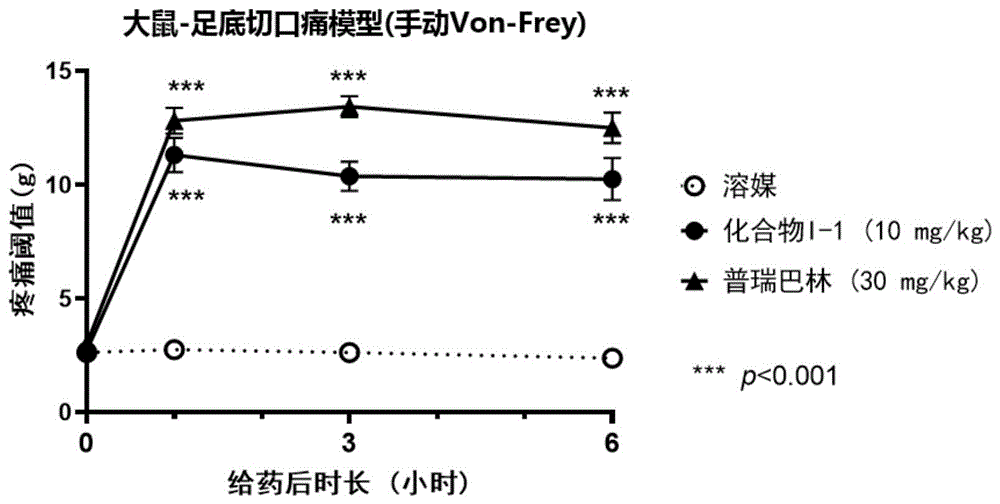
图1:化合物对大鼠足底切口痛疼痛阈值的药效。
具体实施方式
下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
如无特别说明,本发明的化合物均是通过核磁共振(NMR)和/或质谱(MS)来确定其结构的。NMR位移的单位为10
本发明的缩写定义如下:
DIEA:二异丙基乙胺,亦即N,N-二异丙基乙胺
DMF:N,N-二甲基甲酰胺
DCM:二氯甲烷
HATU:O-(7-氮杂苯并三唑-1-基)-N,N,N′,N′-四甲基脲六氟磷酸酯
m-CPBA:间氯过氧苯甲酸
NMI:N-甲基咪唑
PE:石油醚
TCFH:N,N,N',N'-四甲基氯甲脒六氟磷酸盐
TLC:薄层色谱法
LC-MS:液质联用色谱
N:当量浓度,例如2N盐酸表示2mol/L盐酸溶液
IC
制备例1:中间体INT-1的制备
化合物INT-1的合成路线如下所示:
第一步:2,2-二氟苯并[d][1,3]二氧杂环戊烯-4-酚(2)的合成
将化合物1(25.0g,8.20mmol)溶于无水四氢呋喃(250mL)中,然后抽真空,氮气置换,反复三次。氮气保护下,反应液冷却至-78℃,缓慢加入仲丁基锂(134mL,173.94mmol,1.3M),维持反应温度不超过-70℃。加完后,反应液在该温度下继续反应2小时,缓慢滴加硼酸三甲酯(19.72g,189.75mmol),维持温度不超过-65℃。加完后反应液缓慢升至室温并继续反应1小时。再分别加入双氧水(10.76g,316.25mmol,30%)和氢氧化钠(6.32g,158.12mmol)。加完后,反应液在室温条件下反应16hr。TLC(PE:EA=5:1)显示反应完全。加入水(100mL)稀释,然后用EtOAc(100mL×3)萃取,合并有机相,用2N NaOH水溶液(50mL×2)洗涤,合并水相,用2N HCl水溶液调pH在3左右,再用EtOAc(100mL×3)萃取,合并有机相,饱和食盐水(100mL)洗涤,无水硫酸钠干燥,过滤,滤液浓缩旋干,粗品通过正相硅胶柱层析分离(EtOAc/PE=0%-20%)得到2,2-二氟苯并[d][1,3]二氧杂环戊烯-4-酚(2)(31.6g)。
第二步:叔丁基((2,2-二氟苯并[d][1,3]二氧杂环戊烯-4-基)氧基)二甲基硅烷(4)
将2,2-二氟苯并[d][1,3]二氧杂环戊烯-4-酚(31.6g,181.50mmol)溶于无水DMF(300mL),然后加入咪唑(29.66g,435.60mmol)和化合物3(32.83g,217.80mmol)。加完后,反应液加热至80℃,并在该条件下反应10小时。TLC(PE:EA=3:1)显示反应完全。反应液冷却至室温,加入水(300mL)稀释,用EtOAc(150mL×3)萃取,合并有机相,分别用水(100mL×2)洗涤,饱和食盐水(100mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,粗品通过正相硅胶柱层析分离(EtOAc/PE=0%-20%)得到叔丁基((2,2-二氟苯并[d][1,3]二氧杂环戊烯-4-基)氧基)二甲基硅烷(4)(34.0g;产率:64.96%)。
第三步:叔丁基二甲基((2,2,7-三氟苯并[d][1,3]二氧杂环戊烯-4-基)氧基)硅烷(5)
将化合物4(34.0g,117.91mmol)溶于无水四氢呋喃(250mL)中,然后抽真空,氮气置换,反复三次。氮气保护下,反应液冷却至-78℃,缓慢加入正丁基锂(58.95mL,147.38mmol,2.5M),维持反应温度不超过-70℃。加完后,反应液在该温度下继续反应2小时,缓慢滴加NFSI(46.47g,147.38mmol)的四氢呋喃(100mL)溶液,维持温度不超过-65℃。加完后反应液缓慢升至室温并继续反应10小时。LCMS显示少许原料剩余。加入饱和氯化铵水溶液(100mL)淬灭反应,然后用EtOAc(100mL×3)萃取,合并有机相,饱和食盐水(100mL)洗涤,无水硫酸钠干燥,过滤,滤液浓缩旋干,粗品通过正相硅胶柱层析分离(EtOAc/PE=0%-20%)得到叔丁基二甲基((2,2,7-三氟苯并[d][1,3]二氧杂环戊烯-4-基)氧基)硅烷(5)(32.20g;产率:89.14%)。
第四步:2,2,7-三氟苯并[d][1,3]二氧杂环戊烯-4-酚(INT-1)
称取叔丁基二甲基((2,2,7-三氟苯并[d][1,3]二氧杂环戊烯-4-基)氧基)硅烷(32.20g,105.11mmol)溶于无水乙醇(300mL),然后加入氢氧化钾(8.85g,157.66mmol),加完后,反应液在室温下反应10hr。TLC(PE:EA=5:1)显示反应完全。冰浴下,加入2N HCl调节pH至3左右,然后用EtOAc(150mL×3)萃取,合并有机相,饱和食盐水(100mL)洗涤,无水硫酸钠干燥,过滤,滤液浓缩旋干,粗品通过反相硅胶柱层析分离(HCOOH;ACN/H
实施例1:目标化合物I-1的制备
(R)-3-氯-N-(2-(2,3-二羟基丙氧基)吡啶-4-基)-2-氟-6-((2,2,7-三氟苯并[d][1,3])二氧戊环-4-基)氧基)-4-(三氟甲基)苯甲酰胺(目标化合物I-1)
化合物I-1的合成路线如下所示:
第一步:6-溴-3-氯-2-氟-4-三氟甲基苯甲酸(1B)的合成
将6-溴-3-氯-2-氟-4-三氟甲基苯甲酸甲酯(1.00g,2.98mmol)溶于无水四氢呋喃(6mL)和甲醇(2mL),然后称取氢氧化钠(0.60g,14.90mmol)于水(3毫升)中,并加入到上述溶液中。加完后,反应液在室温下反应16小时。LC-MS显示反应完成。反应液用EtOAc(15mL×3)萃取,合并有机相,用水(20mL×2)洗涤两次,水相合并,并用6N盐酸调节pH到1,然后用EtOAc(20mL×3)萃取,合并有机相,饱和食盐水(15mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,粗品直接用于下一步。
第二步:(S)-2-(2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-4-胺(1C)的合成
氮气保护,冰浴条件下,将(S)-(2,2-二甲基-1,3-二氧戊环-4-基)甲醇(2.06g,15.56mmol)溶于N-甲基吡咯烷酮(20mL),然后缓慢加入钠氢(0.62g,15.56mmol,60%),加完后,反应液在冰浴条件下反应30min。加入2-氯吡啶-4-胺(1.0g,7.78mmol),然后反应液升温至120℃下反应10hr。TLC(PE:EtOAc=1:1)监测反应完全。降至室温,并与条件下用饱和氯化铵水溶液(20mL)淬灭反应。反应液用EtOAc(30mL×3)萃取,合并有机相,并用饱和食盐水(20mL)洗涤1次,无水Na
第三步:(S)-6-溴-3-氯-N-(2-((2,2-二甲基-1,3-二氧戊環-4-基)甲氧基)吡啶-4-基)-2-氟-4-(三氟甲基)苯甲酰胺(1D)
冰浴条件下,将中间体1B(0.97g,3.02mmol),中间体1C(0.74g,3.32mmol),DIEA(1.17g,9.05mmol)和HATU(1.72g,4.53mmol)加入到DMF(10mL)中,反应液在室温下反应16小时。LC-MS显示反应完成。向反应液中加入水(20mL),再用EtOAc(30mL×3)萃取,合并有机相,分别用水(20mL×2)和饱和食盐水(20mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,粗品通过正相硅胶制备板分离(EtOAc/PE=1:2)得到淡黄色固体(S)-6-溴-3-氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-4-基)-2-氟-4-(三氟甲基)苯甲酰胺(1D)(0.26g;产率16.3%)。
第四步:(S)-3-氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-4-基)-2-氟-6-((2,2,7-三氟苯并[d][1,3]二氧戊环-4-基)氧基)-4-(三氟甲基)苯甲酰胺(1F)
室温下,将中间体1D(0.26g,0.49mmol),中间体1E(94.65mg,0.49mmol),碳酸铯(401.33mg,1.23mmol),碘化亚铜(18.77mg,0.098mmol)和甲苯(3mL)加入到20毫升微波管中,通入氮气,鼓泡五分钟,密封微波管,在100℃下反应30min。冷却至室温,向反应液中加入水(10mL),用EtOAc(20mL×3)萃取,有机相用饱和食盐水(20mL)洗1次,并用Na
第五步:(R)-3-氯-N-(2-(2,3-二羟基丙氧基)吡啶-4-基)-2-氟-6-((2,2,7-三氟苯并[d][1,3])二氧戊环-4-基)氧基)-4-(三氟甲基)苯甲酰胺(I-1)
冰浴条件下,将中间体1F(70.0mg,0.11mmol)溶于DCM(2mL)中,然后加入三氯化硼(38.51mg,0.33mmol,1.0M)。反应液在冰浴条件下反应2小时。LC-MS监测反应完成后,反应液用饱和碳酸氢钠水溶液淬灭,调至pH到8左右,然后用二氯甲烷(10mL×3)萃取,合并有机相用饱和食盐水(20mL)洗1次,再用无水Na
1
LC-MS,M/Z(ESI):599.4[M+H]
实施例2:目标化合物I-2的制备
(R)-5-氯-N-(2-(2,3-二羟基丙氧基)吡啶-4-基)-2-((2,2,7-三氟苯并[d][1,3]二氧杂环戊烯-4-基)氧基)-4-三氟甲基苯甲酰胺(目标化合物I-2)
化合物I-2的合成路线如下所示:
第一步:2-溴-5-氯-4-三氟甲基苯甲酸(2B)的合成
将2-溴-5-氯-4-三氟甲基苯甲酸甲酯(0.41g,1.29mmol)溶于无水四氢呋喃(4mL)和甲醇(1mL),然后称取一水合氢氧化锂(0.16g,3.87mmol)于水(2毫升)中,并加入到上述溶液中。加完后,反应液在室温下反应16小时。LC-MS显示反应完成。反应液用EtOAc(15mL×3)萃取,合并有机相,用水(20mL×2)洗涤两次,水相合并,并用6N盐酸调节pH到1,然后用EtOAc(20mL×3)萃取,合并有机相,饱和食盐水(15mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,得到2-溴-5-氯-4-三氟甲基苯甲酸(2B)(0.97g;粗品),粗品直接用于下一步。
第二步:(S)-2-溴-5-氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-4-基)-4-(三氟甲基)苯甲酰胺(2D)
冰浴条件下,将化合物2B(0.37g,1.22mmol),化合物2C(0.33g,1.46mmol),DIEA(0.47g,3.66mmol)和HATU(0.70g,1.83mmol)加入到DMF(5mL)中,加完后,反应液在室温下反应16小时。LC-MS显示反应完成。向反应液中加入水(20mL),再用EtOAc(30mL×3)萃取,合并有机相,分别用水(20mL×2)和饱和食盐水(20mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,粗品通过正相硅胶制备板分离(EtOAc/PE=1:2)得到淡黄色固体(S)-2-溴-5-氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-4-基)-4-(三氟甲基)苯甲酰胺(2D)(60mg;产率:9.65%)。
第三步:(S)-5-氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-4-基)-2-((2,2,7-三氟苯并[d][1,3]二氧戊环-4-基)氧基)-4-(三氟甲基)苯甲酰胺(2F)
室温下,将中间体2D(60.0mg,0.12mmol),中间体2E(27.13mg,0.14mmol),碳酸铯(95.88mg,0.29mmol),碘化亚铜(4.5mg,0.024mmol)和甲苯(3mL)加入到10毫升微波管中,通入氮气,鼓泡五分钟,密封微波管,在100℃下反应30min。冷却至室温,向反应液中加入水(10mL),用EtOAc(20mL×3)萃取,有机相用饱和食盐水(20mL)洗1次,并用Na
第四步:(R)-5-氯-N-(2-(2,3-二羟基丙氧基)吡啶-4-基)-2-((2,2,7-三氟苯并[d][1,3]二氧杂环戊烯-4-基)氧基)-4-三氟甲基苯甲酰胺(I-2)
冰浴条件下,将中间体2F(30.0mg,0.05mmol)溶于DCM(2mL)中,然后加入三氯化硼(16.98mg,0.14mmol,1.0M)。反应液在冰浴条件下反应2小时。LCMS监测反应完成后,反应液用饱和碳酸氢钠水溶液淬灭,调至pH到8左右,然后用二氯甲烷(10mL×3)萃取,合并有机相用饱和食盐水(20mL)洗1次,再用无水Na
1
LC-MS,M/Z(ESI):581.3[M+H]
实施例3:目标化合物I-3的制备
(R)-4,5-二氯-N-(2-(2,3-二羟基丙氧基)吡啶-4-基)-2-((2,2,7-三氟苯并[d][1,3]二氧杂环己烷-4-基)氧基)苯甲酰胺(目标化合物I-3)
化合物I-3的合成路线如下所示:
第一步:4,5-二氯-2-((2,2,7-三氟苯并[d][1,3]二氧杂酚-4-基)氧基)苯甲酸甲酯(3C)的合成
将化合物3A(1.0g,4.48mmol)溶于乙腈(10mL),然后加入化合物3B(0.95g,4.93mmol)和碳酸铯(3.65g,11.21mmol)。加完后,反应液在80℃下反应16小时。TLC(PE:EtOAc=5:1)显示反应完成。反应液冷却至室温,用EtOAc(20mL×3)萃取,合并有机相,用饱和食盐水(15mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,得到4,5-二氯-2-((2,2,7-三氟苯并[d][1,3]二氧杂酚-4-基)氧基)苯甲酸甲酯(3C)(1.70g;粗品),粗品直接用于下一步。
第二步:4,5-二氯-2-((2,2,7-三氟苯并[d][1,3]二氧杂酚-4-基)氧基)苯甲酸(3D)的合成
将化合物3C(0.70g,1.77mmol)溶于无水四氢呋喃(4mL)和甲醇(1mL),然后称取一水合氢氧化锂(0.23g,5.31mmol)于水(2毫升)中,并加入到上述溶液中。加完后,反应液在室温下反应16小时。LC-MS显示反应完成。反应液用EtOAc(10mL×3)萃取,合并有机相,用水(15mL×2)洗涤,水相合并,并用6N盐酸调节pH到1,然后用EtOAc(20mL×3)萃取,合并有机相,饱和食盐水(10mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,得到4,5-二氯-2-((2,2,7-三氟苯并[d][1,3]二氧杂酚-4-基)氧基)苯甲酸(3D)(0.62g;粗品),粗品直接用于下一步。
第三步:(S)-3,4-二氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-4-基)-2-氟-6-((2,2,7-三氟苯并[d][1,3]二氧杂环戊醇-4-基)氧基)苯甲酰胺(3F)
冰浴条件下,将化合物3D(0.32g,0.84mmol)加入到无水乙腈(5mL)中,然后加入TCFH(0.28g,1.01mmol)和NMI(0.21g,2.52mmol)。加完后,反应液在冰浴条件下反应10分钟,再加入化合物3E(0.23g,1.01mmol),加完后再在室温下反应16小时。LC-MS显示反应完成。向反应液中加入水(10mL),再用EtOAc(20mL×3)萃取,合并有机相,用饱和食盐水(10mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,粗品通过正相硅胶制备板分离(EtOAc/PE=1:2)得到(S)-3,4-二氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-4-基)-2-氟-6-((2,2,7-三氟苯并[d][1,3]二氧杂环戊醇-4-基)氧基)苯甲酰胺(3F)(0.46g;产率:93.27%)。
第四步:(R)-4,5-二氯-N-(2-(2,3-二羟基丙氧基)吡啶-4-基)-2-((2,2,7-三氟苯并[d][1,3]二氧杂环己烷-4-基)氧基)苯甲酰胺(I-3)
冰浴条件下,将化合物3F(0.46g,0.78mmol)溶于DCM(5mL)中,然后加入三氯化硼(0.28g,2.35mmol,1.0M)。反应液在冰浴条件下反应2小时。LCMS监测反应完成后,反应液用饱和碳酸氢钠水溶液淬灭,调至pH到8左右,然后用二氯甲烷(20mL×3)萃取,合并有机相用饱和食盐水(20mL)洗1次,再用无水Na
1
LC-MS,M/Z(ESI):547.30[M+H]
实施例4:目标化合物I-4的制备
(R)-5-氯-N-(6-(2,3-二羟基丙氧基)吡啶-2-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(目标化合物I-4)
化合物I-4的合成路线如下所示:
第一步:(S)-6-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-2-胺(4E)的合成
将化合物4E-1(1.0g,8.92mmol)溶于无水DMF(10mL),然后加入化合物4E-2(1.30g,9.81mmol)和碳酸铯(8.72g,26.76mmol)。加完后,反应液加热到80℃,并在该温度下反应10小时。TLC(PE:EA=3:1)显示反应完成。反应液加入水(15mL)稀释,用EtOAc(15mL×3)萃取,合并有机相,再分别用水(15mL×2)和饱和食盐水(15mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,粗品通过正相硅胶柱层析分离(EtOAc/PE=0%-50%)得到(S)-6-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-2-胺(4E)(1.92g;产率:95.98%)。
第二步:5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸甲酯(4C)的合成
将5-氯-2-氟-4-(三氟甲基)苯甲酸甲酯(0.70g,2.73mmol)溶于乙腈(5mL),然后加入化合物4B(0.41g,3.27mmol)和碳酸铯(2.67g,8.18mmol)。加完后,反应液加热到85℃,并在该温度下反应10小时。TLC(PE:EA=3:1)显示反应完成。反应液直接减压浓缩旋干,加入水(5mL)稀释,并用6N盐酸调节pH到3,然后用EtOAc(15mL×3)萃取,合并有机相,饱和食盐水(15mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,粗品通过正相硅胶柱层析分离(EtOAc/PE=0%-50%)得到5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸甲酯(4C)(0.50g;产率:50.5%)。
第三步:5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸(4D)
将5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸甲酯(0.50g,1.38mmol)溶于无水四氢呋喃(4mL)和甲醇(1mL),然后称取一水合氢氧化锂(0.18g,4.14mmol)于水(2毫升)中,并加入到上述溶液中。加完后,反应液在室温下反应16小时。LC-MS显示反应完成。反应液用EtOAc(15mL×3)萃取,合并有机相,用水(10mL×2)洗涤两次,水相合并,并用6N盐酸调节pH到1,然后用EtOAc(15mL×3)萃取,合并有机相,饱和食盐水(10mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,得到5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸(4D)(0.42g;粗品),粗品直接用于下一步。
第四步:(S)-5-氯-N-(6-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-2-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(4F)
冰浴条件下,将5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸(0.10g,0.69mmol)溶于乙腈(3mL),随后加入TCFH(0.23,0.83mmol)和NMI(0.17,2.06mmol),10分钟后再加入中间体4E(0.19g,0.83mmol),加完后升至室温并在室温下反应10小时。向反应液中加入水(10mL),用EtOAc(20mL×3)萃取,有机相用饱和食盐水(20mL)洗1次,并用Na
第五步:(R)-5-氯-N-(6-(2,3-二羟基丙氧基)吡啶-2-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(目标化合物I-4)
冰浴条件下,将(S)-5-氯-N-(6-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-2-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(4F)(0.15g,0.27mmol)溶于DCM(2mL)中,然后加入三氯化硼(47.50mg,0.41mmol,1.0M)。反应液在冰浴条件下反应2小时。LCMS监测反应完成后,反应液用饱和碳酸氢钠水溶液淬灭,调至pH到8左右,然后用二氯甲烷(10mL×3)萃取,合并有机相用饱和食盐水(20mL)洗1次,再用无水Na
1
LC-MS,M/Z(ESI):515.2[M+H]
实施例5:目标化合物I-9的制备
(R)-5-氯-N-(2-(2,3-二羟基丙氧基)嘧啶-4-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(目标化合物I-9)
化合物I-9的合成路线如下所示:
第一步:(S)-2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)嘧啶-4-胺(9B)的合成
将化合物9B-1(1.0g,7.72mmol)溶于无水DMF(10mL),然后加入化合物9B-2(1.12g,8.49mmol)和碳酸铯(7.55g,23.16mmol)。加完后,反应液加热到120℃,并在该温度下反应10小时。TLC(PE:EA=3:1)显示反应完成。反应液加入水(15mL)稀释,用EtOAc(15mL×3)萃取,合并有机相,再分别用水(15mL×2)和饱和食盐水(15mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,粗品通过正相硅胶柱层析分离(EtOAc/PE=0%-50%)得到(S)-2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)嘧啶-4-胺(9B)(1.72g;产率:98.92%)。
第二步:(S)-5-氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)嘧啶-4-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(9C)
冰浴条件下,将5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸(0.10g,0.69mmol)溶于乙腈(3mL),随后加入TCFH(0.23,0.83mmol)和NMI(0.17,2.06mmol),10分钟后再加入中间体9B(77.52mg,0.34mmol),加完后升至室温并在室温下反应10小时。向反应液中加入水(10mL),用EtOAc(20mL×3)萃取,有机相用饱和食盐水(20mL)洗1次,并用Na
第五步:(R)-5-氯-N-(2-(2,3-二羟基丙氧基)嘧啶-4-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(目标化合物I-9)
冰浴条件下,将(S)-5-氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)嘧啶-4-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(9C)(0.16g,0.29mmol)溶于DCM(2mL)中,然后加入三氯化硼(50.58mg,0.43mmol,1.0M)。反应液在冰浴条件下反应2小时。LCMS监测反应完成后,反应液用饱和碳酸氢钠水溶液淬灭,调至pH到8左右,然后用二氯甲烷(10mL×3)萃取,合并有机相用饱和食盐水(20mL)洗1次,再用无水Na
1
LC-MS,M/Z(ESI):516.2[M+H]
实施例6:目标化合物I-10的制备
(R)-4-(5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰氨基)-2-(2,3-二羟基丙氧基)吡啶-1-氧化物(目标化合物I-10)
化合物I-10的合成路线如下所示:
第一步:5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸甲酯(10C)的合成
将5-氯-2-氟-4-(三氟甲基)苯甲酸甲酯(0.70g,2.73mmol)溶于乙腈(5mL),然后加入化合物10B(0.41g,3.27mmol)和碳酸铯(2.67g,8.18mmol)。加完后,反应液加热到85℃,并在该温度下反应10小时。TLC(PE:EA=3:1)显示反应完成。反应液直接减压浓缩旋干,加入水(5mL)稀释,并用6N盐酸调节pH到3,然后用EtOAc(15mL×3)萃取,合并有机相,饱和食盐水(15mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,粗品通过正相硅胶柱层析分离(EtOAc/PE=0%-50%)得到5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸甲酯(10C)(0.50g;产率:50.5%)。
第二步:5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸(10D)
将5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸甲酯(0.50g,1.38mmol)溶于无水四氢呋喃(4mL)和甲醇(1mL),然后称取一水合氢氧化锂(0.18g,4.14mmol)于水(2毫升)中,并加入到上述溶液中。加完后,反应液在室温下反应16小时。LC-MS显示反应完成。反应液用EtOAc(15mL×3)萃取,合并有机相,用水(10mL×2)洗涤两次,水相合并,并用6N盐酸调节pH到1,然后用EtOAc(15mL×3)萃取,合并有机相,饱和食盐水(10mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,得到5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸(10D)(0.42g;粗品),粗品直接用于下一步。
第三步:(S)-5-氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-4-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(10F)
冰浴条件下,将5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸(0.24g,0.69mmol)溶于乙腈(3mL),随后加入TCFH(0.23,0.83mmol)和NMI(0.17,2.06mmol),10分钟后再加入中间体10E(0.19g,0.83mmol),加完后升至室温并在室温下反应10小时。向反应液中加入水(10mL),用EtOAc(20mL×3)萃取,有机相用饱和食盐水(20mL)洗1次,并用Na
第四步:(S)-4-(5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰氨基)-2-((2,2-2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-1-氧化物(10G)
/>
冰浴下,称取(S)-5-氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-4-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(10F)(0.10g,0.18mmol)溶于DCM(3mL),然后加入m-CPBA(91.5mg,85%,0.45mmol)。加完后,反应液在室温条件下反应10小时。LC-MS显示反应不完全,有部分原料剩余,反应液用二氯甲烷(20mL)稀释,然后用饱和碳酸氢钠水溶液(10mL×2)洗涤,有机相用Na
第五步:(R)-4-(5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰氨基)-2-(2,3-二羟基丙氧基)吡啶-1-氧化物(目标化合物I-10)
冰浴条件下,将(S)-4-(5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰氨基)-2-((2,2-2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-1-氧化物(10G)(50.0mg,0.08mmol)溶于DCM(2mL)中,然后加入三氯化硼(30.78mg,0.26mmol,1.0M)。反应液在冰浴条件下反应2小时。LC-MS监测反应完成后,反应液用饱和碳酸氢钠水溶液淬灭,调至pH到8左右,然后用二氯甲烷(10mL×3)萃取,合并有机相用饱和食盐水(20mL)洗1次,再用无水Na
1
LC-MS,M/Z(ESI):531.3[M+H]
实施例7:目标化合物I-11的制备
(R)-4-(4,5-二氯-2-(4-氟-2-甲基苯氧基)苯甲酰胺基)-2-(2,3-二羟基丙氧基)吡啶1-氧化物(目标化合物I-11)
化合物I-11的合成路线如下所示:
第一步:(S)-4,5-二氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-4-基)-2-(4-(三氟甲氧基)苯氧基)苯甲酰胺(11C)
冰浴条件下,将化合物11A(0.30g,0.82mmol)溶于乙腈(3mL),随后加入TCFH(0.34,1.23mmol)和NMI(0.20,2.45mmol),10分钟后再加入中间体11B(0.22g,0.98mmol),加完后升至室温并在室温下反应10小时。向反应液中加入水(10mL),用EtOAc(20mL×3)萃取,有机相用饱和食盐水(20mL)洗1次,并用Na
第二步:(S)-4-(4,5-二氯-2-(4-(三氟甲氧基)苯氧基)苯甲酰胺基)-2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶1-氧化物(11D)
冰浴下,称取(S)-4,5-二氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶-4-基)-2-(4-(三氟甲氧基)苯氧基)苯甲酰胺(11C)(0.47g,0.82mmol)溶于DCM(3mL),然后加入m-CPBA(0.42g,85%,2.05mmol)。加完后,反应液在室温条件下反应10小时。LCMS显示反应不完全,有部分原料剩余,反应液用二氯甲烷(20mL)稀释,然后用饱和碳酸氢钠水溶液(10mL×2)洗涤,有机相用Na
第三步:(R)-4-(4,5-二氯-2-(4-(三氟甲氧基)苯氧基)苯甲酰胺基)-2-(2,3-二羟基丙氧基)吡啶1-氧化物(目标化合物I-11)
冰浴条件下,将(S)-4-(4,5-二氯-2-(4-(三氟甲氧基)苯氧基)苯甲酰胺基)-2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)吡啶1-氧化物(0.25g,0.42mmol)溶于DCM(5mL)中,然后加入三氯化硼(124.25mg,1.06mmol,1.0M)。反应液在冰浴条件下反应2小时。LCMS监测反应完成后,反应液用饱和碳酸氢钠水溶液淬灭,调至pH到8左右,然后用二氯甲烷(10mL×3)萃取,合并有机相用饱和食盐水(20mL)洗1次,再用无水Na
1
LC-MS,M/Z(ESI):549.19[M+H]
实施例8:目标化合物I-19的制备
(R)-5-氯-N-(2-(2,3-二羟基丙氧基)-3-氟吡啶-4-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(目标化合物I-19)
化合物I-19的合成路线如下所示:
第一步:(S)-2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)-3-氟吡啶-4-胺(19B)的合成
将化合物19B-1(0.20g,1.37mmol)溶于无水DMF(3mL),然后加入化合物19B-2(0.20g,1.51mmol)和碳酸铯(1.34g,4.11mmol)。加完后,反应液加热到80℃,并在该温度下反应10小时。TLC(PE:EA=3:1)显示反应完成。反应液加入水(15mL)稀释,用EtOAc(15mL×3)萃取,合并有机相,再分别用水(15mL×2)和饱和食盐水(15mL)洗涤,有机相用无水硫酸钠干燥,过滤,滤液浓缩旋干,粗品通过正相硅胶柱层析分离(EtOAc/PE=0%-50%)得到(S)-2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)-3-氟吡啶-4-胺(19B)(0.30g;产率:90.49%)。
第二步:(S)-5-氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)-3-氟吡啶-4-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(19C)
冰浴和氮气条件下,将5-氯-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酸甲基)苯甲酸(0.10g,0.29mmol)溶于无水二氯甲烷(3mL),随后加入草酰氯(43.7mg,0.34mmol),加完后在冰浴和氮气条件下反应30分钟。然后将反应液减压浓缩旋干,再在冰浴和氮气条件下分别加入无水四氢呋喃(3mL),三乙胺(87.1g,0.87mmol)和化合物19B(83.4g,0.34mmol),加完后,反应液在该温度下反应2小时。TLC(PE:EA=3:1)显示反应完成。反应液加入水(10mL)稀释,用DCM(20mL×3)萃取,有机相用饱和食盐水(10mL)洗1次,并用Na
第三步:(R)-5-氯-N-(2-(2,3-二羟基丙氧基)-3-氟吡啶-4-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(目标化合物I-19)
冰浴条件下,将(S)-5-氯-N-(2-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)-3-氟吡啶-4-基)-2-(4-氟-2-甲基苯氧基)-4-(三氟甲基)苯甲酰胺(19C)(0.11g,0.19mmol)溶于DCM(2mL)中,然后加入三氯化硼(67.48mg,0.58mmol,1.0M)。反应液在冰浴条件下反应2小时。LCMS监测反应完成后,反应液用饱和碳酸氢钠水溶液淬灭,调至pH到8左右,然后用二氯甲烷(10mL×3)萃取,合并有机相用饱和食盐水(10mL)洗1次,再用无水Na
1
LC-MS,M/Z(ESI):531.14[M-H]
以下目标化合物参照化合物I-1的合成方法类似制备得到。
/>
/>
测试例1:化合物对Nav1.8离子通道抑制活性检测
试剂除用于酸碱滴定的NaOH和KOH外,均从Sigma(St.Louis,MO)公司购买。测试化合物的最终浓度均在当天配制,再溶于细胞外液。细胞外液(mM)为:NaCl,137;KCl,4;CaCl
测试化合物溶于二甲基亚砜(DMSO),浓度为9mM。测试当天再溶于细胞外液,配制成要求浓度。
电生理实验步骤:
将细胞转移到灌流槽中,用细胞外液进行灌流。细胞内液实验当天融化。电极用PC–10(Narishige,Japan)拉制。全细胞膜片钳记录,噪音用采样频率的五分之一进行过滤。电极内加入1/4电极管长的细胞内液,将电极安装在探针上。设置好所需要的Protocol,将界面调成Membrane test,Stage调成Bath。电极内施加正压,将电极尖端接触到细胞,抽气装置三通阀调成三通状态,然后对电极施加负压,使得电极与细胞形成高阻封接。Stage调成Patch,leak控制在-200pA内,继续施加负压,使得细胞膜破裂,形成电流通路。打开抽滤装置和细胞外液阀门进行灌流,观察细胞电流,待细胞电流稳定开始加药(至少3个sweep的电流曲线重叠)。从低浓度往高浓度加药,每个浓度加药时间不少于2min且等到电流稳定再更换浓度加药。
供试品给药采用利用自身重力的灌流系统进行灌流。在初始记录期间,观察峰值电流幅度至少1分钟直到其稳定。在此期间,所有峰值电流幅度的CV%应小于10%以排除初始电流的上下波动。初始记录期间最后10次记录的峰值电流幅度的平均值作为阴性对照的电流峰值。待初始电流稳定后,试验样品从低浓度开始给药直到10次记录的峰值电流再次稳定或者持续给药5min后,给药后和给药前峰值电流“不变”。我们将下面两种情况定义为“稳定”或者“不变”:1)如果连续10次扫描的峰值电流的绝对平均值超过200pA而CV值小于10%,2)或者连续10次扫描的峰值电流的平均值在200pA和50pA之间而且CV值小于30%。然后给予下一个更高浓度的检测。
每个浓度最后10次扫描的峰值电流平均值作为该浓度的峰值电流,用于数据分析。如果5分钟内不能达到稳定状态,那么此时的最后10次扫描的峰值电流平均值作为该浓度的峰值电流用于数据分析。同时该细胞要丢弃,不再用于更高浓度的检测。化合物每个浓度至少测试两个细胞。
电压脉冲程序:
将细胞钳制在–80mV,然后用持续10毫秒方波去极化到10mV,以得到NaV1.8电流。这一程序每5秒重复一次。检测方波引发的最大电流,待其稳定后,灌流测试化合物,当反应稳定后,计算阻断的强度。
数据处理和拟合
资料采集和分析将用pCLAMP 10(Molecular Devices,Union City,CA)。电流稳定指的是电流随时间变化在有限的范围内。通过绘制药物的梯度稀释系列浓度和其作用在HEK293/Nav1.8上产生的稳定电流值之间的量效关系,
进而计算该药物对Nav1.8离子通道的抑制活性(IC
试验结果表明,本发明化合物具有较强的对Nav1.8离子通道抑制活性。
测试例2:小鼠药代动力学试验
小鼠药代动力学试验,采用雄性ICR小鼠3只,禁食过夜,口服灌胃给药10mg/kg。在给药前和在给药后15、30分钟以及1、2、4、6、8、24小时采血。血液样品8000转/分钟4℃离心6分钟,收集血浆,于-20℃保存。取各时间点血浆,加入3-5倍量含内标的乙腈溶液混合,涡旋混合1分钟,13000转/分钟4℃离心10分钟,取上清液加入3倍量水混合,取适量混合液进行LC-MS/MS分析。主要药代动力学参数用WinNonlin 7.0软件非房室模型分析。
试验结果表明,本发明化合物在小鼠上药代动力学特性好,成药性优良。
测试例3:大鼠药代动力学试验
大鼠药代动力学试验,采用雄性SD大鼠3只,180-240g,禁食过夜,口服灌胃给药10mg/kg。在给药前和在给药后15、30分钟以及1、2、4、6、8、24小时采血。血液样品8000转/分钟4℃离心6分钟,收集血浆,于-20℃保存。取各时间点血浆,加入3-5倍量含内标的乙腈溶液混合,涡旋混合1分钟,13000转/分钟4℃离心10分钟,取上清液加入3倍量水混合,取适量混合液进行LC-MS/MS分析。主要药代动力学参数用WinNonlin 70软件非房室模型分析。
试验结果表明,本发明化合物在大鼠上药代动力学特性好,成药性优良。
测试例4:大鼠足底切口痛模型
将体重为200~250g的雄性SD大鼠,麻醉后俯卧位固定,将其侧后肢脚掌朝上展平,手术胶带固定脚趾,消毒。在动物足底脚后跟0.5cm处用手术刀向趾端划开皮肤筋膜,作一1cm左右的纵向切口。用手术弯镊抬起趾短屈肌后,用手术刀对肌肉腹部进行纵向切口,不将肌肉完全切断。对皮肤进行缝合,消毒。造模第二天,将动物随机分为3组,每组8只,分别口服灌胃溶媒、化合物I-1(10mg/kg)、普瑞巴林(30mg/kg),在每只动物的给药前及给药后1、3、6小时分别用Von-Frey纤维丝检测动物的机械痛阈。
机械痛阈检测方法:将试验动物用Von-Frey纤维丝持续刺激待测后肢足底使纤维丝弯曲,观察动物缩足反应。按照纤维丝克数从小到大的顺序逐一刺激受试动物,每一个克数的纤维丝连续刺激5次。若出现的阳性反应小于3次,则使用较大一级的纤维丝重复上述操作,当测试第一次出现3次或3次以上的阳性反应,则该纤维丝为该动物的疼痛阈值(每只动物测3次测试,取其平均值)。纤维丝克数:0.6,1.0,1.4,2.0,4.0,6.0,8.0,10.0,15.0;切断值为15.0g。各组数据采用GraphPad Prism 8.0进行数据统计,统计方法采用单因素方法分析法(One-way ANOVA),比较各组之间有无统计学差异,P<0.05具有统计学差异。
如图1所示,试验结果表明,本发明化合物可以抑制大鼠足底切口疼痛反应,显示出明显抗急性疼痛效果。
以上对本公开技术方案的实施方式进行了示例性的说明。应当理解,本公开的保护范围不拘囿于上述实施方式。凡在本公开的精神和原则之内,本领域技术人员所做的任何修改、等同替换、改进等,均应包含在本申请权利要求书的保护范围之内。
- 作为NAV1.8抑制剂的环烷基3-氧代哌嗪甲酰胺和环杂烷基3-氧代哌嗪甲酰胺
- 作为NAV1.8抑制剂的5-氧代-吡咯烷-3-甲酰胺