一种高效脱汞催化剂设计优化方法
文献发布时间:2024-05-31 01:29:11
技术领域
本发明涉及金属化合物材料设计技术领域,特别是涉及一种高效脱汞催化剂设计优化方法。
背景技术
重金属汞作为一种具有强神经毒性、高生物蓄积性的污染物,一直以来都是污染物脱除方向的研究重点。其中,燃煤排放的汞占全世界人为汞排放的三分之一,控制火力发电厂的汞排放显然是解决全球汞污染问题的关键所在。汞主要以单质汞Hg
目前,最常见的单质汞脱除技术是活性炭喷射技术,但由于活性炭的吸附容量有限,这使得它的使用成本高昂,因此,开发性能优异、成本低廉且具有循环回收特性的脱汞催化剂势在必行。而对于新型材料的研发绝大多数依靠试错法,通过大量的实验试错,才有可能得出一条新的制备工艺。
机器学习可以通过采集大量数据建立数据库,并根据需求进行信息筛分从而建立源域模型,将相关知识迁移到目标域模型可以强化目标域的建立。结合集成学习算法和机器学习算法,可以优化模型,降低模型方差,提高模型泛化能力、稳定性及准确性。机器学习的兴起带来了一种全新的材料研发思路,结合密度泛函理论的模拟计算,在总结归纳前人经验的基础上,来逆向预测材料的制备工艺,可以减少人工试错带来的资源浪费。
发明内容
发明目的:为了开发一种用于脱除单质汞的脱汞催化剂,同时降低催化剂的开发成本,本发明提供了一种高效脱汞催化剂设计优化方法,本发明基于机器学习和密度泛函理论进行设计。
技术方案:一种高效脱汞催化剂设计优化方法,包括以下步骤:
步骤一、利用量子化学软件构建催化剂系列基础及改性模型;
所述催化剂系列基础及改性模型用于计算催化剂的体系能量、吸附单质汞后催化剂的体系能量、单质汞的体系能量以及Hirshfeld电荷;
步骤二、通过步骤一构建的催化剂系列基础及改性模型和密度泛函理论计算单质汞吸附能,并构建脱汞催化剂的源域数据集;
所述源域数据集包括:催化剂结构模型信息,催化剂结构模型对应的特征变量、单质汞吸附能和Hirshfeld电荷;所述催化剂结构模型信息包括催化剂的基础构型、活性点位信息、掺杂元素、掺杂位型信息;所述特征变量是指原子半径、元素周期性、价电子数目、第一电离能和Pauling电负性特征;
步骤三、对步骤二中构建的源域数据集进行预处理和规范化,通过权重分析确定对催化剂结构模型的单质汞吸附能影响显著的特征变量,形成最优特征集;
步骤四、采用机器学习方法,建立最优特征集与脱汞性能指标间的定量关系模型;所述脱汞性能指标包括催化剂的单质汞吸附能和Hirshfeld电荷;
步骤五、使用R
步骤六、选取多个材料构建具有脱汞性能的催化剂目标域数据集;
所述目标域数据集包括选取材料的原子半径、元素周期性、价电子数目、第一电离能和Pauling电负等特征,以及对应的基础构型、活性点位信息、掺杂元素、掺杂位点;
步骤七、使用步骤五中确定的定量关系模型,预测步骤二中目标域数据集中的材料的脱汞性能指标,根据脱汞性能指标,筛选出具有最佳脱汞性能指标的材料,即为优化设计的脱汞催化剂。
进一步的,所述步骤二的具体操作步骤如下:
步骤2.1)、以原子半径、元素周期性、价电子数目、第一电离能和Pauling电负性等特征为依据,选取具有脱汞性能的催化剂,并收集这些催化剂的基础构型、活性点位信息、掺杂元素和掺杂位型,建立脱汞系列的催化剂结构模型;
所述催化剂结构模型中,包括催化剂的基础构型、活性点位信息、掺杂元素、掺杂位型信息;
步骤2.2)、将步骤2.1)建立的脱汞系列的催化剂结构模型代入通过S1建立的催化剂系列基础及改性模型中,获取催化剂结构模型的体系能量、吸附单质汞后催化剂的体系能量、单质汞的体系能量以及Hirshfeld电荷;
步骤2.3)、将步骤2.2)中得到的催化剂结构模型的体系能量、吸附单质汞后催化剂的体系能量、单质汞的体系能量,采用密度泛函理论计算得到催化剂结构模型的单质汞吸附能;
单质汞吸附能是指“单质汞在催化剂表面的吸附能”,记为E
其中,
步骤2.4)、建立脱汞系列催化剂结构模型的源域数据集。
进一步的,所述步骤四中,所述步骤四中,采用的机器学习算法包括但不限于:随机森林回归、K近邻法、随机树法、随机森林法、随机委员会法、缩减误差修减树法、梯度下降回归树法、极端随机森林回归法和XGBRegressor法。
进一步的,所述步骤四中,所述脱汞性能指标包括催化剂的单质汞吸附能和Hirshfeld电荷;
1)单质汞吸附能:吸附能为负值,其绝对值越大,则吸附越可靠;
当吸附能在-0.25eV~0eV时,为物理吸附,主要通过范德华力实现,吸附能较小;而当吸附能小于-0.25eV时,为化学吸附,此时存在化学键的形成,吸附能较大;
2)Hirshfeld电荷:Hirshfeld电荷的绝对值越大,电子转移能力越大,脱汞能力越好。
进一步的,所述步骤四中,所述定量关系模型的输入为催化剂结构模型的特征变量;所述定量关系模型的输出为预测的脱汞性能指标。
进一步的,所述步骤五中,脱汞性能指标为单质汞吸附能时,R
其中,Y
其中,Y
进一步的,所述步骤六的具体操作如下:
步骤1)、对步骤二中建立的源域数据集,进行分析,收集对单质汞有高效脱除能力的催化剂结构模型;
步骤2)、选取成分或性能与步骤1)中选取出的催化剂结构模型接近的多个材料数据,构建目标域数据集;
所述目标域数据集中包括选取材料的原子半径、元素周期性、价电子数目、第一电离能和Pauling电负性特征,以及对应的基础构型、活性点位信息、掺杂元素、掺杂位点。
进一步的,所述步骤七的具体操作如下:
1)将目标域数据集中材料的原子半径、元素周期性、价电子数目、第一电离能和Pauling电负性等特征,作为输入参数输入至步骤五中确定的定量关系模型中,输出预测的脱汞性能指标;
2)根据预测的脱汞性能指标,筛选出具有最佳脱汞性能指标的材料,即为优化设计的脱汞催化剂;
3)根据目标域数据集中材料对应的基础构型、活性点位信息、掺杂金属和非金属元素的数据,获得优化设计的脱汞催化剂的基础构型、活性位点及掺杂元素的脱汞催化剂信息。
进一步的,还包括,步骤八、通过化学合成步骤七中筛选出的脱汞催化剂,并结合材料表征技术分析其脱汞活性和稳定性,将结果与定量关系模型输出的预测结果进行比较,以此为基础对定量关系模型的模型参数进行迭代优化,从而指导催化剂的进一步改进。
有益效果:
相比现有催化剂研发工艺,本发明基于大量数据构建的机器学习模型和密度泛函理论计算辅助设计了高效脱汞性能的催化剂,极大的节省了材料合成开发过程中人力、物力、时间的消耗,必将会促进催化剂合成领域的蓬勃发展。
附图说明
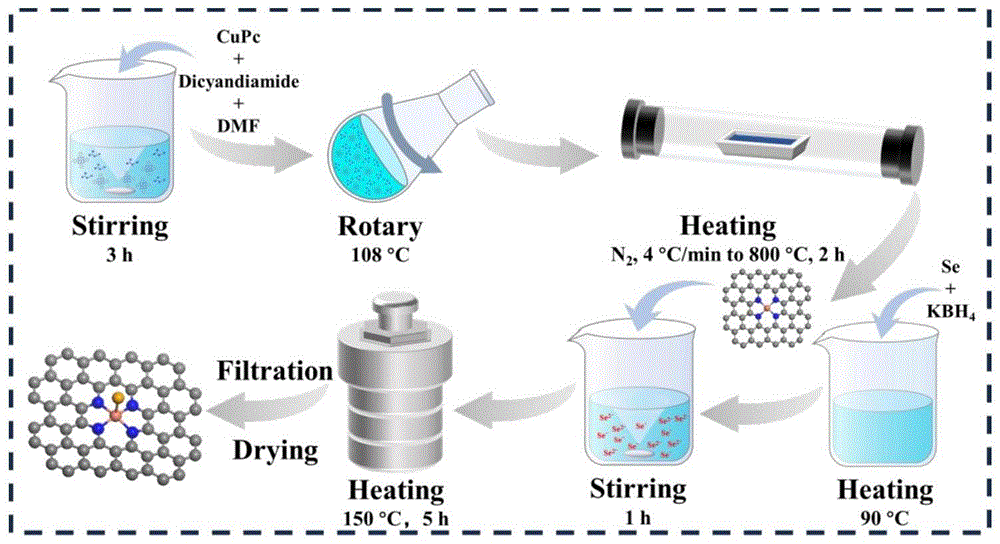
图1是实施例1的制备工艺流程图;
图2是不同Se掺杂点位的分子模型图;
图3是不同活性位点吸附Hg的分子模型图;
图4是Cu-NC、Cu-NSeC、Cu-NC-Se
图5是不同样品的瞬时脱汞效率曲线图。
具体实施方式
下面通过附图对本发明技术方案进行详细说明,但是本发明的保护范围不局限于所述实施例。
本发明公开了一种高效脱汞催化剂设计优化方法,包括以下步骤:
步骤一、利用量子化学软件构建催化剂系列基础及改性模型;
所述量子化学软件,如Materials Studio、Vasp、Gaussian等;所述催化剂系列基础及改性模型用于计算催化剂的体系能量、吸附单质汞后催化剂的体系能量、单质汞的体系能量以及Hirshfeld电荷;
步骤二、通过步骤一构建的催化剂系列基础及改性模型和密度泛函理论计算单质汞吸附能,并构建脱汞催化剂的源域数据集;
所述步骤二的具体操作步骤如下:
步骤2.1)、以原子半径、元素周期性、价电子数目、第一电离能和Pauling电负性等特征为依据,选取具有脱汞性能的催化剂,并收集这些催化剂的基础构型、活性点位信息、掺杂元素和掺杂位型,建立脱汞系列的催化剂结构模型;
所述催化剂结构模型中,包括催化剂的基础构型、活性点位信息、掺杂元素、掺杂位型信息;
步骤2.2)、将步骤2.1)建立的脱汞系列的催化剂结构模型代入通过S1建立的催化剂系列基础及改性模型中,获取催化剂结构模型的体系能量、吸附单质汞后催化剂的体系能量、单质汞的体系能量以及Hirshfeld电荷;
步骤2.3)、将步骤2.2)中得到的催化剂结构模型的体系能量、吸附单质汞后催化剂的体系能量、单质汞的体系能量,采用密度泛函理论(DFT)计算得到催化剂结构模型的单质汞吸附能;
单质汞吸附能是指“单质汞在催化剂表面的吸附能(E
其中,
步骤2.4)、建立脱汞系列催化剂结构模型的源域数据集;
所述源域数据集包括,催化剂结构模型信息,催化剂结构模型对应的特征变量、单质汞吸附能和Hirshfeld电荷;
所述催化剂结构模型信息包括催化剂的基础构型、活性点位信息、掺杂元素、掺杂位型信息;所述特征变量是指原子半径、元素周期性、价电子数目、第一电离能和Pauling电负性等特征。
步骤三、对步骤二中构建的源域数据集进行预处理和规范化,通过权重分析确定对催化剂结构模型的单质汞吸附能影响显著的特征变量,形成最优特征集;
所述预处理和规范化是指对源域数据集,进行分类和初筛,删除无差异的特征变量和删除异常值。
步骤四、采用机器学习方法,建立最优特征集与脱汞性能指标间的定量关系模型;
所述步骤四中,采用的机器学习算法包括但不限于:随机森林回归、K近邻法、随机树法、随机森林法、随机委员会法、缩减误差修减树法、梯度下降回归树法、极端随机森林回归法和XGBRegressor法;
所述脱汞性能指标包括催化剂的单质汞吸附能和Hirshfeld电荷;
1)单质汞吸附能:一般来说,吸附能为负值,其绝对值越大,则吸附越可靠。当吸附能在-0.25eV~0eV时,为物理吸附,主要通过范德华力实现,吸附能较小。而当吸附能小于-0.25eV时,为化学吸附,此时存在化学键的形成,吸附能较大。
2)Hirshfeld电荷:一般而言,Hirshfeld电荷的绝对值越大,电子转移能力越大,脱汞能力越好。
所述定量关系模型的输入为催化剂结构模型的特征变量,所述特征变量包括原子半径、元素周期性、价电子数目、第一电离能和Pauling电负性等特征。所述定量关系模型的输出为预测的脱汞性能指标。
步骤五、使用R
所述步骤五的步骤具体包括:将源域数据集中催化剂结构模型的特征变量输入至步骤四中建立的定量关系模型中,获得催化剂结构模型预测的脱汞性能指标,比较预测的脱汞性能指标与对应催化剂结构模型的实际脱汞性能指标的相对误差;
1)若误差未超出允许范围,则定量关系模型的模型参数无需调整,即该定量关系模型为具有最佳预测结果的定量关系模型;
2)若误差超出允许范围,迁移催化剂实际的脱汞性能指标至定量关系模型中,获得调整后的模型参数,进而确定具有最佳预测结果的定量关系模型。
3)所述步骤六中,脱汞性能指标为单质汞吸附能时,R
其中,Y
其中,Y
脱汞性能指标为Hirshfeld电荷时,R
步骤六、选取多个材料构建具有脱汞性能的催化剂目标域数据集;
所述步骤六的具体操作步骤如下:
步骤1)、对步骤二中建立的源域数据集,进行分析,收集对单质汞有高效脱除能力的催化剂结构模型;
在判断单质汞是否高效脱除能力时,主要参考单质汞吸附能。吸附能用来评价脱汞能力的优异性,当单质汞吸附能达到化学吸附的标准后,该催化剂就认定为具有高效脱除能力。
步骤2)、选取成分或性能与步骤1)中选取出的催化剂结构模型接近,数据量充足的材料数据,构建目标域数据集。
所述目标域数据集中包括选取材料的原子半径、元素周期性、价电子数目、第一电离能和Pauling电负性等特征,以及对应的基础构型、活性点位信息、掺杂元素、掺杂位点。
步骤七、使用步骤五中确定的定量关系模型,预测步骤二中目标域数据集中的材料的脱汞性能指标,根据脱汞性能指标,筛选出具有最佳脱汞性能指标的材料,即为优化设计的脱汞催化剂;
所述步骤七的具体操作为:
1)将目标域数据集中材料的原子半径、元素周期性、价电子数目、第一电离能和Pauling电负性等特征,作为输入参数输入至步骤五中确定的定量关系模型中,输出预测的脱汞性能指标;
2)根据预测的脱汞性能指标,筛选出具有最佳脱汞性能指标的材料,即为优化设计的脱汞催化剂;
3)根据目标域数据集中材料对应的基础构型、活性点位信息、掺杂金属和非金属元素的数据,获得优化设计的脱汞催化剂的基础构型、活性位点及掺杂元素的脱汞催化剂信息。
步骤八、通过化学合成步骤七中筛选出的脱汞催化剂,并结合材料表征技术分析其脱汞活性和稳定性,将结果与定量关系模型输出的预测结果进行比较,以此为基础对定量关系模型的模型参数进行迭代优化,从而指导催化剂的进一步改进。
根据本发明方法步骤一至步骤七,筛选出的最佳脱汞性能指标的材料Cu-NC-Se,其基础构型为中心原子为Cu的氮化碳材料,表面掺杂的非金属为硒元素,掺杂位型为Cu位,将该材料的原子半径、元素周期性、价电子数目、第一电离能和Pauling电负性输入作为摄像参数输入至最佳预测结果的定量关系模型中,以评估是否具有良好的脱汞能力,结果显示,该材料存在一个良好的脱汞性能。
下面通过将得到的催化剂进行化学合成验证筛选出的催化剂性能,具体通过如下是实施例进行说明。
实施例1
Cu-NC-Se的制备参数如图1所示,具体步骤如下:
称取一定量的CuPc和双氰胺(Cu和N的化学计量比为1:4),加入50ml的DMF中,常温下连续搅拌3h。之后在108℃旋转蒸发,收集粉末,在N
通过二次浸渍硒化法制备Cu-NC-Se催化剂:称取一定量的Se粉、Cu-NC和NaBH
本发明中用到的化学试剂有酞菁铜(β型,90.0%)、双氰胺(98%)、DMF(AR,99.5%)、硼氢化钾(94%)、硒粉(99.9%)、二苯基二硒醚(96%),除二甲基甲酰胺和硼氢化钾来自国药集团,其他试剂均来自阿拉丁。所有试剂都直接使用,没有经过其他处理。
图2是不同Se掺杂点位的分子模型图,图3是不同活性位点吸附Hg的分子模型图;
图2中的(a)为不掺杂非金属Se元素的Cu-NC分子模型图,(b)为Se元素掺杂在Cu点位上的分子模型图,(c)为Se元素掺杂在N点位上的分子模型图,(d)为Se元素掺杂在C点位上的分子模型图。
图3中的(a)为Hg吸附在Cu点位上的吸附分子模型图,(b)为Hg吸附在Se点位上的吸附分子模型图,(c)为Hg吸附在N点位上的吸附分子模型图,(d)为Hg吸附在C1点位上的吸附分子模型图,(e)为Hg吸附在C2点位上的吸附分子模型图,(f)为Hg吸附在C3点位上的吸附分子模型图,(g)为Hg吸附在C4点位上的吸附分子模型图,(h)为Hg吸附在C5点位上的吸附分子模型图。C1-C5为五个典型的C点位上的吸附情况。
经过密度泛函理论计算,不同掺杂位型的催化剂结构模型的E
实施例2(对照实验)
作为对照实验,称取一定量的Se粉、Cu-NC和NaBH
作为对照实验,称取一定量的Se粉、Cu-NC和NaBH
作为对照实验,称取一定量的Se粉、Cu-NC和NaBH
作为对照实验,称取一定量的CuPc、双氰胺和二苯二硒(Cu、N、Se的化学计量比为1:4:1),加入50ml的DMF中,常温下连续搅拌3h;之后在108℃旋转蒸发,收集粉末,在N
图4展示了Cu-NC、Cu-NSeC、Cu-NC-Se
从图4中的(a)中可以看出,Cu-NC是由超薄的不规则薄片堆叠而成,总体上呈现出蓬松多孔的结构。这是因为CuPc在高温下会受到热拉伸,与双氰胺热缩聚产生的气泡结合形成超薄薄膜。图4中的(b)、(c)和(d)都显示了大量的亮点,这是原始Cu-NC中所没有的,可以推测这些亮点是掺杂的硒。在图4中的(b)中,Cu-NSeC表面观察到许多无序分布的硒亮点,并有轻微的聚集现象。与图4中的(b)相比,图4中的(c)和(d)表面的亮点分布更为均匀,尤其是图4(d)。由于Se的添加量不足,Cu-NC-Se
图5中的(a)是在90℃的反应温度下不同制备工艺制备的样品的瞬时脱汞效率曲线图,图5中的(b)是在90℃的反应温度下不同掺杂比的瞬时脱汞效率曲线图。图5给出了不同样品的瞬时Hg
由此可见,本发明基于机器学习和密度泛函理论计算得到的脱汞催化剂确实有良好的脱汞性能。
以上所述描述了本发明的优选实施方式,应理解的是以上所述仅为本发明的具体实施例,并不用于限制本发明,凡是在本发明的原则范围内所做的任何修改和改进等,均应包含在本发明的保护范围内。
- 一种同时脱硝脱汞脱二噁英稀土基催化剂及制备方法和应用
- 一种燃煤电厂用脱硝脱汞催化剂及其制备方法
- 一种用于脱汞的钴改性多孔生物炭催化剂的制备方法
- 一种烟气脱硫脱汞催化剂及其制备方法和应用
- 一种高含汞凝析油脱汞方法
- 一种低温高效抗硫抗水协同脱硝脱汞催化剂的制备方法
- 一种用于烟气高效脱汞的催化剂及其制备方法