固体制剂
文献发布时间:2024-01-17 01:27:33
技术领域
本发明涉及药物制剂,该药物制剂包含:活性药物成分;和甲基丙烯酸共聚物、羟丙基甲基纤维素或它们的组合,以及包含所述药物制剂的固体剂型。本发明还涉及制备此类药物制剂的方法,以及此类药物制剂用于预防或治疗疾病、综合征、病况或病症的用途。
背景技术
由蚊子或蜱虫传播的黄病毒在人类中引起威胁生命的感染,诸如脑炎和出血热。已知黄病毒登革热(登革热病毒)的四种不同但密切相关的血清型。WO 2016/180696公开了对登革热病毒的所有四(4)种血清型显示出高效活性的活性药剂。
许多活性药物成分(API)具有诸如疏水性和不稳定性的性质,导致给提供合适的药物制剂带来了挑战。
存在对活性药物成分(诸如WO 2016/180696中描述的登革热病毒复制抑制剂)的改进药物制剂的需要。
发明内容
本发明涉及一种药物制剂,该药物制剂包含:
a)活性药物成分(API);和
b1)甲基丙烯酸共聚物,或
b2)纤维素衍生物,诸如甲基纤维素(MC)、乙基纤维素(EC)、羟乙基纤维素(HEC)、羟丙基纤维素(HPC)、羧甲基纤维素(CMC)、羧甲基纤维素钠(NaCMC)或羟丙基甲基纤维素(HPMC);
其中API是登革热病毒复制抑制剂。
本发明的实施方案包括如本文所述的药物制剂,其中API是登革热病毒复制抑制剂。
本发明还提供了包含如本文所述的药物制剂的固体剂型。
在其中API是登革热病毒复制抑制剂的实施方案中,本发明提供了用于使用本文所述的药物制剂和固体剂型来治疗或预防受试者的疾病、综合征、病况或病症的方法,该受试者包括其疾病、综合征、病况或病症是登革热病毒感染的哺乳动物和/或人。
在某些实施方案中,本发明涉及用于在感染登革热病毒或有感染登革热病毒风险的哺乳动物和/或人中抑制登革热病毒复制的方法。
本发明还涉及此类药物制剂和/或固体剂型在制备药物中的用途,其中该药物被制备用于治疗或预防登革热病毒感染。
本发明还涉及将此类药物制剂和/或固体剂型用作药物。在另一个实施方案中,本发明涉及用于治疗或预防登革热病毒感染的本文所述的药物制剂和/或固体剂型。
在某些实施方案中,本发明涉及用于在哺乳动物和/或人中抑制登革热病毒复制的本文所述的药物制剂和/或固体剂型。
本发明还提供了一种用于制备如本文所述的药物制剂的方法,该方法包括以下步骤:
a)将API溶解于溶剂中以形成溶液;
b)将甲基丙烯酸共聚物或羟丙基甲基纤维素或它们的组合与步骤a)中形成的溶液混合,从而获得混合物;
c)将混合物喷雾干燥以获得固体分散体;
d)任选地将该固体分散体与至少一种药学上可接受的赋形剂共混;
以提供如本文所述的药物制剂。
本发明还提供了一种用于制备本文所述的固体剂型的方法,该方法包括以下步骤:
a)将API溶解于溶剂中以形成溶液;
b)将甲基丙烯酸共聚物或羟丙基甲基纤维素或它们的组合与步骤a)中形成的溶液混合,从而获得混合物;
c)将混合物喷雾干燥以获得固体分散体;
d)任选地将该固体分散体与至少一种药学上可接受的赋形剂共混;
e)将共混物压制成片剂;
以提供如本文所述的固体剂型。
在某些实施方案中,本发明提供了一种用于制备如本文所述的药物制剂或固体剂型的方法,该方法包括将活性药物成分与甲基丙烯酸共聚物或与纤维素衍生物(例如,羟丙基甲基纤维素)组合以形成药物制剂或固体剂型。
附图说明
当结合附图阅读时,进一步理解发明内容以及下文的具体实施方式。出于示出本发明的目的,附图中所示的是本发明的示例性实施方案;然而,本发明并不限于附图的具体公开内容。
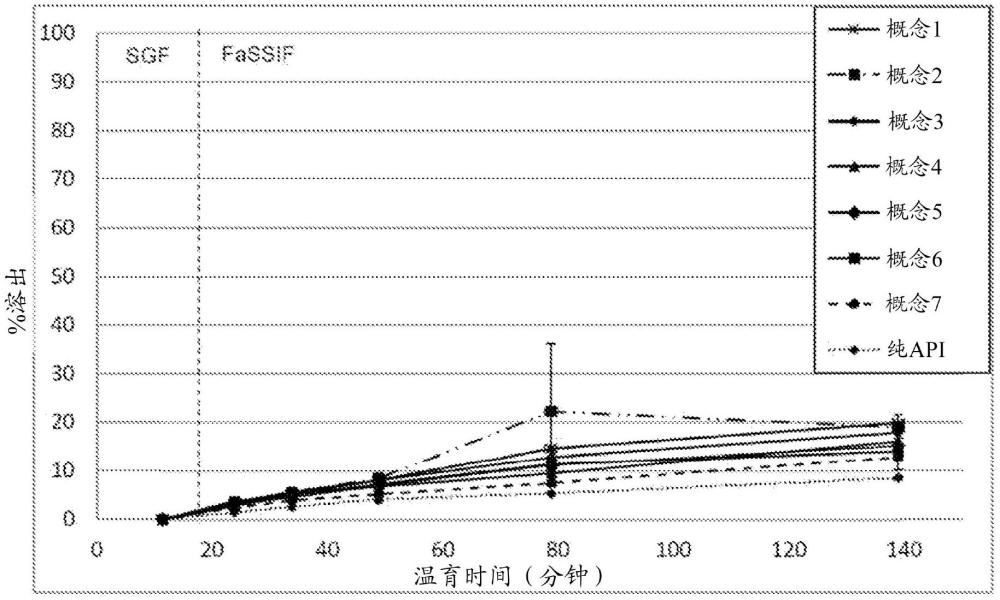
图1是实施例1中制备的概念1至7的固体分散体的溶出的API%对温育时间的曲线图。该曲线图示出了在模拟胃液(SGF)和空腹状态模拟肠液(FaSSIF)中的2相溶出曲线。
图2是实施例1中制备的概念8至12的固体分散体的溶出的API%对温育时间的曲线图。该曲线图示出了在模拟胃液(SGF)和空腹状态模拟肠液(FaSSIF)中的2相溶出曲线。
图3A和图3B是实施例5B中制备的固体分散体的溶出的API%对温育时间(图3A)和溶出的API的重量(以mg计)对温育时间(图3B)的曲线图。该曲线图示出了在模拟胃液(SGF-垂直虚线左侧)和空腹状态模拟肠液(FaSSIF-垂直虚线右侧)中的2相溶出曲线。
图4是实施例5B中制备的固体分散体的溶出的API%对温育时间的曲线图。该曲线图示出了在模拟胃液(SGF-垂直虚线左侧)和进食状态模拟肠液(FeSSIF-垂直虚线右侧)中的2相溶出曲线。
具体实施方式
通过参考以下描述,包括以下术语表和结论性实施例,可更完全地理解本公开。应当理解,所公开的药物制剂和方法的某些特征为清楚起见在单独方面的上下文中进行描述,但也可以组合形式在单个方面中进行提供。相反地,所公开的药物制剂和方法的各种特征为简明起见在单个方面的上下文中进行描述,也可分开地或以任何子组合形式进行提供。
本文给出的一些数量表述没有用术语“约”修饰。应当理解,无论是否明确地使用了术语“约”,本文所给出的每个量都意在指代实际的给定值,并且还意在指代由本领域的普通技术人员可合理推测出的这些给定值的近似值,包括这些给定值的由实验和/或测量条件所产生的近似值。如本文所用的术语“约”或“大约”在涉及数值或范围时允许该值或范围的一定程度的波动,例如,在所述值或所述范围界限的10%内(即,±10%)、5%内(即,±5%)或2.5%内(即,±2.5%)。
在本说明书的描述和权利要求书通篇中,字词“包含(comprise)”和“含有(contain)”以及这些字词的变型例如“包含(comprising)”和“包含(comprises)”意指“包括但不限于”,并且不旨在(并且不)排除其他成分。
通过端点表述的数值范围包括所有整数,并且在适当的情况下,包括该范围内包含的分数(例如,当涉及例如多个要素时,1至5可包括1、2、3、4,并且当涉及例如测量结果时,还可包括1.5、2、2.75和3.80)。端点的表述还包括端点值本身(例如,1.0至5.0包括1.0和5.0两者)。本文所表述的任何数值范围旨在包括其中包含的所有子范围。
本说明书中引用的所有参考文献据此全文以引用方式并入。具体地,本文特别提及的所有参考文献的教导内容以引用方式并入。
在整个说明书中引用的“一个实施方案”或“实施方案”是指结合实施方案描述的特定特征、结构或特性包括在本发明的至少一个实施方案中。因此,在整个说明书中各处出现的短语“在一个实施方案中”或“在实施方案中”不一定都是指同一实施方案,但是可以指同一实施方案。此外,在一个或多个实施方案中,特定特征、结构或特性可以任何合适的方式组合,如本领域技术人员根据本公开将显而易见的那样。此外,虽然本文所述的一些实施方案包括一些特征,但不包括其他实施方案中所包括的其他特征,但不同实施方案的特征的组合意味着在本发明的范围内,并且形成不同的实施方案,如本领域技术人员将理解的那样。
对于在医学上的使用,API的共晶体或盐诸如本文所公开的式(I)化合物的盐是指无毒的“药学上可接受的盐”。“药学上可接受的”可指经联邦或州政府的监管机构或美国以外的国家的相应机构批准或可被其批准或在美国药典或其他公认的药典中被列为用于动物,尤其是人类。
然而,其他盐也可用于制备API诸如式(I)的化合物或其药学上可接受的盐形式。API诸如式(I)的化合物的合适的药学上可接受的盐包括酸加成盐,该酸加成盐可例如通过将化合物的溶液与药学上可接受的酸(诸如盐酸、硫酸、富马酸、马来酸、琥珀酸、乙酸、苯甲酸、柠檬酸、酒石酸、碳酸或磷酸)的溶液混合来形成。此外,如果API诸如式(I)的化合物带有酸性部分,则其合适的药学上可接受的盐可包括碱金属盐,诸如钠盐或钾盐;碱土金属盐,诸如钙盐或镁盐;以及与合适的有机配体形成的盐,诸如季铵盐。因此,代表性药学上可接受的盐包括乙酸盐、苯磺酸盐、苯甲酸盐、碳酸氢盐、硫酸氢盐、酒石酸氢盐、硼酸盐、溴化物、依地酸钙、樟脑磺酸盐、碳酸盐、氯化物、克拉维酸盐、柠檬酸盐、二盐酸盐、依地酸盐、乙二磺酸盐、丙酸酯十二烷基硫酸盐、乙磺酸盐、延胡索酸盐、葡庚糖酸盐、葡糖酸盐、谷氨酸盐、对α-羟乙酰氨基苯砷酸盐、己基间苯二酚盐、海巴明、氢溴酸盐、盐酸盐、羟萘酸盐、碘化物、异硫代硫酸盐、乳酸盐、乳糖醛酸盐、月桂酸盐、苹果酸盐、马来酸盐、扁桃酸盐、甲磺酸盐、甲基溴化物、甲基硝酸盐、甲基硫酸盐、粘液酸盐、萘磺酸盐、硝酸盐、N-甲基葡糖胺铵盐、油酸盐、双羟萘酸盐(扑酸盐)、棕榈酸盐、泛酸盐、磷酸盐/二磷酸盐、聚半乳糖醛酸盐、水杨酸盐、硬脂酸盐、硫酸盐、碱式乙酸盐、琥珀酸盐、鞣酸盐、酒石酸盐、茶氯酸盐、甲苯磺酸盐、三乙基碘化物和戊酸盐。
可用于制备药学上可接受的盐的代表性酸和碱包括:酸,包括乙酸、2,2-二氯乙酸、乙酰化的氨基酸、己二酸、藻酸、抗坏血酸、L-天冬氨酸、苯磺酸、苯甲酸、4-乙酰氨基苯甲酸、(+)-樟脑酸、樟脑磺酸、(+)-(1S)-樟脑-10-磺酸、癸酸、己酸、辛酸、肉桂酸、柠檬酸、环拉酸、十二烷基硫酸、乙烷-1,2-二磺酸、乙磺酸、2-羟基-乙磺酸、甲酸、富马酸、半乳糖二酸、龙胆酸、葡庚糖酸、D-葡糖酸、D-葡萄糖醛酸、L-谷氨酸、α-氧代-戊二酸、乙醇酸、马尿酸、氢溴酸、盐酸、(+)-L-乳酸、(±)-DL-乳酸、乳糖酸、马来酸、(-)-L-苹果酸、丙二酸、(±)-DL-扁桃酸、甲磺酸、萘-2-磺酸、萘-1,5-二磺酸、1-羟基-2-萘甲酸、烟酸、硝酸、油酸、乳清酸、草酸、棕榈酸、扑酸、磷酸、L-焦谷氨酸、水杨酸、4-氨基-水杨酸、癸二酸、硬脂酸、琥珀酸、硫酸、鞣酸、(+)-L-酒石酸、硫氰酸、对甲苯磺酸和十一碳烯酸;以及碱,包括氨、L-精氨酸、苯乙苄胺、苄星、氢氧化钙、胆碱、丹醇、二乙醇胺、二乙胺、2-(二乙基氨基)-乙醇、乙醇胺、乙二胺、N-甲基-葡糖胺、海巴明、1H-咪唑、L-赖氨酸、氢氧化镁、4-(2-羟乙基)-吗啉、哌嗪、氢氧化钾、1-(2-羟乙基)-吡咯烷、氢氧化钠、三乙醇胺、氨基丁三醇和氢氧化锌。
如果API诸如式(I)的化合物具有至少一个手性中心,则它们可以对映体形式相应地存在。如果化合物具有两个或更多个手性中心,则它们还可以非对映体形式存在。应当理解,所有的此类异构体及其混合物涵盖在本发明的范围内。此外,化合物中的一些化合物可作为多晶型物存在,并因此也旨在包括在本发明内。此外,化合物中的一些化合物可与水形成溶剂化物(即,水合物)或与有机溶剂形成溶剂化物,并且此类溶剂化物也旨在涵盖在本发明的范围内。技术人员将理解,本文所使用的术语化合物旨在包括溶剂化的式(I)的化合物。
如果用于制备API诸如式(I)的化合物的方法产生立体异构体的混合物,则这些异构体可通过常规技术诸如制备型色谱法来分离。该化合物可以外消旋形式制备,或者可通过对映体特异性合成或通过拆分来制备单独的对映体。例如,可通过标准技术,如通过与光学活性酸(诸如(-)-二对甲苯酰-d-酒石酸和/或(+)-二对甲苯酰-l-酒石酸)形成盐来形成非对映体对,然后分级结晶并再生游离碱,来将化合物拆分成它们的组分对映体。该化合物还可通过形成非对映体酯或酰胺,随后进行色谱分离并去除手性助剂来拆分。另选地,可使用手性固定相或手性柱拆分该化合物。
在本发明的药物制剂的一个实施方案中,API是式(I)的化合物,其中所述化合物是包含(+)-对映体、由(+)-对映体组成以及/或者基本上由(+)-对映体组成的化合物,其中所述化合物基本上不含(-)-异构体。在本发明的上下文中,基本上不含意指少于约25重量%、优选地少于约10重量%、更优选地少于约5重量%、甚至更优选地少于约2重量%并且甚至更优选地少于约1重量%的(-)-异构体,其计算方式如下:
在本发明的药物制剂的另一个实施方案中,API是式(I)的化合物,其中所述化合物是包含(-)-对映体、由(-)-对映体组成以及基本上由(-)-对映体组成的化合物,其中所述化合物基本上不含(+)-异构体。在本发明的上下文中,基本上不含意指少于约25重量%、优选地少于约10重量%、更优选地少于约5重量%、甚至更优选地少于约2重量%并且甚至更优选地少于约1重量%的(+)-异构体,其计算方式如下:
在用于制备本发明多个实施方案的化合物的任何工艺过程中,可能有必要和/或期望保护所关注的任何分子上的敏感性或反应性基团。这可通过常规保护基团来实现,诸如在《有机化学中的保护性基团(Protective Groups in Organic Chemistry)》,第二版,J.F.W.McOmie,美国殷实出版社(Plenum Press),1973;T.W.Greene和P.G.M.Wuts,《有机合成中的保护性基团(Protective Groups in Organic Synthesis)》,约翰·威利父子出版公司(John Wiley&Sons),1991;以及T.W.Greene和P.G.M.Wuts,《有机合成中的保护性基团(Wuts,Protective Groups in Organic Synthesis)》,第三版,约翰·威利父子出版公司,1999中所描述的那些保护性基团。可使用本领域已知的方法在方便的后续阶段去除保护基团。
除非另有说明,否则术语“重量%”和“重量百分比”可互换使用,指成分(例如,赋形剂或活性药物成分)按药物制剂重量计的浓度。
术语“室温”(RT)是指约15℃至约30℃,特别是约20℃至约30℃的温度。优选地,室温是约25℃的温度。
平均分子量可例如是指数均分子量或重均分子量。平均分子量可例如使用凝胶渗透色谱法或质谱法来测量。
术语“受试者”是指已经是治疗、观察或实验的对象的动物,优选指哺乳动物,最优选指人。
术语“活性化合物”、“活性成分”和“活性药物成分”在本文中可互换使用。
术语“治疗有效量”是指活性化合物或药剂的量,该量引起研究者、兽医、医生或其他临床医生所追求的组织系统、动物或人的生物学或医学响应,这包括降低或抑制酶或蛋白质活性,或改善症状,减轻病况,减缓或延迟疾病进展,或预防疾病。
在其中API是登革热病毒复制抑制剂的实施方案中,术语“治疗有效量”可指本发明的制剂在施用于受试者时有效地至少部分地减轻、抑制、预防和/或改善所述受试者中由登革热病毒引起的病况或病症或疾病的量。
如本文所用,术语“登革热病毒”是指黄病毒科的单正链RNA病毒;已知黄病毒登革热的四种不同但密切相关的血清型,即所谓的DENV-1、DENV-2、DENV-3和DENV-4。由蚊子或蜱虫传播的黄病毒在人类中引起威胁生命的感染,诸如脑炎和出血热。
如本文所用,术语“登革热病毒复制抑制剂”是指抑制或降低由登革热病毒引起的至少一种病况、综合征、病症和/或疾病的试剂。
除非另外指明,否则如本文所用,术语“影响”或“受影响的”(当涉及受登革热病毒复制的抑制影响的疾病、综合征、病况或病症时)包括所述疾病、综合征、病况或病症的一种或多种症状或临床表现的频率和/或严重性的降低;并且/或者包括防止所述疾病、综合征、病况或病症的一种或多种症状或临床表现的发展或者所述疾病、病况、综合征或病症的发展。
如本文所用,在一个实施方案中,术语任何疾病、病况、综合征或病症的“治疗”是指改善疾病、病况、综合征或病症(即,减慢或阻止或减缓疾病或其临床症状中的至少一种的发展)。在另一个实施方案中,“治疗”是指减轻或改善至少一种身体参数,包括患者可能无法察觉的那些。在另一个实施方案中,“治疗”是指在身体上(例如,可察觉的症状的稳定)、生理上(例如,身体参数的稳定)或者两者兼有地调节疾病、病况、综合征或病症。在又一个实施方案中,“治疗”是指预防或延缓疾病、病况、综合征或病症的发作或发展或进展。
如本文所用,术语“预防”是指降低获得或发展给定病况、疾病或疾病症状的风险,或者降低或抑制未患病但有患病风险的受试者的所述病况、疾病或疾病症状的复发。例如,所述受试者处于或曾去过高风险区域,或者曾经或可能靠近患有疾病的人。
如本文所用,“赋形剂”是药物制剂中的非活性成分。赋形剂的示例包括填充剂、润湿剂(例如,表面活性剂)、粘结剂、助流剂、润滑剂、崩解剂等。
如本文所用,“崩解剂”是使药物制剂水合并帮助片剂分散的赋形剂。崩解剂的示例包括交联羧甲基纤维素钠、交聚维酮(即,交联的聚乙烯基N-吡咯烷酮)、羧甲淀粉钠或它们的任何组合。
如本文所用,“填充剂”或“填料”是增加药物制剂的体积的赋形剂。填充剂的示例包括乳糖、山梨糖醇、纤维素、磷酸钙、淀粉、糖(例如,甘露糖醇、蔗糖等)或它们的任何组合。
如本文所用,“润湿剂”或“表面活性剂”是赋予药物制剂增强的溶解度和/或可润湿性的赋形剂。润湿剂的示例包括月桂基硫酸钠(SLS)、硬脂酰富马酸钠(SSF)、聚氧乙烯20单油酸脱水山梨糖醇酯(即聚山梨酸酯20)(例如,Tween
如本文所用,“粘结剂”是赋予药物制剂增强的内聚力或拉伸强度(例如,硬度)的赋形剂。粘结剂的示例包括磷酸氢钙、蔗糖、玉米(玉蜀黍)淀粉、微晶纤维素和改性纤维素(例如,羟甲基纤维素)。
如本文所用,“助流剂”是赋予药物制剂增强的流动特性的赋形剂。助流剂的示例包括胶态二氧化硅和/或滑石。
如本文所用,“着色剂”是赋予药物制剂期望颜色的赋形剂。着色剂的示例包括可商购获得的颜料,诸如FD&C蓝#1铝色淀、FD&C蓝#2、其他FD&C蓝色料、二氧化钛、氧化铁和/或它们的组合。其他着色剂包括可商购获得的颜料,诸如FD&C绿#3。
如本文所用,“润滑剂”是添加到压制成片剂的药物制剂中的赋形剂。润滑剂有助于将颗粒压制成片剂并将药物制剂的片剂从模压机中顶出。润滑剂的示例包括硬脂酸镁、硬脂酸(硬脂精)、氢化油、硬脂酰富马酸钠或它们的任何组合。
本发明的药物制剂、用途和方法的优选陈述(特征)和实施方案在下文中阐述。除非有明确相反的说明,否则如此定义的本发明的每个陈述和实施方案可以与任何其他陈述和/或实施方案组合。具体地,任何被指示为优选或有利的特征可与任何其他被指示为优选或有利的一个或多个特征或陈述组合。至此,本发明特别地被以下编号的方面和实施方案中的一者或多者的任何一个或任何组合以及任何其他陈述和/或实施方案所捕集。
1.一种药物制剂,所述药物制剂包含:
a)活性药物成分(API);和
b1)甲基丙烯酸共聚物,或
b2)纤维素衍生物,诸如甲基纤维素(MC)、乙基纤维素(EC)、羟乙基纤维素(HEC)、羟丙基纤维素(HPC)、羧甲基纤维素(CMC)、羧甲基纤维素钠(NaCMC)或羟丙基甲基纤维素(HPMC)或它们的组合。优选地,所述纤维素衍生物是HPMC。
其中所述API是登革热病毒复制抑制剂。
2.根据陈述1所述的药物制剂,其中所述药物制剂是固体制剂。
3.根据陈述1或2中任一项所述的药物制剂,其中所述API和所述甲基丙烯酸共聚物以下列比率存在于所述制剂中:4:1w/w至1:9w/w,优选地3.9:1w/w至1:8w/w;优选地3.8:1w/w至1:7w/w;3.7:1w/w至1:6w/w;优选地3.6:1w/w至1:5w/w,优选地3.5:1w/w至1:4.5w/w;优选地3:1w/w至1:4w/w;优选地2.5:1w/w至1:3.5w/w;优选地2:1w/w至1:3w/w;优选地2.5:1w/w至1:2.5w/w,优选地2:1w/w至1:2w/w;优选地1.5:1w/w至1:1.5w/w或包含在本文提及的任何两个比率内的比率,或本文提及的任何两个比率内的比率范围或子范围。
4.根据陈述1或2中任一项所述的药物制剂,其中所述API和所述纤维素衍生物(例如,羟丙基甲基纤维素)以下列比率存在于所述制剂中:4:1w/w至1:5w/w,优选地3.5:1w/w至1:4.5w/w;优选地3:1w/w至1:4w/w;优选地2.5:1w/w至1:3.5w/w;优选地2:1w/w至1:3w/w;优选地2.5:1w/w至1:2.5w/w,优选地2:1w/w至1:2w/w;优选地1.5:1w/w至1:1.5w/w或包含在本文提及的任何两个比率内的比率,或本文提及的任何两个比率内的比率范围或子范围。
5.根据陈述1至4中任一项所述的药物制剂,其中所述甲基丙烯酸共聚物是丙烯酸和/或甲基丙烯酸/酯的共聚物,诸如以商品名
6.根据陈述1至5中任一项所述的药物制剂,其中所述甲基丙烯酸共聚物选自包括以下项的组:甲基丙烯酸和甲基丙烯酸甲酯的共聚物;甲基丙烯酸和丙烯酸乙酯的共聚物;以及它们的任何混合物。
7.根据陈述1至6中任一项所述的药物制剂,其中所述甲基丙烯酸共聚物是甲基丙烯酸和甲基丙烯酸甲酯的共聚物。
8.根据陈述7所述的药物制剂,其中所述共聚物中甲基丙烯酸与甲基丙烯酸甲酯的摩尔比为0.5:2至0.6:1.5,优选地0.8:1.3至1.2:1,更优选地约1:1。
9.根据陈述1至8中任一项所述的药物制剂,其中所述甲基丙烯酸共聚物是聚(甲基丙烯酸-共-甲基丙烯酸甲酯)1:1(
10.根据陈述1至9中任一项所述的药物制剂,其中纤维素衍生物是羟丙基甲基纤维素,诸如HPMC E5、HPMC E6、HPMC E15、HPMC E50、HPMC K3、HPMC K4M、HPMC-AS以及它们的混合物,所述羟丙基甲基纤维素在25℃下在2重量%的H
11.根据陈述1至10中任一项所述的药物制剂,其中所述纤维素衍生物是羟丙基甲基纤维素,诸如HPMC E5、HPMC E6、HPMC E15、HPMC E50以及它们的混合物,所述羟丙基甲基纤维素在25℃下在2重量%的H
12.根据陈述1至11中任一项所述的药物制剂,其中所述纤维素衍生物在25℃下在2重量%的H
13.根据陈述1至12中任一项所述的药物制剂,其中相对于所述制剂的总重量,所述制剂包含至多50重量%、优选地至多40重量%、更优选地至多30重量%、最优选地至多25重量%、甚至最优选地20重量%的所述API。
14.根据陈述1至13中任一项所述的药物制剂,其中相对于所述制剂的总重量,所述制剂包含0.1重量%至50重量%、优选地1重量%至40重量%、更优选地2.5重量%至30重量%、最优选地5重量%至25重量%的所述API。
15.根据陈述1至14中任一项所述的药物制剂,其中所述制剂还包含一种或多种药学上可接受的赋形剂,所述药学上可接受的赋形剂选自崩解剂、粘结剂、填充剂、润滑剂、稳定剂、润湿剂、助流剂、渗透剂、着色剂、增塑剂和包衣。
16.根据陈述1至15中任一项所述的药物制剂,其中所述制剂还包含一种或多种赋形剂;其中相对于所述制剂的总重量,所述制剂包含至多80重量%、优选地至多70重量%、优选地至多60重量%的所述一种或多种赋形剂;并且/或者其中相对于所述制剂的总重量,所述制剂包含至少10重量%、优选地至少20重量%、优选地至少30重量%的所述一种或多种赋形剂。
17.根据陈述1至16中任一项所述的药物制剂,其中所述制剂还包含一种或多种填充剂;其中相对于所述制剂的总重量,所述制剂包含至多95重量%、优选地至多80重量%、优选地至多75重量%的所述填充剂;并且/或者其中相对于所述制剂的总重量,所述制剂包含至少5重量%、优选地至少10重量%、优选地至少15重量%的所述填充剂。
18.根据陈述1至17中任一项所述的药物制剂,其中所述制剂还包含一种或多种崩解剂;其中相对于所述制剂的总重量,所述制剂包含至多30重量%、优选地至多20重量%、优选地至多15重量%、优选地至多10重量%的所述崩解剂;并且/或者其中相对于所述制剂的总重量,所述制剂包含至少1重量%、优选地至少2.5重量%、优选地至少5重量%、优选地至少8重量%的所述崩解剂。
19.根据陈述1至18中任一项所述的药物制剂,其中所述制剂还包含一种或多种粘结剂;其中相对于所述制剂的总重量,所述制剂包含至多50重量%、优选地至多45重量%、优选地至多40重量%、更优选地至多35重量%的所述粘结剂;并且/或者其中相对于所述制剂的总重量,所述制剂包含至少5重量%、优选地至少10重量%、优选地至少15重量%、更优选地至少20重量%、最优选地25重量%的所述粘结剂。
20.根据陈述1至19中任一项所述的药物制剂,其中所述制剂还包含一种或多种润滑剂;其中相对于所述制剂的总重量,所述制剂包含至多5.5重量%、优选地至多3.5重量%、优选地至多2重量%的所述润滑剂;并且/或者其中相对于所述制剂的总重量,所述制剂包含至少0.5重量%、优选地至少1重量%、优选地至少1.5重量%的所述润滑剂。
21.根据陈述1至20中任一项所述的药物制剂,其中所述制剂还包含一种或多种润湿剂;其中相对于所述制剂的总重量,所述制剂包含至多5.5重量%、优选地至多3.5重量%、优选地至多2重量%的所述润湿剂;并且/或者其中相对于所述制剂的总重量,所述制剂包含至少0.5重量%、优选地至少1重量%、优选地至少1.5重量%的所述润湿剂。
22.根据陈述1至21中任一项所述的药物制剂,其中所述制剂还包含一种或多种助流剂;其中相对于所述制剂的总重量,所述制剂包含至多10重量%、优选地至多8重量%、优选地至多5重量%的所述助流剂;并且/或者其中相对于所述制剂的总重量,所述制剂包含至少0.1重量%、优选地至少1重量%、优选地至少2重量%的所述助流剂。
23.根据陈述1至22中任一项所述的药物制剂,其中所述制剂包含形成所述制剂的颗粒内相的多个颗粒和形成所述制剂的颗粒外相的一种或多种赋形剂。
24.根据陈述23所述的药物制剂,其中所述制剂包含按所述药物制剂的重量计至少15重量%;至少20重量%;至少28重量%;至少34重量%的颗粒内相。
25.根据陈述23或24中任一项所述的药物制剂,其中所述制剂包含按所述药物制剂的重量计至多99重量%、至多93重量%、至多85重量%、至多80重量%、至多74重量%;至多73重量%;至多67重量%;至多63重量%;至多60重量%、至多53重量%的颗粒内相。
26.根据陈述23至25中任一项所述的药物制剂,其中所述制剂包含颗粒内相,并且其中所述颗粒内相包含按所述药物制剂的总重量计1重量%至70重量%;2重量%至60重量%;3重量%至55重量%;4重量%至50重量%;5重量%至45重量%;优选地5重量%至40重量%;优选地10重量%至35重量%;优选地15重量%至30重量%的API。
27.根据陈述23至26中任一项所述的药物制剂,其中所述制剂包含颗粒内相,并且其中所述颗粒内相包含按所述药物制剂的总重量计5重量%至60重量%;优选地8重量%至50重量%;优选地15重量%至45重量%的甲基丙烯酸共聚物或羟丙基甲基纤维素或它们的混合物。
28.根据陈述23至27中任一项所述的药物制剂,其中所述制剂包含颗粒内相,并且其中所述颗粒内相包含按所述药物制剂的总重量计5重量%至60重量%;优选地10重量%至60重量%;优选地10重量%至50重量%;优选地15重量%至50重量%的填料。
29.根据陈述23至28中任一项所述的药物制剂,其中所述制剂包含颗粒内相,所述颗粒内相包含按所述药物制剂的总重量计1重量%至10重量%;优选地1重量%至9重量%、优选地1.7重量%至8重量%的崩解剂。
30.根据陈述23至29中任一项所述的药物制剂,其中所述制剂包含颗粒内相,所述颗粒内相包含按所述药物制剂的总重量计0.1重量%至5重量%;优选地0.2重量%至4.7重量%;优选地0.5重量%至4.5重量%的助流剂。
31.根据陈述23至30中任一项所述的药物制剂,其中所述制剂包含颗粒内相,所述颗粒内相包含按所述药物制剂的总重量计0.1重量%至5重量%;优选地0.2重量%至4.7重量%;优选地0.5重量%至4.5重量%的表面活性剂。
32.根据陈述23至31中任一项所述的药物制剂,其中所述制剂包含颗粒内相,所述颗粒内相包含按所述药物制剂的总重量计0.1重量%至3重量%;优选地0.2重量%至2.7重量%;优选地0.5重量%至2.5重量%的润滑剂。
33.根据陈述23至32中任一项所述的药物制剂,其中所述制剂包含颗粒内相,所述颗粒内相包含:(i)按所述药物制剂的总重量计,5重量%至45重量%;优选地5重量%至40重量%;优选地10重量%至35重量%;优选地15重量%至30重量%的API;(ii)按所述药物制剂的总重量计,5重量%至60重量%;优选地8重量%至50重量%;优选地15重量%至45重量%的甲基丙烯酸共聚物或羟丙基甲基纤维素或它们的混合物;(iii)按所述药物制剂的总重量计,5重量%至60重量%;优选地10重量%至60重量%;优选地10重量%至50重量%;优选地15重量%至50重量%的填料;(iv)按所述药物制剂的总重量计,1重量%至10重量%;优选地1重量%至9重量%、优选地1.7重量%至8重量%的崩解剂;(v)按所述药物制剂的总重量计,0.1重量%至5重量%;优选地0.2重量%至4.7重量%;优选地0.5重量%至4.5重量%的助流剂;(vi)按所述药物制剂的总重量计,0.1重量%至5重量%;优选地0.2重量%至4.7重量%;优选地0.5重量%至4.5重量%的表面活性剂;以及(vii)按所述药物制剂的总重量计,0.1重量%至3重量%;优选地0.2重量%至2.7重量%;优选地0.5重量%至2.5重量%的润滑剂。
34.根据陈述23至33中任一项所述的药物制剂,其中所述药物制剂包含按所述药物制剂的总重量计至少5重量%;至少7重量%;至少10重量%;至少15重量%;至少20重量%;至少28重量%;至少34重量%的颗粒外相。
35.根据陈述23至34中任一项所述的药物制剂,其中所述药物制剂包含按所述药物制剂的总重量计至多74重量%;至多73重量%;至多67重量%;至多63重量%;至多53重量%、至多40重量%的颗粒外相。
36.根据陈述23至35中任一项所述的药物制剂,其中所述制剂包含颗粒外相,所述颗粒外相包含按所述药物制剂的总重量计0.1重量%至5.0重量%;优选地0.2重量%至4.7重量%;优选地0.5重量%至3.5重量%的崩解剂。
37.根据陈述23至36中任一项所述的药物制剂,其中所述制剂包含颗粒外相,所述颗粒外相包含按所述药物制剂的总重量计0.1重量%至3重量%;优选地0.2重量%至2.7重量%;优选地0.5重量%至2.5重量%的润滑剂。
38.根据陈述23至37中任一项所述的药物制剂,其中所述制剂包含颗粒外相,所述颗粒外相包含按所述药物制剂的总重量计0.1重量%至55重量%;0.2重量%至50重量%;0.3重量%至45重量%;0.4重量%至40重量%;0.5重量%至35重量%;0.6重量%至30重量%;0.7重量%至25重量%;0.8重量%至20重量%;1重量%至15重量%;1.3重量%至12重量%;2重量%至10重量%;2.5重量%至9重量%;3重量%至8重量%;4重量%至7重量%;5重量%至6重量%的填料。
39.根据陈述23至38中任一项所述的药物制剂,其中所述制剂包含颗粒外相,所述颗粒外相包含:(a)按所述药物制剂的总重量计,0.1重量%至5.0重量%;优选地0.2重量%至4.7重量%;优选地0.5重量%至3.5重量%;更优选地1重量%至3重量%的崩解剂;(b)按所述药物制剂的总重量计,0.1重量%至3重量%;优选地0.2重量%至2.7重量%;优选地0.5重量%至2.5重量%的润滑剂;以及(c)按所述药物制剂的总重量计,1重量%至15重量%;优选地1.3重量%至12重量%;优选地2.5重量%至8重量%;更优选地3重量%至5重量%的填料。
40.根据陈述1至39中任一项所述的药物制剂,其中所述API是式(I)的化合物
其立体异构形式、药学上可接受的盐、溶剂化物或多晶型物;所述化合物选自以下组,其中:
R
R
R
R
R
R
R
41.根据陈述40所述的药物制剂,其中所述式(I)的化合物是:
或其立体异构形式、药学上可接受的盐、溶剂化物或多晶型物。
42.一种固体剂型,所述固体剂型包含根据陈述1至41中任一项所述的药物制剂。
43.根据陈述42所述的固体剂型,其中所述剂型是口服剂型。
44.根据陈述42或43所述的固体剂型,其中所述剂型是片剂。
45.根据陈述42至44中任一项所述的固体剂型,其中所述制剂包含0.5mg至1000mg的所述API;优选地1mg至900mg的所述API;优选地2mg至800mg的所述API;优选地3mg至700mg的所述API;优选地4mg至600mg的所述API;优选地5mg至500mg的所述API;优选地6mg至400mg的所述API;优选地7mg至300mg的所述API;优选地8mg至200mg的所述API;优选地9mg至100mg的所述API;优选地10mg至50mg的所述API。
46.根据陈述42至45中任一项所述的固体剂型,其中所述制剂包含至少0.1mg、至少5mg、至少10mg、至少20mg、至少50mg、至少100mg、至少200mg、至少300mg、至少400mg、至少500mg或至少1000mg的所述API;优选地,所述API是:
47.一种治疗或预防登革热病毒感染的方法,所述方法包括向对其有需要的受试者施用治疗有效量的根据陈述1至41中任一项所述的药物制剂,或者向对其有需要的受试者施用治疗有效量的根据陈述42至46中任一项所述的固体剂型。
48.根据陈述1至41中任一项所述的药物制剂在制备用于治疗或预防登革热病毒感染的药物中的用途。
49.根据陈述42至46中任一项所述的固体剂型在制备用于治疗或预防登革热病毒感染的药物中的用途。
50.根据陈述1至41中任一项所述的药物制剂,所述药物制剂用作药物。
51.根据陈述42至46中任一项所述的固体剂型,所述固体剂型用作药物。
52.根据陈述1至41中任一项所述的药物制剂,所述药物制剂在用于治疗或预防登革热病毒感染的方法中使用。
53.根据陈述42至46中任一项所述的固体剂型,所述固体剂型在用于治疗或预防登革热病毒感染的方法中使用。
54.一种用于制备根据陈述1至41中任一项所述的药物制剂的方法,所述方法包括以下步骤:
a)将API溶解于溶剂中以形成溶液;
b)将所述甲基丙烯酸共聚物或羟丙基甲基纤维素或它们的组合与步骤a)中形成的所述溶液混合,从而获得混合物;
c)将所述混合物喷雾干燥以获得固体分散体;
d)任选地将所述固体分散体与至少一种药学上可接受的赋形剂共混;
以提供根据陈述1至41中任一项所述的药物制剂。
55.根据陈述54中任一项所述的方法,其中所述API是式(I)的化合物
其立体异构形式、药学上可接受的盐、溶剂化物或多晶型物;所述化合物选自以下组,其中:
R
R
R
R
R
R
R
56.根据陈述54至55中任一项所述的方法,其中所述API是:
57.一种用于制备根据陈述1至41中任一项所述的药物制剂或根据陈述42至46中任一项所述的固体剂型的方法,所述方法包括将所述活性药物成分与甲基丙烯酸共聚物或与纤维素衍生物(例如,羟丙基甲基纤维素)组合以形成所述药物制剂或所述固体剂型。
本发明提供了一种药物制剂,该药物制剂包含:
a)活性药物成分(API),该API是登革热病毒复制抑制剂;和
b1)甲基丙烯酸共聚物,或
b2)纤维素衍生物,诸如甲基纤维素(MC)、乙基纤维素(EC)、羟乙基纤维素(HEC)、羟丙基纤维素(HPC)、羧甲基纤维素(CMC)、羧甲基纤维素钠(NaCMC)或羟丙基甲基纤维素(HPMC)或它们的组合。
在一些实施方案中,纤维素衍生物是HPMC。
在一些实施方案中,根据本发明的药物制剂包含API和至少一种甲基丙烯酸共聚物,该API为登革热病毒复制抑制剂。
如本文所用,术语“甲基丙烯酸共聚物”优选地是指丙烯酸和/或甲基丙烯酸/酯的共聚物,诸如以商品名
可使用不同类型的甲基丙烯酸共聚物,并且它们包括聚(甲基丙烯酸-共-甲基丙烯酸甲酯)1:1(诸如
在一些实施方案中,甲基丙烯酸共聚物选自包括以下项的组:甲基丙烯酸和甲基丙烯酸甲酯的共聚物;甲基丙烯酸和丙烯酸乙酯的共聚物;以及它们的混合物。
在一些实施方案中,甲基丙烯酸共聚物是甲基丙烯酸和甲基丙烯酸甲酯的共聚物。优选地,共聚物中甲基丙烯酸与甲基丙烯酸甲酯的摩尔比为0.5:2至2:0.5,优选地0.8:1至1.2:1(例如,1:1)。
在一些实施方案中,甲基丙烯酸共聚物是聚(甲基丙烯酸-共-甲基丙烯酸甲酯)1:1(
在一些实施方案中,根据本发明的药物制剂包含API和羟丙基甲基纤维素,该API为登革热病毒复制抑制剂。
在一些实施方案中,根据本发明的药物制剂包含结晶速率抑制剂。术语“结晶速率抑制剂”是指当将该制剂施用于受试者时,为了抑制API的结晶而将其添加到该制剂中的赋形剂,例如聚合物赋形剂。结晶速率抑制剂可用于提高API的生物利用度,其中结晶形式的生物利用度与无定形/溶解状态相比显著较低。结晶速率抑制剂可被称为结晶抑制剂或稳定剂。
在一个实施方案中,结晶速率抑制剂选自聚乙烯吡咯烷酮(PVP)、聚乙烯吡咯烷酮-乙酸乙烯酯共聚物(PVPVA)、聚(甲基)丙烯酸酯聚合物(例如甲基丙烯酸-甲基丙烯酸甲酯共聚物)、环糊精或环糊精衍生物(例如(2-羟丙基)-β-环糊精(HPBCD))、羟丙基纤维素、羟乙基纤维素、甲基纤维素、羟丙基甲基纤维素(HPMC)、乙酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)、聚乙二醇-聚乙酸乙烯酯-聚乙烯基己内酰胺接枝共聚物、聚(乙烯醇)、泊洛沙姆(例如泊洛沙姆188、338或407)以及它们的任何组合。
在一个实施方案中,结晶速率抑制剂选自羟丙基甲基纤维素(HPMC)、乙酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)、聚乙二醇-聚乙酸乙烯酯-聚乙烯基己内酰胺接枝共聚物、聚乙烯吡咯烷酮(PVP)和聚乙烯吡咯烷酮-乙酸乙烯酯共聚物(PVPVA)以及它们的组合。在另一个实施方案中,结晶速率抑制剂选自羟丙基甲基纤维素(HPMC)和聚乙烯吡咯烷酮-乙酸乙烯酯共聚物(PVPVA)。PVPVA可以是质量比为6:4的1-乙烯基-2-吡咯烷酮和乙酸乙烯酯的共聚物(PVPVA64)。
聚乙烯吡咯烷酮-乙酸乙烯酯共聚物的名称和缩写包括但不限于PVPVA、PVP-Vac-共聚物和聚(1-乙烯基吡咯烷酮-共-乙酸乙烯酯)。
质量比为6:4的1-乙烯基-2-吡咯烷酮和乙酸乙烯酯的共聚物(PVPVA64)的名称和缩写包括但不限于共聚维酮(copolyvidone)、copovidum和共聚维酮(copovidone)。可商购获得的PVPVA64的示例是
聚乙烯吡咯烷酮的名称和缩写包括但不限于PVP、聚维酮和交聚维酮。交聚维酮是乙烯基吡咯烷酮的交联均聚物。可商购获得的PVP的示例是
羟丙基甲基纤维素(也称为羟丙甲纤维素(HPMC))是一种脱水葡萄糖,其中的羟基基团中的一些羟基基团被甲基基团取代以形成甲基醚部分,并且其他羟基基团被羟丙基基团或甲氧基丙基基团取代以形成羟丙基醚或甲氧基丙基醚部分。
羟丙基甲基纤维素聚合物(HPMC)可以不同粘度等级从若干来源获得,诸如以商品名
在一些实施方案中,羟丙基甲基纤维素在25℃下在2重量%的H
在一个实施方案中,本发明提供了一种药物制剂,该药物制剂包含a)活性药物成分(API),该API是登革热病毒复制抑制剂,其是如上文所定义的式(I)的化合物,并且优选地
b1)甲基丙烯酸共聚物,或
b2)纤维素衍生物,诸如甲基纤维素(MC)、乙基纤维素(EC)、羟乙基纤维素(HEC)、羟丙基纤维素(HPC)、羧甲基纤维素(CMC)、羧甲基纤维素钠(NaCMC)或羟丙基甲基纤维素(HPMC)或它们的组合。优选地,纤维素衍生物是HPMC。
相对于制剂的总重量,本发明的药物制剂可包含至多50重量%、至多40重量%、至多35重量%、至多30重量%或至多25重量%的API。相对于制剂的总重量,药物制剂可包含至少0.1重量%、至少0.5重量%、至少1重量%、至少5重量%、至少10重量%、至少15重量%、至少17重量%或至少20重量%的API。相对于制剂的总重量,药物制剂可包含0.1重量%至45重量%、0.5重量%至40重量%、1重量%至40重量%、5重量%至35重量%或10重量%至35重量%的API。
本发明的药物制剂可包含0.1mg至3000mg的API、1mg至2000mg的API、5mg至1500mg的API、5mg至1000mg的API、10mg至500mg的API、15mg至400mg的API、20mg至350mg的API或其中包含的任何特定量或范围。所述API的治疗有效量将随着所预防或治疗的疾病、综合征、病况和病症而变化。
本发明的药物制剂可包含:
20mg至6000mg的甲基丙烯酸共聚物、30mg至4000mg的甲基丙烯酸共聚物、40mg至2000mg的甲基丙烯酸共聚物、50mg至1500mg的甲基丙烯酸共聚物、60mg至1000mg的甲基丙烯酸共聚物、70mg至1000mg的甲基丙烯酸共聚物、80mg至600mg的甲基丙烯酸共聚物或其中包含的任何特定量或范围;或者
20mg至6000mg的羟丙基甲基纤维素、30mg至4000mg的羟丙基甲基纤维素、40mg至2000mg的羟丙基甲基纤维素、50mg至1500mg的羟丙基甲基纤维素、60mg至1000mg的羟丙基甲基纤维素、70mg至1000mg的羟丙基甲基纤维素、80mg至600mg的羟丙基甲基纤维素或其中包含的任何特定量或范围。
本发明的药物制剂还可包含一种或多种填充剂;其中制剂包含20mg至7500mg的填充剂,优选地30mg至6500mg、优选地40mg至4500mg、优选地50mg至2500mg、优选地60mg至2000mg、优选地80mg至1000mg、优选地90mg至550mg的填充剂,或其中包含的任何特定量或范围。
本发明的药物制剂还可包含一种或多种表面活性剂;其中制剂包含0.5mg至300mg的表面活性剂,优选地0.6mg至250mg、优选地0.8mg至200mg、优选地1mg至150mg、优选地1.2mg至100mg、优选地1.5mg至80mg、优选地2mg至30mg的表面活性剂,或其中包含的任何特定量或范围。
本发明的药物制剂还可包含一种或多种崩解剂;其中制剂包含3mg至900mg的崩解剂,优选地4mg至850mg、优选地5mg至600mg、优选地6mg至500mg、优选地7mg至400mg、优选地7.5mg至200mg、优选地8mg至100mg的崩解剂,或其中包含的任何特定量或范围。
本发明的药物制剂还可包含一种或多种助流剂;其中制剂包含1mg至400mg的助流剂,优选地2mg至350mg、优选地3mg至300mg、优选地4mg至200mg、优选地5mg至100mg、优选地6mg至50mg、优选地8.5mg至35mg的助流剂,或其中包含的任何特定量或范围。
本发明的药物制剂还可包含一种或多种润滑剂;其中制剂包含0.5mg至200mg的润滑剂,优选地1mg至150mg、优选地3mg至100mg、优选地4mg至90mg、优选地7mg至90mg、优选地8mg至80mg、优选地10mg至50mg的润滑剂,或其中包含的任何特定量或范围。
本发明的药物制剂可包含:
相对于制剂的总重量,至多60重量%、至多50重量%、至多45重量%、至多40重量%或至多35重量%的甲基丙烯酸共聚物;或者
相对于制剂的总重量,至多60重量%、至多50重量%、至多45重量%、至多40重量%或至多35重量%的羟丙基甲基纤维素。
本发明的药物制剂可包含:
相对于制剂的总重量,至少0.2重量%、至少1重量%、至少5重量%、至少10重量%、至少20重量%的甲基丙烯酸共聚物;或者
相对于制剂的总重量,至少0.2重量%、至少1重量%、至少5重量%、至少10重量%、至少20重量%的羟丙基甲基纤维素。
药物制剂可包含:
相对于制剂的总重量,0.2重量%至60重量%、1重量%至50重量%或5重量%至40重量%的甲基丙烯酸共聚物;或者
相对于制剂的总重量,0.2重量%至60重量%、1重量%至50重量%或5重量%至40重量%的羟丙基甲基纤维素。
在一些实施方案中,API和甲基丙烯酸共聚物以下列比率存在于本发明的药物制剂中:4:1w/w的比率;3.8:1w/w的比率;3.5:1w/w的比率;3.3:1w/w的比率;3:1w/w的比率;2.8:1w/w的比率;2.5:1w/w的比率;2.3:1w/w的比率;2:1w/w的比率;1.8:1w/w的比率;1.5:1w/w的比率;1:5w/w的比率;1:4.8w/w的比率;1:4.5w/w的比率;1:4.3w/w的比率;1:4w/w的比率;1:3.8w/w的比率;1:3.5w/w的比率;1:3.3w/w的比率;1:3w/w的比率;1:2.8w/w的比率;1:2.5w/w的比率;1:2.3w/w的比率;1:2w/w的比率;1:1.5w/w的比率或1:1.2w/w的比率或包含在本文提及的任何两个比率内的比率,或本文提及的任何两个比率内的比率范围或子范围。
在一些实施方案中,API和羟丙基甲基纤维素以下列比率存在于本发明的药物制剂中:4:1w/w的比率;3.8:1w/w的比率;3.5:1w/w的比率;3.3:1w/w的比率;3:1w/w的比率;2.8:1w/w的比率;2.5:1w/w的比率;2.3:1w/w的比率;2:1w/w的比率;1.8:1w/w的比率;1.5:1w/w的比率;1:5w/w的比率;1:4.8w/w的比率;1:4.5w/w的比率;1:4.3w/w的比率;1:4w/w的比率;1:3.8w/w的比率;1:3.5w/w的比率;1:3.3w/w的比率;1:3w/w的比率;1:2.8w/w的比率;1:2.5w/w的比率;1:2.3w/w的比率;1:2w/w的比率;1:1.5w/w的比率或1:1.2w/w的比率或包含在本文提及的任何两个比率内的比率,或本文提及的任何两个比率内的比率范围或子范围。
如本文更详细描述的,本发明的药物制剂还可包含一种或多种药学上可接受的赋形剂。药学上可接受的赋形剂包括但不限于崩解剂、粘结剂、填充剂、润滑剂、稳定剂、渗透剂、着色剂、增塑剂、包衣等。其他合适的药物赋形剂和它们的性质可以在文本诸如药物赋形剂手册(Handbook of Pharmaceutical Excipients),由R.C.Rowe,P.J.Sheskey和P.J.Weller编,第六版(由英国皇家药物学会分会的药物出版社出版(PharmaceuticalPress,a Division of Royal Pharmaceutical Society of Great Britain))中找到。
可用于本发明的填充剂(或填料)包括微晶纤维素(例如,
崩解剂增强药物制剂的分散。可用于本发明的崩解剂的非限制性示例包括交联羧甲基纤维素(例如,交联羧甲基纤维素钠)、交聚维酮、羟乙酸金属淀粉(例如,羟乙酸淀粉钠)以及它们的任何组合。崩解剂的其他示例包括交联羧甲基纤维素钠(例如,
粘结剂可包括在通过将粘结剂与填充剂和活性药物成分混合而制备活性药物成分的颗粒时使用的试剂。可用于本发明的粘结剂的非限制性示例包括聚乙烯吡咯烷酮、糖、改性纤维素(例如,羟丙基甲基纤维素(HPMC)、羟丙基纤维素(HPC)和羟乙基纤维素(HEC))以及它们的任何组合。粘结剂的其他示例包括聚乙烯吡咯烷酮(PVP)。HPC的示例包括低粘度聚合物HPC-SL。PVP可通过其“K值”来表征,该“K值”是聚合物组合物粘度的有用量度。PVP可以以商品名
润滑剂用于改善药物制剂从例如模压机中的压制和顶出。可用于本发明的润滑剂的非限制性示例包括硬脂酸镁、硬脂酸(硬脂精)、氢化油、硬脂酰富马酸钠、compritol(山嵛酸甘油酯)以及它们的任何组合。在一个示例中,润滑剂包括硬脂酰富马酸钠。在另一个示例中,润滑剂包括硬脂酸镁。本发明的药物制剂可包含一种或多种润滑剂,其组合(或总)浓度为按药物制剂的重量计0.1重量%至10重量%、0.5重量%至6重量%、0.8重量%至3.5重量%、1重量%至3重量%、1.5重量%至5.5重量%、2重量%至4重量%或0.25重量%至5.25重量%。在一些实施方案中,药物制剂包含0.5重量%至5.5重量%、优选地1重量%至2.5重量%的润滑剂(例如,硬脂酸镁)。
一种或多种润湿剂可用于本发明的药物制剂中。适用于本发明的润湿剂通常提高药物制剂的溶解度。润湿剂包括表面活性剂,诸如非离子表面活性剂和阴离子表面活性剂。可用于本发明的表面活性剂的非限制性示例包括月桂基硫酸钠(SLS)、聚氧乙烯脱水山梨糖醇脂肪酸(例如,聚山梨酸酯20(例如,TWEEN 20
助流剂增强制剂在加工成最终药物产品形式过程中的流动特性。可用于本发明的助流剂的非限制性示例包括二氧化硅(例如,胶态热解法二氧化硅、胶态无水二氧化硅)和/或滑石。助流剂的具体示例包括胶态热解法二氧化硅(例如,
片剂剂型还可包含包衣。合适的包衣是成膜性聚合物,例如来自以下组的那些:纤维素衍生物(诸如HPC(羟丙基纤维素)、HPMC(羟丙基甲基纤维素)、MC(甲基纤维素)、HPMCAS(乙酸羟基丙氧基甲基纤维素琥珀酸酯))、糊精、淀粉、
在一些实施方案中,根据本发明的药物制剂包含形成制剂的颗粒内相的多个颗粒和形成制剂的颗粒外相的一种或多种赋形剂。优选地,药物制剂包括片剂,所述片剂包含颗粒内相和颗粒外相。
如本文所用,术语“颗粒内相”是指制剂的与颗粒一起和/或在颗粒内的那些组分。如本文所用,术语“颗粒外相”是指制剂中的在颗粒之外的那些组分。
在一些实施方案中,根据本发明的药物制剂的颗粒内相包含活性药物成分,以及甲基丙烯酸共聚物或纤维素衍生物(诸如甲基纤维素(MC)、乙基纤维素(EC)、羟乙基纤维素(HEC)、羟丙基纤维素(HPC)、羧甲基纤维素(CMC)、羧甲基纤维素钠(NaCMC)或羟丙基甲基纤维素(HPMC))中的一种或组合。在一些实施方案中,根据本发明的药物制剂的颗粒内相包含活性药物成分,以及甲基丙烯酸共聚物或羟丙基甲基纤维素(HPMC)中的一种或组合,以及一种或多种赋形剂。
在一些实施方案中,药物制剂包含按药物制剂的总重量计至少15重量%;至少20重量%;至少25重量%;至少28重量%;至少30重量%;至少34重量%;至少40重量%;至少45重量%,至少50重量%;至少55重量%;至少60重量%;至少65重量%的颗粒内相。在一些实施方案中,药物制剂包含按药物制剂的总重量计至多99重量%;至多95重量%;至多93重量%;至多90重量%;至多85重量%;至多80重量%;至多75重量%;至多74重量%;至多73重量%;至多70重量%;至多67重量%;至多63重量%;至多60重量%;至多53重量%的颗粒内相。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含按药物制剂的总重量计优选地1重量%至70重量%;优选地2重量%至69重量%;优选地3重量%至68重量%;优选地4重量%至67重量%;优选地5重量%至66重量%;优选地6重量%至65重量%;优选地10重量%至64重量%;优选地15重量%至60重量%;优选地20重量%至55重量%;优选地25重量%至50重量%;优选地30重量%至45重量%;优选地35重量%至40重量%的API。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含按药物制剂的总重量计5重量%至60重量%;优选地7重量%至55重量%;优选地8重量%至50重量%;优选地17重量%至45重量%;优选地15重量%至45重量%的甲基丙烯酸共聚物或羟丙基甲基纤维素或它们的组合。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含按药物制剂的总重量计5重量%至60重量%;优选地10重量%至60重量%;优选地12重量%至60重量%;优选地15重量%至50重量%;优选地18重量%至45重量%的填料。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含按药物制剂的总重量计1重量%至10重量%;优选地1重量%至9重量%,优选地1.7重量%至8重量%;优选地1.6重量%至7.5重量%;优选地2重量%至5重量%的崩解剂。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含按药物制剂的总重量计0.1重量%至5重量%;优选地0.2重量%至4.7重量%;优选地0.5重量%至4.5重量%;优选地0.8重量%至4重量%;优选地1重量%至3.5重量%的助流剂。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含按药物制剂的总重量计0.1重量%至5重量%;优选地0.2重量%至4.7重量%;优选地0.5重量%至4.5重量%;优选地0.6重量%至4.5重量%;优选地0.8重量%至4.0重量%;优选地1重量%至3.5重量%的表面活性剂。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含按药物制剂的总重量计0.1重量%至3重量%;优选地0.2重量%至2.7重量%;优选地0.5重量%至2.5重量%;优选地0.7重量%至2重量%的润滑剂。
在一些实施方案中,药物制剂包含按药物制剂的总重量计至少5重量%;至少7重量%;至少10重量%;至少15重量%;至少20重量%;至少25重量%;至少28重量%;至少30重量%;至少34重量%;至少35重量%的颗粒外相。在一些实施方案中,药物制剂包含按药物制剂的总重量计至多75重量%;至多74重量%;至多73重量%;至多70重量%;至多67重量%;至多63重量%;至多65重量%;至多60重量%;至多55重量%、至多53重量%、至多40重量%的颗粒外相。在一些实施方案中,药物制剂包含按药物制剂的总重量计至少15重量%至最多75重量%;至少25重量%至最多70重量%;至少30重量%至最多65重量%;至少35重量%至最多60重量%;至少35重量%至最多70重量%的颗粒外相。
在一些实施方案中,药物制剂包含按药物制剂的总重量计至少15重量%;至少20重量%;至少25重量%;至少28重量%;至少30重量%;至少34重量%;至少35重量%的颗粒内相。在一些实施方案中,药物制剂包含按药物制剂的总重量计至多99重量%;至多93重量%;至多85重量%;至多80重量%;至多75重量%;至多74重量%;至多73重量%;至多70重量%;至多67重量%;至多65重量%;至多63重量%;至多60重量%;至多55重量%;至多53重量%的颗粒内相。在一些实施方案中,药物制剂包含按药物制剂的总重量计至少15重量%至最多75重量%;至少25重量%至最多70重量%;至少30重量%至最多65重量%;至少35重量%至最多60重量%;至少35重量%至最多70重量%的颗粒内相。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含:
(i)按药物制剂的总重量计,5重量%至45重量%、10重量%至40重量%、15重量%至35重量%的API;
(ii)按药物制剂的总重量计,5重量%至60重量%、10重量%至50重量%、15重量%至55重量%的甲基丙烯酸共聚物或羟丙基甲基纤维素或它们的组合;
(iii)按药物制剂的总重量计,5重量%至60重量%、10重量%至50重量%、15重量%至40重量%的填料(例如,甘露糖醇、微晶纤维素);
(iv)按药物制剂的总重量计,1重量%至10重量%、1.5重量%至9重量%、1.5重量%至8重量%的崩解剂(例如,交联羧甲基纤维素钠);
(v)按药物制剂的总重量计,0.1重量%至5重量%、0.2重量%至4.5重量%、0.5重量%至4重量%的助流剂(例如,胶态热解法二氧化硅);
(vi)按药物制剂的总重量计,0.1重量%至5重量%、0.2重量%至4.5重量%、0.5重量%至4重量%的表面活性剂(例如,月桂基硫酸钠);以及
(vii)按药物制剂的总重量计,0.1重量%至3重量%、0.2重量%至2.5重量%、0.5重量%至2重量%的润滑剂(例如,硬脂酸镁)。
在一些实施方案中,药物制剂包含颗粒外相,该颗粒外相包含按药物制剂的总重量计0.1重量%至5.0重量%;优选地0.2重量%至4.7重量%;优选地0.5重量%至4.5重量%;优选地1重量%至3.5重量%;优选地1.5重量%至3重量%的崩解剂。
在一些实施方案中,药物制剂包含颗粒外相,该颗粒外相包含按药物制剂的总重量计0.1重量%至3重量%;优选地0.2重量%至2.7重量%;优选地0.5重量%至2.5重量%;优选地0.8重量%至2重量%的润滑剂。
在一些实施方案中,药物制剂包含颗粒外相,该颗粒外相包含按药物制剂的总重量计0.1重量%至55重量%;0.2重量%至50重量%;0.3重量%至45重量%;0.4重量%至40重量%;0.5重量%至35重量%;0.6重量%至30重量%;0.7重量%至25重量%;0.8重量%至20重量%;1重量%至15重量%;1.3重量%至12重量%;2重量%至10重量%;2.5重量%至9重量%;3重量%至8重量%;4重量%至7重量%;5重量%至6重量%的填料。
在一些实施方案中,药物制剂包含颗粒外相,该颗粒外相包含:
(a)按药物制剂的总重量计,0.1重量%至5.0重量%、0.2重量%至4.5重量%、0.5重量%至3重量%的崩解剂(例如,交联羧甲基纤维素钠);
(b)按药物制剂的总重量计,0.1重量%至3重量%、0.2重量%至2.5重量%、0.5重量%至2重量%的润滑剂(例如,硬脂酸镁);以及
(c)按药物制剂的总重量计,1重量%至15重量%、1.5重量%至10重量%或2重量%至8重量%的填料(例如,羟丙基甲基纤维素)。
在一些实施方案中,本发明的药物制剂包含颗粒内相和颗粒外相;其中制剂包含50mg至17000mg的颗粒内相,优选地80mg至10000mg、优选地100mg至5000mg、优选地120mg至3000mg、优选地150mg至2500mg、优选地200mg至2000mg、优选地250mg至1200mg的颗粒内相,或其中包含的任何特定量或范围。
在一些实施方案中,本发明的药物制剂包含颗粒内相和颗粒外相;其中制剂包含4mg至1500mg的颗粒外相,优选地6mg至1000mg、优选地8mg至500mg、优选地10mg至300mg、优选地15mg至250mg、优选地20mg至200mg、优选地30mg至100mg的颗粒外相,或其中包含的任何特定量或范围。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含1mg至2000mg;优选地5mg至1500mg;优选地5mg至1000mg;优选地10mg至500mg;优选地15mg至400mg;优选地20mg至350mg的API,或其中包含的任何特定量或范围。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含20mg至6000mg;优选地30mg至4000mg;优选地40mg至2000mg;优选地70mg至1000mg的甲基丙烯酸共聚物或羟丙基甲基纤维素或它们的组合中的一种,或其中包含的任何特定量或范围。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含10mg至6500mg、优选地12mg至5000mg、优选地15mg至4000mg、优选地15mg至2000mg、优选地18mg至1000mg、优选地18mg至500mg的填充剂/填料,或其中包含的任何特定量或范围。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含0.5mg至300mg、优选地0.6mg至250mg、优选地0.8mg至200mg、优选地1mg至150mg、优选地1.2mg至100mg、优选地1.5mg至80mg、优选地2mg至30mg的表面活性剂,或其中包含的任何特定量或范围。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含2mg至720mg、优选地4mg至650mg、优选地5mg至400mg、优选地6mg至300mg、优选地7mg至200mg、优选地7.5mg至100mg、优选地8mg至60mg的崩解剂,或其中包含的任何特定量或范围。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含1mg至400mg、优选地2mg至350mg、优选地3mg至300mg、优选地4mg至200mg、优选地5mg至100mg、优选地6mg至50mg、优选地8.5mg至35mg的助流剂,或其中包含的任何特定量或范围。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含0.3mg至90mg;优选地0.5mg至70mg;优选地0.6mg至50mg;优选地0.7mg至25mg;优选地0.8mg至10mg的润滑剂,或其中包含的任何特定量或范围。
在一些实施方案中,药物制剂包含颗粒内相,该颗粒内相包含:
(i)1mg至2000mg;优选地5mg至1500mg;优选地5mg至1000mg;优选地10mg至500mg;优选地15mg至400mg;优选地20mg至350mg的API;
(ii)20mg至6000mg;优选地30mg至4000mg;优选地40mg至2000mg;优选地70mg至1000mg的甲基丙烯酸共聚物或羟丙基甲基纤维素或它们的组合中的一种;
(iii)10mg至6500mg、优选地12mg至5000mg、优选地15mg至4000mg、优选地15mg至2000mg、优选地18mg至1000mg、优选地18mg至500mg的填充剂/填料(例如,甘露糖醇、微晶纤维素);
(iv)2mg至720mg、优选地4mg至650mg、优选地5mg至400mg、优选地6mg至300mg、优选地7mg至200mg、优选地7.5mg至100mg、优选地8mg至60mg的崩解剂(例如,交联羧甲基纤维素钠);
(v)1mg至400mg、优选地2mg至350mg、优选地3mg至300mg、优选地4mg至200mg、优选地5mg至100mg、优选地6mg至50mg、优选地8.5mg至35mg的助流剂(例如,胶态热解法二氧化硅);
(vi)0.5mg至300mg、优选地0.6mg至250mg、优选地0.8mg至200mg、优选地1mg至150mg、优选地1.2mg至100mg、优选地1.5mg至80mg、优选地2mg至30mg的表面活性剂(例如,月桂基硫酸钠);以及
(vii)0.3mg至90mg;优选地0.5mg至70mg;优选地0.6mg至50mg;优选地0.7mg至25mg;优选地0.8mg至10mg的润滑剂(例如,硬脂酸镁)。
在一些实施方案中,药物制剂包含颗粒外相,该颗粒外相包含0.6mg至180mg;优选地1mg至100mg;优选地2mg至80mg;优选地3mg至50mg;优选地5mg至20mg的崩解剂。
在一些实施方案中,药物制剂包含颗粒外相,该颗粒外相包含3.5mg至1100mg;优选地5mg至500mg;优选地10mg至250mg;优选地15mg至100mg;优选地25mg至90mg的填充剂/填料。
在一些实施方案中,药物制剂包含颗粒外相,该颗粒外相包含0.3mg至90mg;优选地0.5mg至70mg;优选地1mg至50mg;优选地1.5mg至20mg;优选地2mg至10mg的润滑剂。
在一些实施方案中,药物制剂包含颗粒外相,该颗粒外相包含:
(a)0.6mg至180mg;优选地1mg至100mg;优选地2mg至80mg;优选地3mg至50mg;优选地5mg至20mg的崩解剂(例如,交联羧甲基纤维素钠);
(b)0.3mg至90mg;优选地0.5mg至70mg;优选地1mg至50mg;优选地1.5mg至20mg;优选地2mg至10mg的润滑剂(例如,硬脂酸镁);以及
(c)3.5mg至1100mg;优选地5mg至500mg;优选地10mg至250mg;优选地15mg至100mg;优选地25mg至90mg的填充剂/填料(例如,羟丙基甲基纤维素)。
本领域技术人员将容易地认识到,选择合适的药学上可接受的赋形剂,使得它们与其他赋形剂相容并且不与活性药物成分结合或相互作用或者不引起活性成分或药物制剂的降解。
应当理解,涉及药物制剂组分的任何上述描述可适用于本发明的任何其他方面和实施方案。
合适的活性药物成分(API)是发挥药理学、免疫学或代谢作用以恢复、纠正或改变生理功能或进行医学诊断的那些。其非限制性示例包括止痛药和抗炎药;抗心律失常药;抗细菌剂和抗原虫剂;抗凝剂;抗抑郁药;抗糖尿病药;抗癫痫药;抗真菌剂;抗组胺药;抗高血压药;抗毒蕈碱剂;抗肿瘤剂和抗代谢药;抗偏头痛药;抗帕金森病药物;抗精神病剂、催眠剂和镇静剂;抗卒中剂;镇咳药;抗病毒药;β-肾上腺素受体阻断剂;心肌收缩剂;皮质类固醇;消毒剂;利尿剂;酶;精油;胃肠剂;脂质调节剂;局部麻醉剂;阿片类止痛药;拟副交感神经药和抗痴呆药物;性激素;刺激剂和血管扩张剂。
本发明提供了一种药物制剂,该药物制剂包含:
a)API;和
b1)甲基丙烯酸共聚物,或
b2)纤维素衍生物,诸如甲基纤维素(MC)、乙基纤维素(EC)、羟乙基纤维素(HEC)、羟丙基纤维素(HPC)、羧甲基纤维素(CMC)、羧甲基纤维素钠(NaCMC)或羟丙基甲基纤维素(HPMC)或它们的组合;
其中API是登革热病毒复制抑制剂。
具体地,API在药物制剂中为无定形形式或溶解状态(即分子分散体)。
在一个实施方案中,API是登革热病毒复制抑制剂。本发明的实施方案包括如本文所述的药物制剂,其中API是式(I)的化合物
其立体异构形式、药学上可接受的盐、溶剂化物或多晶型物;所述化合物选自以下组,其中:
R
R
R
R
R
R
R
本发明的另外的实施方案包括如本文所述的药物制剂,其中API是选自由以下项组成的组的式(I)的化合物:
或其立体异构形式、药学上可接受的盐、溶剂化物或多晶型物。
具体地,API是式(I)的化合物或其对映体、非对映体或药学上可接受的盐形式。
具体地,API是呈无定形状态或溶解状态(即分子分散体)的式(I)的化合物或其对映体、非对映体或药学上可接受的盐形式。
具体地,在制备如本文所述的药物制剂的方法中用作起始物质的API是式(I)的化合物或其对映体、非对映体、溶剂化物或药学上可接受的盐形式;而如本文所定义的最终药物制剂或固体剂型中的API是呈无定形形式或溶解状态的式(I)的化合物或其对映体、非对映体或药学上可接受的盐形式。
在一个优选的实施方案中,式(I)的化合物为
API可以是化合物(a)或其溶剂化物或药学上可接受的盐形式。API可以是化合物(a)或其药学上可接受的盐形式。API可以是呈溶剂化形式的化合物(a),例如作为一水合物。优选地,API是化合物(a)。优选地,API是化合物(a)的(S)-对映体。优选地,API是呈无水形式的化合物(a)。优选地,API是呈无定形形式的化合物(a)。优选地,API是呈无定形形式或溶解状态的化合物(a)或其药学上可接受的盐形式。优选地,API是呈无定形形式或溶解状态的化合物(a)。优选地,API是呈无定形形式的化合物(a)的(S)-对映体。优选地,API是呈无水形式的化合物(a)的(S)-对映体。
具体地,在制备如本文所述的药物制剂的方法中用作起始物质的API是化合物(a)、其溶剂化形式或药学上可接受的盐形式;而最终药物制剂或固体剂型中的API是呈无定形形式或溶解状态的化合物(a)或其药学上可接受的盐形式。
具体地,在制备如本文所述的药物制剂的方法中用作起始物质的API是呈溶剂化形式的化合物(a)或其药学上可接受的盐形式;而最终药物制剂或固体剂型中的API是呈无定形形式或溶解状态(即分子分散体)的化合物(a)或其药学上可接受的盐形式。
式(I)的化合物可以根据WO 2016/180696中公开的方法合成,其全文以引用方式并入本文。
应当理解,涉及活性药物成分的任何上述描述可适用于本文所述的药物制剂、固体剂型、方法、用途以及预防、治疗和病毒抑制方法的任何实施方案。例如,对登革热病毒复制抑制剂的任何提及可以指式(I)的化合物或其立体异构形式、药学上可接受的盐、溶剂化物、共晶体或多晶型物。
在一个具体实施方案中,如本文所述的药物制剂中的API是化合物(a)或其立体异构形式、药学上可接受的盐、溶剂化物或多晶型物。在一个具体实施方案中,如本文所述的药物制剂中的API是化合物(a)。
在一个具体实施方案中,如本文所述的药物制剂中的API是呈无定形形式或溶解状态的登革热病毒复制抑制剂。在一个具体实施方案中,如本文所述的药物制剂中的API是呈无定形形式或溶解状态的化合物(a)或其药学上可接受的盐形式。在一个具体实施方案中,如本文所述的药物制剂中的API是呈无定形形式或溶解状态的化合物(a)。
本发明还提供了包含如本文所述的药物制剂的固体剂型。
剂型可以是口服剂型(例如用于口服施用的胶囊)。另选地,剂型可以是肠内剂型。另选地,固体剂型可以是片剂。
如本文所述的固体剂型(例如片剂)可包含0.1mg至3000mg的API、1mg至2000mg的API、5mg至1000mg的API、10mg至500mg的API、20mg至400mg的API、30mg至300mg的API、50mg至200mg的API、70mg至150mg的API、100mg至120mg的API或其中包含的任何特定量或范围。所述API的治疗有效量将随着所预防或治疗的疾病、综合征、病况和病症而变化。
如本文所述的固体剂型(例如片剂)可包含0.5mg至1000mg的API。在一些实施方案中,固体剂型可包含0.5mg至800mg,例如1.0mg至600mg,例如2.0mg至450mg;优选地,API是
在一个具体实施方案中,固体剂型是片剂,该片剂包含:
a)API;和
b1)甲基丙烯酸共聚物,或
b2)纤维素衍生物,诸如甲基纤维素(MC)、乙基纤维素(EC)、羟乙基纤维素(HEC)、羟丙基纤维素(HPC)、羧甲基纤维素(CMC)、羧甲基纤维素钠(NaCMC)或羟丙基甲基纤维素(HPMC)或它们的组合;
其中API是登革热病毒复制抑制剂。
在一个具体实施方案中,固体剂型是片剂,该片剂包含:
a)API;
b1)甲基丙烯酸共聚物,或
b2)羟丙基甲基纤维素(HPMC);以及
d)一种或多种药学上可接受的赋形剂,该药学上可接受的赋形剂选自崩解剂、粘结剂、填充剂、润滑剂、稳定剂、润湿剂、助流剂、渗透剂、着色剂、增塑剂和包衣;
其中API是登革热病毒复制抑制剂。
在一个具体实施方案中,固体剂型是包含本发明的药物制剂的片剂。
在一个实施方案中,固体剂型包含药物制剂,其中制剂包含至少5mg、至少15mg、至少25mg、至少50mg、至少55mg、至少100mg、至少150mg、至少200mg、至少250mg、至少300mg、至少350mg、至少400mg、至少450mg或至少500mg的API;优选地,API是
或其药学上可接受的盐形式。
对于口服施用,固体剂型具体地以片剂形式提供,该片剂包含至少1.0mg、至少0.5mg、至少1mg、至少5mg、至少10mg、至少20mg、至少30mg、至少40mg、至少50mg、至少60mg、至少70mg、至少80mg、至少90mg、至少100mg、至少110mg、至少120mg、至少130mg、至少140mg、至少150mg、至少160mg、至少170mg、至少180mg、至少190mg、至少200mg、至少210mg、至少220mg、至少230mg、至少240mg、至少250mg、至少260mg、至少270mg、至少280mg、至少290mg、至少300mg、至少310mg、至少320mg、至少330mg、至少340mg、至少350mg、至少360mg、至少370mg、至少380mg、至少390mg、至少400mg、至少410mg、至少420mg、至少430mg、至少440mg、至少450mg、至少460mg、至少470mg、至少480mg、至少490mg、至少500mg、至少510mg、至少520mg、至少530mg、至少540mg、至少550mg、至少560mg、至少570mg、至少580mg、至少590mg、至少600mg、至少610mg、至少620mg、至少630mg、至少640mg、至少650mg、至少660mg、至少670mg、至少680mg、至少690mg、至少700mg、至少710mg、至少720mg、至少730mg、至少740mg、至少750mg、至少760mg、至少770mg、至少780mg、至少790mg、至少800mg、至少810mg、至少820mg、至少830mg、至少840mg、至少850mg、至少860mg、至少870mg、至少880mg、至少890mg、至少900mg、至少910mg、至少920mg、至少930mg、至少940mg、至少950mg、至少960mg、至少970mg、至少980mg、至少990mg、至少1000mg、至少1100mg、至少1200mg、至少1210mg、至少1220mg、至少1230mg、至少1240mg、至少1250mg、至少1260mg、至少1270mg、至少1280mg、至少1290mg、至少1300mg、至少1410mg、至少1320mg、至少1330mg、至少1340mg、至少1350mg、至少1360mg、至少1370mg、至少1390mg、至少1400mg的API,或包含在前述值中的任何值。具体地,固体剂型包含15mg至500mg的API。
有利地,API或固体剂型可以单次日剂量施用,或者总日剂量可以每日两次、三次、四次或五次的分剂量施用。日剂量可以在治疗或预防期的所有天或几天内保持不变。所述日剂量可以在治疗或预防期的各天中变化,诸如其在所述治疗或预防期的各天期间增加和/或减少。例如,所述日剂量对于前1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40天或更多天可以是不变的,随后对于治疗或预防期的剩余天是较低和/或较高的日剂量。治疗或预防期的所述剩余天可以是1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60天或更多天。API或固体剂型也可以每周至少一次、每两周至少一次、每三周至少一次、每四周或每月至少一次、每两个月至少一次、每三个月至少一次、每四个月至少一次、每五个月至少一次、每六个月至少一次或每年至少一次施用。API或固体剂型也可以每天施用,持续1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60天或更多天,随后至少每周至少一次、每两周至少一次、每三周至少一次、每四周或每月至少一次、每两个月至少一次、每三个月至少一次、每四个月至少一次、每五个月至少一次、每六个月至少一次或每年至少一次施用。
在某些实施方案中,API或固体剂型可以在第一持续时间(例如,加载阶段)以第一剂量施用,并且在第二持续时间(例如,维持阶段)以第二剂量施用。加载阶段可包括施用本文所述的任何剂量(例如,约10mg至约1000mg、约25mg至约800mg或约50mg至约400mg)。加载阶段中的第一施用持续时间可以是本文所设想的任何时间段(例如,约1天至约40天、约3天至约20天或约5天至约10天)。维持阶段可包括施用本文所述的任何剂量(例如,约10mg至约1000mg、约25mg至约800mg或约50mg至约400mg)。维持阶段中的第二施用持续时间可以是本文所设想的任何时间段(例如,约1天至约60天、约5天至约45天或约10天至约30天)。
待施用的药物制剂的最佳剂量可容易确定,并且将随所使用的具体化合物、施用模式、制剂强度以及疾病、综合征、病况或病症的进程而变化。因此,上述剂量为一般情况的示例。当然,可能会存在其中较高或较低剂量范围是有益的个别情况,并且这类情况也在本发明的范围内。
本发明还提供了用于制备如本文所述的药物制剂的方法。该方法包括以下步骤:
a)将API溶解于溶剂中以形成溶液;
b)将甲基丙烯酸共聚物或羟丙基甲基纤维素或它们的组合与步骤a)中形成的溶液混合,从而获得混合物,并且例如进一步搅拌该混合物;
c)将所述混合物喷雾干燥以获得固体分散体;
d)任选地将固体分散体与一种或多种药学上可接受的赋形剂共混;
以提供如本文所述的药物制剂。
如本文所用,术语“固体分散体”意指API在固体基质中的分散体,其中基质包含小分子或聚合物或它们的组合。
根据本发明的药物制剂包括固体分散体,其中基质是聚合物,并且该聚合物选自甲基丙烯酸共聚物或纤维素衍生物诸如甲基纤维素(MC)、乙基纤维素(EC)、羟乙基纤维素(HEC)、羟丙基纤维素(HPC)、羧甲基纤维素(CMC)、羧甲基纤维素钠(NaCMC)或羟丙基甲基纤维素(HPMC)或它们的组合。优选地,纤维素衍生物是HPMC。
本发明还提供了一种用于制备本文所述的固体剂型的方法,方法包括以下步骤:
a)将API溶解于溶剂中以形成溶液;
b)将甲基丙烯酸共聚物或纤维素衍生物诸如甲基纤维素(MC)、乙基纤维素(EC)、羟乙基纤维素(HEC)、羟丙基纤维素(HPC)、羧甲基纤维素(CMC)、羧甲基纤维素钠(NaCMC)或羟丙基甲基纤维素或它们的组合与步骤a)中形成的溶液混合,从而获得混合物,并且任选地进一步搅拌该混合物;
c)将混合物喷雾干燥以获得固体分散体;
d)任选地将固体分散体与一种或多种药学上可接受的赋形剂共混;例如,药学上可接受的赋形剂可选自包括以下项的组:填料、表面活性剂、崩解剂、助流剂、润滑剂以及它们的混合物;
e)将共混物压制成片剂;
以提供如本文所述的固体剂型。
在一些实施方案中,制备本文所述的固体剂型的方法包括以下步骤:
a)将API溶解于溶剂中以形成溶液;
b)将甲基丙烯酸共聚物或羟丙基甲基纤维素或它们的组合与步骤a)中形成的溶液混合,从而获得混合物,并且任选地进一步搅拌该混合物;
c)将混合物喷雾干燥以获得固体分散体;
d)将固体分散体与至少一种填料、至少一种表面活性剂、至少一种崩解剂、至少一种助流剂和至少一种润滑剂共混;
e)将共混物制粒;
f)将步骤e)中获得的混合物与至少一种崩解剂、填料和至少一种润滑剂共混;
g)将共混物压制成片剂;
以提供如本文所述的固体剂型。
在一些实施方案中,可使用热熔挤出获得固体分散体。在一些实施方案中,将共混物制粒的步骤使用辊式压实机或通过重压进行。
本发明的另一方面提供了包装的药物制剂,其中药物制剂是密封在泡罩膜中的本文所述的任何制剂(例如,片剂),其中泡罩膜包括被构造成容纳一种或多种药物制剂(例如,片剂)的制剂保持层和被构造成覆盖保持层以将药物制剂密封在保持层内的密封层,其中密封层包括铝箔和干燥材料。如本文所用,术语“干燥材料”是指可用作干燥剂的任何吸湿性物质。干燥材料的示例包括但不限于二氧化硅(例如,硅胶)、活性炭、硫酸钙、氯化钙和沸石材料。
在一些实施方案中,保持层包括一个或多个室,其中每个室被构造成容纳一种或多种药物制剂(诸如本文所述的任何药物制剂(例如,一个或多个片剂)),并且每个室由密封层密封。在一些实施方案中,保持层包含透明或不透明材料(例如,透明或不透明聚乙烯材料)。在一些实施方案中,密封层与保持层和设置在保持层中的任何室完全重叠。
可用于本发明的可商购获得的泡罩膜的示例包括购自Amcor plc.的DessiflexPlus和Dessiflex Ultra。在一些实施方案中,包装的药物制剂由密封在泡罩膜中的1个或多个(例如,1、2、3、4、5、6、7、8、9、10、1至4、2至10或1至10个)片剂组成,其中泡罩膜包含一个卡片。与将药物制剂包装在具有不含干燥材料的密封层的泡罩膜(例如,Aclar 400泡罩膜)中相比,使用其中密封层包含干燥材料的本发明泡罩膜包装改善了本发明的药物制剂的稳定性和保存期限。
本发明的另一方面提供了试剂盒,该试剂盒包括包装的药物制剂,诸如本文所述的任何包装的药物制剂,以及该包装的药物制剂的施用说明书。
应当理解,涉及固体剂型及其制备方法的任何上述讨论可适用于本文所述的固体剂型、方法、预防和治疗的任何实施方案。
本发明还包括将本文所述的药物制剂和固体剂型用作药物。本发明还包括用于预防登革热病毒感染或用于治疗登革热病毒感染或用于抑制登革热病毒在生物体外样品或受试者中的病毒复制的方法。该方法包括向体外样品或受试者(对其有需要)施用有效量的本文所述的药物制剂和固体剂型。体外样品或受试者处于感染登革热病毒的风险中或已感染登革热病毒。
每当随其有需要的受试者需要使用该药物制剂时,本文所述的药物制剂可以前述剂型和方案中的任一者或通过本领域中已确立的那些剂型和方案施用。
本发明的药物制剂和剂型可用于在对其有需要的受试者中治疗、改善和/或预防疾病、综合征、病况或病症的方法中。此类方法包括以下步骤、由以下步骤组成和/或基本上由以下步骤组成:向受试者(包括需要此类治疗、改善和/或预防的动物、哺乳动物和人)施用治疗有效量的本文所述的制剂或剂型。在其中活性药物成分是登革热病毒复制抑制剂的实施方案中,本发明的药物制剂和剂型可用于治疗、改善和/或预防受登革热病毒复制的抑制影响的疾病、综合征、病况的方法中。
本发明的一个实施方案涉及在对其有需要的受试者(包括需要预防登革热病毒感染的动物、哺乳动物和人)中预防登革热病毒感染的方法,该方法包括向受试者施用治疗有效量的本文所述的药物制剂或剂型。
本发明的一个实施方案涉及在对其有需要的受试者(包括需要治疗登革热病毒感染的动物、哺乳动物和人)中治疗登革热病毒感染的方法,该方法包括向受试者施用治疗有效量的本文所述的药物制剂或剂型。
本发明的一个实施方案涉及在对其有需要的受试者(包括需要抑制登革热病毒的病毒复制的动物、哺乳动物和人)中抑制登革热病毒的病毒复制的方法,该方法包括向受试者施用治疗有效量的本文所述的药物制剂或剂型。
在某些实施方案中,API的血浆水平例如在治疗方案(用于治疗或用于预防)的持续时间内处于约5ng/ml至约10,000ng/ml、约10ng/ml至约8,000ng/ml、约15ng/ml至约6,500ng/ml、约20ng/ml至约5,000ng/ml、约25ng/ml至约4,500ng/ml、约30ng/ml至约3,000ng/ml、约40ng/ml至约2,000ng/ml、或约50ng/ml至约1,000ng/ml范围内、或其中的任何单个值或子范围的水平。在某些实施方案中,API的最大血浆水平为至多约10,000ng/ml、至多约8,000ng/ml、至多约6,500ng/ml、至多约4,500ng/ml、至多约3,000ng/ml、至多约2,000ng/ml、至多约1,000ng/ml或其中的任何单个值或子范围。在某些实施方案中,API的最小血浆水平例如在治疗方案(用于治疗或用于预防)的持续时间内为至少约5ng/ml、至少约10ng/ml、至少约15ng/ml、至少约20ng/ml、至少约25ng/ml、至少约30ng/ml、至少约40ng/ml、至少约50ng/ml或其中的任何单个值或子范围。此处提及的血浆水平可以用本文所述的任何剂量和/或给药方案获得。
在本发明的另一个实施方案中,本文所述的药物制剂可以与一种或多种其他药剂组合使用,更具体地与其他抗病毒剂组合使用。
应当理解的是,可对本发明的前述实施方案进行变化,同时仍处于本发明的范围内。在本说明书中公开的每个特征,除非另有说明,否则可被用于相同、等效或类似目的的替代特征替换。因此,除非另有说明,否则所公开的每个特征仅是一系列等效或类似特征的一个示例。
上文所述的化合物和药学上可接受的制剂可根据所预防或治疗的感染的严重程度,通过口服给药、直肠给药、颅内给药、阴道内给药、腹膜内给药、局部给药(如通过粉剂、软膏剂或滴剂)、颊给药(以口腔或鼻腔喷雾剂形式)等途径,施用于人和其他动物。
用于口服施用的液体剂型包括但不限于药学上可接受的乳液、微乳液、溶液、悬浮液、糖浆和酏剂。除活性化合物之外,液体剂型还可包含本领域常用的惰性填充剂,例如水或其他溶剂,增溶剂和乳化剂诸如乙醇、异丙醇、碳酸乙酯、乙酸乙酯、苯甲醇、苯甲酸苄酯、丙二醇、1,3-丁二醇、二甲基甲酰胺、油(具体地,棉籽油、落花生油、玉米油、胚芽油、橄榄油、蓖麻油和芝麻油)、甘油、四氢糠醇、聚乙二醇和脱水山梨糖醇的脂肪酸酯,以及它们的混合物。除惰性填充剂之外,口服制剂还可包含助剂,诸如润湿剂、乳化剂和悬浮剂、甜味剂、调味剂和芳香剂。
用于直肠或阴道施用的制剂具体地为可通过将本文所述的化合物与合适的非刺激性赋形剂或载体(诸如可可脂、聚乙二醇或栓剂蜡)混合来制备的栓剂,这些非刺激性赋形剂或载体在室温下为固体,但在体温下为液体,从而在直肠或阴道腔中熔化并释放活性化合物。
用于口服施用的固体剂型包括胶囊剂、片剂、丸剂、粉剂和颗粒剂。在此类固体剂型中,将活性化合物与至少一种惰性的、药学上可接受的赋形剂或载体(诸如柠檬酸钠或磷酸二钙),和/或a)填充剂或增量剂,诸如淀粉、乳糖、蔗糖、葡萄糖、甘露醇和硅酸;b)粘结剂,诸如羧基甲基纤维素、藻酸盐、明胶、聚乙烯吡咯烷酮、蔗糖和阿拉伯树胶;c)湿润剂,诸如甘油;d)崩解剂,诸如琼脂、碳酸钙、马铃薯或木薯淀粉、藻酸、某些硅酸盐和碳酸钠;e)溶液阻滞剂,诸如石蜡;f)吸收促进剂,诸如季铵化合物;g)润湿剂,诸如鲸蜡醇和单硬脂酸甘油酯;h)吸收剂,诸如高岭土和膨润土;以及i)润滑剂,诸如滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、月桂基硫酸钠,以及它们的混合物混合。就胶囊剂、片剂和丸剂而言,剂型还可包含缓冲剂。
类似类型的固体制剂还可用作使用乳糖(lactose或milk sugar)以及高分子量聚乙二醇等赋形剂的软填充明胶胶囊和硬填充明胶胶囊中的填充剂。片剂、糖锭剂、胶囊剂、丸剂和颗粒剂的固体剂型可用包衣和外壳(诸如肠溶包衣)和药物配制领域众所周知的其他包衣制备。它们可以任选地含有乳浊剂,并且还可以是一种仅在或优先在肠道的某一部分任选地以延迟的方式释放活性成分的制剂。可以使用的包埋制剂的示例包括聚合物质和蜡。类似类型的固体制剂还可用作使用乳糖(lactose或milk sugar)以及高分子量聚乙二醇等赋形剂的软填充明胶胶囊和硬填充明胶胶囊中的填充剂。
活性化合物还可为具有如上所述的一种或多种赋形剂的微包封形式。片剂、糖锭剂、胶囊剂、丸剂和颗粒剂的固体剂型可用包衣和外壳(诸如肠溶包衣)、控释包衣和药物配制领域众所周知的其他包衣制备。在此类固体剂型中,将活性化合物与至少一种惰性填充剂(诸如蔗糖、乳糖或淀粉)混合。按照惯例,此类剂型还可包含除惰性填充剂之外的另外的物质,例如压片润滑剂和其他压片助剂,诸如硬脂酸镁和微晶纤维素。就胶囊剂、片剂和丸剂而言,剂型还可包含缓冲剂。它们可以任选地含有乳浊剂,并且还可以是一种仅在或优先在肠道的某一部分任选地以延迟的方式释放活性成分的制剂。可以使用的包埋制剂的示例包括聚合物质和蜡。
本文所述的化合物的局部或经皮施用的剂型包括软膏剂、糊剂、霜剂、洗剂、凝胶剂、粉剂、溶液剂、喷雾剂、吸入剂或贴剂。在无菌条件下,将活性组分与药学上可接受的载体和任何所需的防腐剂或可能需要的缓冲剂混合。眼用制剂、滴耳剂和滴眼剂也被认为是在本发明的范围内。另外,本发明设想使用透皮贴剂,其具有向机体提供化合物的受控递送的附加优势。此类剂型可以通过将化合物溶解或分配在适当的介质中来制备。吸收促进剂也可用于增加化合物跨皮肤的通量。速率可以通过提供速率控制膜或通过将化合物分散在聚合物基质或凝胶中来控制。
本文所述的制剂可通过口服给药、吸入喷雾、局部给药、直肠给药、鼻腔给药、颊给药、阴道给药途径施用或经由植入的储存器施用。
本文所述的药物制剂可以任何口服可接受的剂型口服施用,该剂型包括但不限于胶囊剂、片剂、水性悬浮液或溶液。就用于口服使用的片剂而言,常用的载体包括但不限于乳糖和玉米淀粉。通常还添加润滑剂,诸如硬脂酸镁。对于胶囊形式的口服施用,可用的填充剂包括乳糖和干燥的玉米淀粉。当需要口服使用水性悬浮液时,活性成分与乳化剂和悬浮剂组合。如有需要,也可添加某些甜味剂、调味剂或着色剂。
另选地,本文所述的药物制剂可以用于直肠施用的栓剂的形式施用。这些药物制剂可通过使药剂与合适的非刺激性赋形剂混合来制备,该非刺激性赋形剂在室温下为固体,但在直肠温度下为液体,从而在直肠中熔化以释放药物。此类材料包括但不限于可可脂、蜂蜡和聚乙二醇。
本文所述的药物制剂也可以局部施用,尤其是当预防和/或治疗目标包括通过局部应用容易接近的区域或器官时,包括眼睛、皮肤或下肠道的疾病。易于制备适用于这些区域或器官中的每一者的局部制剂。下肠道的局部应用可以直肠栓剂制剂(参见上文)或合适的灌肠制剂实现。也可以使用局部透皮贴剂。
对于局部应用,药物制剂可被配制成含有悬浮或溶解于一种或多种载体中的活性组分的合适的软膏剂。用于局部施用本发明的化合物的载体包括但不限于矿物油、液体凡士林、白凡士林、丙二醇、聚氧乙烯、聚氧丙烯化合物、乳化蜡和水。另选地,药物制剂可被配制成含有悬浮或溶解于一种或多种药学上可接受的载体中的活性组分的合适的洗剂或霜剂。合适的载体包括但不限于矿物油、脱水山梨糖醇单硬脂酸酯、聚山梨酸酯60、十六烷基酯蜡、鲸蜡硬脂醇、2辛基十二烷醇、苯甲醇和水。
药物制剂还可通过鼻气溶胶或吸入施用。此类制剂根据药物制剂领域中众所周知的技术制备,并且可使用苯甲醇或其他合适的防腐剂、用于提高生物利用率的吸收促进剂、氟碳化合物和/或其他常规增溶剂或分散剂制备成盐溶液。
用于本发明方法的化合物可被配制成单位剂型。术语“单位剂型”是指适合作为经历预防或治疗的受试者的单一剂量的物理离散单元,每个单元含有经计算能产生期望的治疗效果的预定量的活性物质,任选地与合适的药物载体相结合。单位剂型可以是单个日剂量或多次日剂量(例如,每天1至4次或更多次)中的一次。当使用多次日剂量时,单位剂型对于每一剂量可以相同或不同。
上述实施方案的所有可能的组合被认为包含在本发明的范围内。
现在参考以下实施例,这些实施例以非限制性方式说明了本发明。
用于本发明的代表性化合物可根据以下所述的和在之后的方案和实施例中说明的通用合成方法合成。由于所述方案是举例说明性的,所以本发明不应理解为受所述方案和实施例中的化学反应和条件的限制。与这些示例的目标化合物类似的化合物可按相似的路径制得。所公开的化合物可如本文中所述用作药物。用于方案和实施例中的不同原料可商购获得或者可通过本领域的技术人员熟知的方法制备。
通式I化合物的合成可以如方案1中所概述的进行。2-(4-氯-2-甲氧基苯基)乙酸(II)可以用氯化试剂(例如亚硫酰氯)转化成对应的2-(4-氯-2-甲氧基苯基)乙酰氯(III)。酰基氯III与通式IV的取代吲哚的Friedel-Crafts反应可以使用Lewis酸试剂例如Et
在一些情况下,经由Friedel-Crafts合成方法合成通式V的中间体受益于在Friedel-Crafts反应步骤期间在吲哚-N处存在保护基团(PG),如方案2中所概述。为此,通式IV的取代吲哚可以使用试剂例如甲苯磺酰氯,在碱例如氢化钠的存在下,首先转化为通式VIII的N-保护的中间体,诸如通式VIII的N-甲苯磺酰化的中间体(PG=Ts)。通式IV的取代吲哚与酰基氯III的Friedel-Crafts反应可使用Lewis酸试剂例如Et
作为替代方法,通式V的中间体还可以如方案3中所概述进行制备:通式X的N-Boc-保护的取代吲哚-3-甲醛可通过与吗啉在试剂例如氰化钠和亚硫酸氢钠的存在下,并且在合适的溶剂例如水和可与水混合的有机溶剂例如二氧杂环己烷的混合物中反应而转化为对应的Strecker型的通式XI的中间体。通式XI的化合物与4-氯-2-甲氧基-苄基氯的烷基化可以在碱例如六甲基二硅氮烷钾的存在下,并且在合适的溶剂例如二甲基甲酰胺(DMF)中完成,得到通式XII的化合物。将通式XII的化合物置于合适的含水酸性水解条件下,例如通过在升高的温度下用盐酸水溶液处理,得到通式V的中间体。
式(I)的化合物可以根据WO 2016/180696中公开的方法合成,其全文以引用方式并入本文。
在以下实施例中,化合物(a)、优选地化合物(a)的(+)-对映体用作活性药物成分(API)。化合物(a)如WO 2016/180696的实施例9中所述的那样合成。其作为白色粉末获得:
通过溶剂蒸发方法在96孔板中制备固体分散体制剂(将100μL含有所需量的API和添加剂的液体转移至每个孔中,将孔板转移至70℃和<2mbar压力的真空炉中一小时,冷却至室温)。将API(呈无定形形式的化合物(a))和赋形剂两者均溶解于二氯甲烷和甲醇(50/50,v/v)的混合物中。使用自动液体处理工作站(Hamilton Microlab STAR plus)制备混合物。分配后,通过有机溶剂的快速蒸发产生无定形API-聚合物膜。这通过使用设定在70℃和200毫巴的真空烘箱在减压下蒸发一小时来实现。所制备的固体分散体示于表A中。在开始溶出测定之前,将每份含有约100μg API的所得膜(每次筛选每种制剂12个平行样)冷却并保持在室温下,第一次筛选保持一天,第二次筛选保持3天。用恒定百分比的API(33重量%)制备膜,而赋形剂的总和为67重量%。作为参考,仅含有API的膜也包括在内。
*如果存在的话
在膜浇铸后、溶出研究开始的那天以及在40℃/75%湿度下稳定一、二和四周后,通过偏振光显微术进行结晶度评估。在膜浇铸后、溶出研究开始的那天以及在40℃/75%相对湿度下稳定一、二和四周后,未检测到结晶材料。
体外进行2相(SGF/FaSSIF)微型化溶出,其中随时间推移来监测溶出的API的量。在开始溶出测定之前,将膜储存在室温下,第一次筛选储存一天,第二次筛选储存3天。通过这样做,大部分残留溶剂被蒸发。使用用于取样和样品制备的Hamilton STAR plus液体处理平台在96个1mL玻璃小瓶中进行实际溶出实验。在添加到膜中之前,将介质预热至37℃。然后将300μL预热的SGF(37℃,pH 1.3)添加到固体分散体中。在SGF中温育15分钟后,向样品中加入600μL预热的浓缩FaSSIF(37℃,pH10.5)。将这种浓缩的FaSSIF添加到SGF中产生了具有与在单相溶出研究中使用的典型FaSSIF介质类似组成的介质。以预定的时间间隔(11、24、34、49、79、139分钟),从溶出介质中取出等分试样并通过0.45μmGHP膜过滤器过滤。随后,将过滤的溶液用N-甲基吡咯烷酮(NMP)定量稀释以防止可能的沉淀。通过UPLC/UV-Vis分析测定溶出的API的量。每次筛选对每种制剂一式两份进行实验。
图1示出了固体分散体1至7(对应于图1中的概念1至7)在SGF-FaSSIF中的溶出曲线。对于纯无定形API参考,测量到膜中存在的API总量的约9%的最终释放。与参考相比,所有其他测试的固体分散体显示出改善的溶出曲线。在胃相中,API不溶于任何固体分散体中。对于固体分散体1,达到20% API的最终释放。
如上所述对固体分散体8至12进行第二次溶解度筛选,并且它们的组成列于表B。这些固体分散体如上所述制备。
*如果存在的话
a在第一次筛选中固体分散体也已进行测试
图2示出了固体分散体8至12(对应于图2中的概念8至12)在SGF-FaSSIF中的溶出曲线。对于纯无定形API参考,测量到膜中存在的API总量的约9%的最终释放。对于分散体8,达到54% API的最终释放,而对于分散体9,达到28% API的最终释放。
为了筛选用于喷雾干燥的适当溶剂,在环境温度下估计起始物质在不同有机溶剂中的近似溶解度。将每种溶剂以50μL或100μL的增量添加到含有约5mg每种样品(API、Eudragit L100和HPMC E5)的2mL玻璃小瓶中,直到固体溶解或达到1mL的总体积。结果汇总于下表中。
*具有非常少颗粒的澄清溶液,可能是由于HPMC E5本身的固有特性。
*
*
通过UPLC测定在MeOH和MeOH/二氯甲烷(DCM)(1/1,v/v)中的平衡溶解度,分别为42mg/mL和317mg/mL。
基于上述部分中提及的溶剂选择和聚合物筛选结果,选择包括Eudragit L100、HPMC AS和HPMC E5的三种聚合物作为载体材料,分别选择丙酮/EtOH(2:1,v/v)(对于Eudragit L100)和MeOH/DCM(1:1,v/v)(对于HPMC E5和HPMC AS)作为用于喷雾干燥的溶剂体系。
将足量的不同聚合物分别称取到合适的玻璃瓶中,添加足量的所选溶剂以溶解。然后称取约25g化合物(a)(无定形状态)并分别溶解于上述各溶液中,分别获得EudragitL100在丙酮/EtOH(2:1,v/v)中的API浓度为14mg/mL的澄清溶液和HPMC E5和HPMC AS在MeOH/DCM(1:1,v/v)中的API浓度为10mg/mL的澄清溶液。还称取约35g化合物(a)并溶解于丙酮中以获得用于制备无定形API的API浓度为50mg/mL的澄清溶液。
固体分散体1(SD1):API+Eudragit L100(1:2w/w)。
固体分散体2(SD2):API+HPMC E5(1:2w/w)。
固体分散体3(SD2):API+HPMC AS(1:2w/w)。
喷雾干燥装置:Buchi B-290。
将喷雾干燥的分散体(SDD)和无定形API喷雾干燥粉末在低于混合物的Tg的温度下进一步真空干燥。
在最终后干燥后,将获得的SD1、SD2和SD3粉末(即基于Eudragit L100的ASD1、基于HPMC E5的ASD2、基于HPMC AS的ASD3)在小瓶中直接称重并设置用于物理和化学稳定性测试。
将合适量的每种SD产物称取到40mL玻璃小瓶中(即12mg用于化学稳定性,50mg用于物理稳定性)。然后将样品小瓶用垫圈和盖子紧密密封并用铝箔包裹,并储存在25℃/60%RH(封闭)和40℃/75%RH(封闭)的稳定室中达不同的时间点(2周、1个月、2个月、3个月和6个月)。对于化学稳定性,在每个时间点在每个条件下设置一式三份样品,并且对于物理稳定性设置单个样品。还将化合物(a)、Eudragit L100和HPMC E5称取到40mL玻璃小瓶中,并且在每个条件和每个时间点设置用于API和赋形剂对照。对于化学稳定性,在每个时间点针对每个条件测试含量和杂质,并且对于物理稳定性,测试XRPD、PLM和mDSC。
通过在如表3的第一列中所述的应力条件下将SD1粉末置于玻璃瓶中来测试实施例3中制备的SD1的稳定性。
表3:SD1的物理和化学稳定性
“RH”:相对湿度;“M”:月;“N”:非双折射;“Am”:无定形;“P1”:白色蓬松粉末;“P2”:白色蓬松粉末,并且一些粉末可能附着到小瓶的底部和壁上;“PLM”:偏振光显微镜。
在25℃和60%相对湿度下在封闭玻璃瓶中储存6个月后,未观察到明显的化学和物理变化。然而,与初始总杂质相比,在40℃和75%相对湿度下在封闭玻璃瓶中储存6个月后观察到总杂质的增加。
通过在如表4的第一列中所述的应力条件下将SD2粉末置于玻璃瓶中来测试实施例3中制备的SD2的稳定性。
“RH”:相对湿度;“M”:月;“N”:非双折射;“Am”:无定形;“P3”:白色粉末,一些粉末可能附着到小瓶的底部和壁上;“PLM”:偏振光显微镜。
与初始相比,在25℃/60%相对湿度和40℃/75%相对湿度下在封闭玻璃瓶中储存6个月后,未观察到明显的化学和物理变化。
通过在如表4A的第一列中所述的应力条件下将SD3粉末置于玻璃瓶中来测试实施例3中制备的SD3的稳定性。
“RH”:相对湿度;“W”:周;“M”:月;“N”:非双折射;“Am”:无定形;“P3”:白色粉末,一些粉末可能附着到小瓶的底部和壁上;“PLM”:偏振光显微镜。
与初始相比,在密闭玻璃瓶中储存期间,特别是在40℃和75%相对湿度下,总杂质增加(从T0的0.08%至3个月时的约0.2%)。
如以上实施例3所述制备包含不同API/HPMC E5重量比(如下列出)的四种固体分散体。
固体分散体2a(SD2a):API+HPMC E5(1:1w/w)
固体分散体2b(SD2b):API+HPMC E5(1:2w/w)
固体分散体2c(SD2c):API+HPMC E5(1:3w/w)
固体分散体2d(SD2d):API+HPMC E5(1:4w/w)
在基于生理学的溶出试验(PBDT)中测试所制备的固体分散体,该试验利用模拟肠液作为介质。在禁食条件下在PBDT中测试SD2a、SD2b、SD2c和SD2d。在进食条件下在PBDT中测试SD2a、SD2b和SD2d。表4B和4B'中分别提供了禁食和进食条件下PBDT的详细信息。
*模拟胃液;**禁食状态模拟肠液
**进食状态模拟肠液
图3A示出了测试的SD粉末的溶出百分比,图3B示出了相同SD粉末的以mg计的溶出。与SD2a和SD2d相比,SD2b和SD2c表现出改善的溶出曲线。在PBDT进食条件下也观察到SD2b的改善的溶出曲线,如图4所示。
另外,通过向150mL预热的SGF pH 1.3中添加50mg每种SD,评价SD2a、SD2b、SD2c和SD2d在SGF-FaSSIF中的API和聚合物溶出。15分钟后,将280mL预热的1.5x浓缩FaSSIFpH10.3添加到SGF pH1.3中,得到FaSSIF pH6.5。在37℃的培养箱中以250rpm搅拌样品。通过在5’-14'-20'-25'-30'-45'-60'-75'-105'-135'后取5mL等分试样进行时间依赖性分析。通过用具有0.45μm PTFE的Millex LCR 25mm过滤器过滤,从溶液中分离未溶解的API(丢弃3mL,并将2mL用于浓度测量)。将溶液在ACN/水50/50v/v中进行2倍稀释以避免沉淀。使用UPLC-UV测定溶液中的API浓度。使用UPLC-RI测定溶液中的HPMC E5浓度。
制备包含SD1的片剂(10mg强度)。片剂的组成示于表5中。
表5:SD1片剂的组成(10mg强度)
API、SD1粉末以及包含SD1的片剂(10mg强度)的稳定性通过将片剂置于高密度聚乙烯(HDPE)瓶中在如表6的第2列所述的应力条件下进行测试。结果示于表6中。
“RH”:相对湿度
“W”:周
“M”:月
“Am”:无定形;
“T1”:白色粉末;
“T2”:白色或灰白色圆形片剂
制备包含SD2的片剂(10mg强度)。片剂的组成示于表7中。
API、SD2以及包含SD2的片剂(10mg强度)的稳定性通过将片剂置于HDPE瓶中在如表7的第2列所述的应力条件下进行测试。结果示于表7中。
“RH”:相对湿度
“W”:周
“M”:月
“Am”:无定形;
“T1”:白色粉末;
“T2”:白色或灰白色圆形片剂
1.将甲醇(对于SD1粉末)或甲醇与二氯甲烷的混合物(对于SD2粉末,1/1,v/v)转移到容器中并使用混合器搅拌。在搅拌的同时,将化合物(a)添加到溶剂中。继续搅拌,直到化合物(a)溶解。过滤所得溶液。
2.将Eudragit L100或羟丙基甲基纤维素E5添加到步骤1的溶液中。搅拌混合物,直到完全混合。
3.使用合适的喷雾干燥器(PSD-1)将两种混合物喷雾干燥,并收集喷雾干燥的产物。
使用托盘干燥器干燥所得粉末并收集。
表9
a在加工过程中去除(通过干燥)
1.将SD1粉末、微晶纤维素、甘露糖醇、交联羧甲基纤维素钠、胶态二氧化硅、硬脂酸镁、月桂基硫酸钠一起过筛,然后在箱式共混器中共混。
2.将共混物在辊式压实机WP-120中辊式压实,并收集所得颗粒。
3.将交联羧甲基纤维素钠和微晶纤维素一起过筛,并添加到步骤2中获得的干燥颗粒中,将混合物在箱式共混器中共混。
4.额外添加筛过的硬脂酸镁,并将所得混合物在箱式共混器中共混。
5.使用XL 100Korsch压片机将最终共混物压制成片剂。
1.将SD2粉末、微晶纤维素、甘露糖醇、交联羧甲基纤维素钠、胶态二氧化硅、硬脂酸镁、月桂基硫酸钠一起过筛,然后在箱式共混器中共混。
2.将共混物在辊式压实机WP-120中辊式压实,并收集所得颗粒。
3.将交联羧甲基纤维素钠和微晶纤维素一起过筛,并添加到步骤2中获得的干燥颗粒中,将混合物在箱式共混器中共混。
4.额外添加筛过的硬脂酸镁,并将所得混合物在箱式共混器中共混。
5.使用XL 100Korsch压片机将最终共混物压制成片剂。
使用上述片剂7和10进行药代动力学(PK)研究。将片剂在禁食条件下对雄性比格犬(N=3)口服施用一次、两次或3次。结果示于表14中。
平均值±标准偏差
AUC=血浆浓度对时间曲线下面积;AUC
使用上述片剂9和12进行药代动力学(PK)研究。在禁食和进食条件下对雄性比格犬(N=3)口服施用三片片剂。结果示于表15中。
平均值±标准偏差
AUC=血浆浓度对时间曲线下面积;AUC
犬只的药代动力学数据清楚地表明禁食和进食状态的AUC值是相似的,表明不存在食物效应。因此,该制剂不依赖于食物的存在或不存在而释放API。这从患者顺从性的观点来看是有益的。因此,与其他制剂相比,根据本发明的制剂可导致降低的食物效应。
虽然本文已示出和描述了本发明的优选实施方案,但对本领域的技术人员将显而易见的是,此类实施方案仅以举例的方式提供。在不脱离本发明的情况下,本领域的技术人员可设想出多种变型、改变和替代形式。应当理解,在实施本发明时可采用本文所述的本发明的实施方案的各种替代方案,并且由此涵盖在这些权利要求及其等同物的范围内的实施方案。
- 包括质子泵抑制剂的口服用固体制剂组合物、包括该组合物的口服用固体制剂及其制造方法
- 药物固体制剂用的包衣剂、药物用膜制剂以及包覆药物固体制剂