用于CRISPR选择的方法和系统
文献发布时间:2023-06-19 13:48:08
相关申请的交叉引用
本申请要求2019年3月18日提交的美国临时申请号62/820,106的权益,该临时申请据此以引用的方式整体并入本文。
背景技术
成簇规律间隔短回文重复序列(CRISPR)-Cas9技术已经彻底改变了基因组工程。在该系统中,向导RNA(gRNA)指导Cas9核酸酶在靶标基因组区域诱导双链断裂。gRNA的5'-末端包括与靶标区域互补的约20个核苷酸的核苷酸序列。当双链断裂通过非同源末端接合(NHEJ)修复时,插入和缺失的发生频率很高,从而有效地敲除靶标基因组基因座。慢病毒递送方法的开发使基因组规模的CRISPR/Cas9敲除文库的产生成为可能。这些文库允许对哺乳动物细胞系进行负向和正向选择筛选。在CRISPR/Cas9敲除筛选中,每个基因都被若干gRNA靶向,并且携带不同基因敲除的突变库可以通过高通量测序来确定。CRISPR活化(CRISPRa)也可以与gRNA文库一起使用,其中活化的基因可以通过高通量测序来确定。
全基因组CRISPR/Cas9敲除或基因活化技术是一种有效的基因扰动筛选技术。目标是鉴定与表型相关的gRNA,从而鉴定相应的受影响的基因。然而,这些筛选产生的数据对计算分析提出了若干挑战。CRISPR研究通常以多次重复方式进行。CRISPR易受差异的影响,因为每个实验可能不会在筛选文库中使用相同的gRNA病毒滴定度,慢病毒感染率可能因实验而异,并且gRNA在实验中可能不会以相同的效率靶向基因。因此,即使是具有相同表型的细胞,所观察到的gRNA丰度在整个实验中也是高度可变的。现有技术依靠读段计数来鉴定与表型相关的gRNA,特别是使用归一化的gRNA读段计数的平均值和方差来测试gRNA丰度在具有或不具有表型的细胞之间是否存在显著差异。
然而,此类技术并未解决上述实验间差异问题,而是假设选择前和选择后实验之间具有高度同一性。这些技术不能解决单个CRISPR实验内和/或CRISPR实验之间的差异。
因此,需要对计算技术进行技术改进,以在通过正向选择和负向选择筛选鉴定基因时解决CRISPR差异问题。
发明内容
应当理解,下文的一般性描述和下文的详细描述都仅为示例性和说明性而非限制性的。
在一个实施方案中,一种方法包括(A)用病毒载体的文库感染cas9阳性细胞的第一培养物,所述文库包含至少3个向导RNA(gRNA),用于切割所述细胞的基因组内的DNA的靶标区域;对所述细胞进行测序以获得所述gRNA中的每个的读段计数;对读段计数超过背景阈值的每个DNA靶标区域的相应gRNA数量进行求和(Σ),其中Σ=n;以及对读段计数超过所述背景阈值的所有靶标区域中的gRNA总数进行求和(Σ),其中Σ=N。所述方法包括(B)用所述病毒载体的文库感染cas9阳性细胞的第二培养物;将所述第二培养物的所述细胞分类为具有指定表型或不具有指定表型;选择具有所述指定表型的细胞并且对所选细胞进行测序以获得所述gRNA中的每个的选择后读段计数;对选择后读段计数超过所述背景阈值的所选细胞中的每个DNA靶标区域的相应gRNA数量进行求和(Σ),其中Σ=n’;以及对读段计数超过所述阈值的所选细胞的所有靶标区域中的gRNA总数进行求和(Σ),其中Σ=N’。所述方法包括(C)对于DNA的靶标区域,根据公式
在一个实施方案中,一种方法包括根据对于DNA的多个靶标区域中的每个,在用包含至少3个向导RNA(gRNA)的文库的载体感染后,对第一细胞群进行的测序,确定读段计数超过背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n),根据对于所述DNA的多个靶标区域中的每个的所述相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第一细胞群中的gRNA的总数(N),根据对于所述DNA的多个靶标区域中的每个,在用包含所述至少3个gRNA的文库的载体感染后,对第二细胞群进行的测序,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n’),根据对于所述DNA的多个靶标区域中的每个的所述相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第二细胞群中的gRNA的总数(N’),对于所述多个靶标区域中的每个靶标区域,根据n、N、n’和N’来确定在所选细胞中偶然观察到所述靶标区域的n’个gRNA的概率,对于包含所关注的序列的靶标区域,根据在所选细胞中偶然观察到所述靶标区域的n’个gRNA的概率来确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率,以及根据在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率来鉴定所关注的序列是正向选择的。
其他优点将在下面的描述中进行部分阐述或者可以通过实践来了解。所述优点将借助于所附权利要求中特别指出的要素和组合来实现和获得。
附图说明
并入本说明书中并构成其一部分的附图说明了实施方案,并与说明书一起用于解释所述方法和系统的原理:
图1是CRISPR正向选择的示例性方法;
图2显示了另一个示例性方法;
图3显示了一个示例性操作环境;
图4显示了一个存储四个假设基因的人工读段计数的示例性数据结构,其中每个基因具有三个(A-C或D-F)或四个(G-J或K-N)gRNA,据此采集每个gRNA的选择前和选择后读段计数,以及据此鉴定出大于阈值30的选择前n和选择后n’读段计数,并且将这些读段计数加合在一起(N和N'),以及根据所示的公式来确定概率;
图5显示了一个存储所公开的方法的人工数据和人工结果的示例性数据结构,据此最初使用gRNA的数量N,在细胞培养10天后提供gRNA的数量N’。如图所示,靶标区域1或靶标区域2具有的gRNA的数量为n,并且提供了在培养10天后细胞中的gRNA的数量为n’的概率(概率以p值表示);
图6显示了涉及大约21,000个基因(G#)的实验结果,其中显示了来自四个选择前和三个选择后实验的每个gRNA(g)的读段计数和加合在一起的读段计数(Sum),以及鉴定出大于阈值30的读段计数(存在之和),并且将这些读段计数加合在一起(Sum);
图7显示了在细胞培养的三天(d03)、六天(d06)和十天(d10)的多个平行实验(xp#)中使用的样品gRNA文库(Gecko A和Geck B),其后确定通过FRET荧光测量的Tau聚集;Gecko A和B文库的构成显示为包括特定基因靶向gRNA、特定微RNA靶向gRNA和非靶向gRNA以及每个靶标的大约gRNA数量;
图8显示了在第3、6和10天,每个实验(xp#)中使用的Gecko A文库中的每个gRNA归一化之前的读段计数,据此gRNA用于指导靶标的失活;
图9显示了图8所示的实验中的Gecko A文库的读段计数的归一化,归一化基于读段计数的中位数,并且第10天是选择后;以及
图10显示,由于靶标可能具有不同数量的gRNA,而且在第10天某些样品的“存在”gRNA比其他样品多得多,因此计算“存在”的频率的另一种方法是计算基因“存在”的概率。使用Gecko A文库gRNA来计算第10天(选择后)筛选的五种不同的基因的概率。
具体实施方式
在公开和描述本发明的方法和系统之前,应当理解,所述方法和系统不限于特定方法、特定部件或特定具体实施方式。还应当理解,本文所用的术语仅仅是为了描述特定实施方案的目的,而非旨在进行限制。
如说明书以及所附权利要求书中所使用,除非上下文另外清楚地规定,否则单数形式的词语“一个”、“一种”和“该”均包括多个指代物。
如本文所用,术语“探针”和“向导RNA(gRNA)”和“向导”可互换使用。在一个实施方案中,gRNA还可以以编码gRNA的DNA的形式提供。
如本文所用,“Cas蛋白”可以是野生型蛋白(即,自然界中存在的那些)、经修饰的Cas蛋白(即,Cas蛋白变体)或者野生型或经修饰的Cas蛋白的片段。就野生型或经修饰的Cas蛋白的催化活性而言,Cas蛋白也可以是活性变体或片段。
在第一方面,本公开的特征在于用于鉴定基因或基因产物的方法,例如,调控其他基因或基因产物的表达的方法。例如,这些方法可用于展示使用CRISPR向导进行扰动后的正向选择。
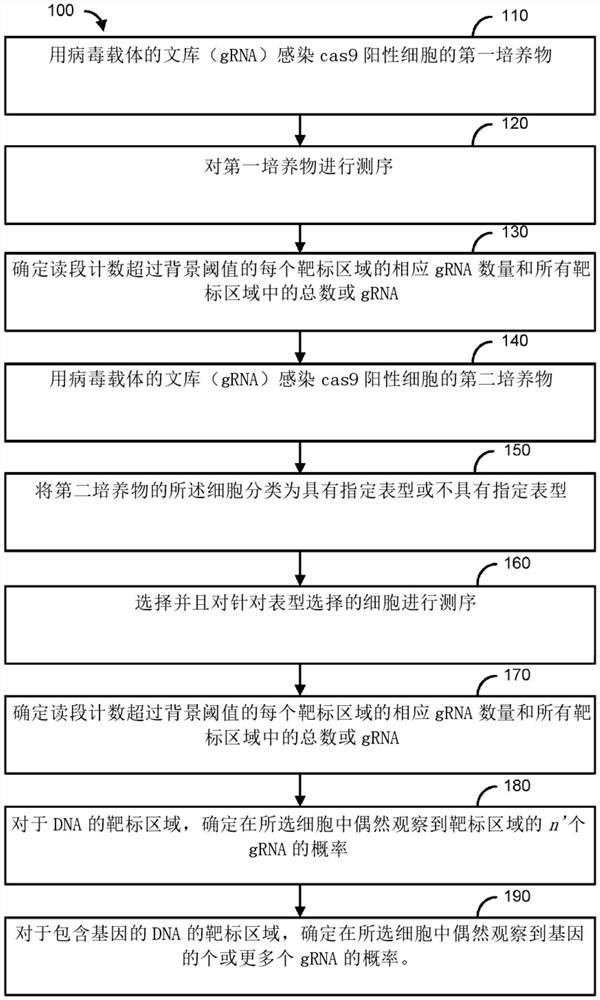
在一个实施方案中(如图1所示),所述方法包括以下步骤:用病毒载体的文库感染cas9阳性细胞的第一培养物110,所述文库包含至少3个向导RNA,用于切割所述细胞的基因组内的DNA的靶标区域,对所述细胞进行测序以获得所述gRNA中的每个的读段计数120,对读段计数超过背景阈值的每个DNA靶标区域的相应gRNA数量进行求和(Σ)(Σ=n)以及对读段计数超过所述背景阈值的所有靶标区域中的gRNA总数进行求和(Σ)(Σ=N)130,用所述病毒载体的文库感染cas9阳性细胞的第二培养物140,将所述第二培养物的所述细胞分类为具有指定表型或不具有指定表型150,选择具有所述指定表型的细胞并且对所选细胞进行测序以获得所述gRNA中的每个的选择后读段计数160,对选择后读段计数超过所述背景阈值的所选细胞中的每个DNA靶标区域的相应gRNA数量进行求和(Σ)(Σ=n’),以及对读段计数超过所述阈值的所选细胞的所有靶标区域中的gRNA总数进行求和(Σ)(Σ=N’)170,对于DNA的靶标区域,根据以下公式来计算在所选细胞中偶然观察到靶标区域的n'个gRNA的概率
其中
在一个实施方案中(也如图1所示),所述方法包括以下步骤:用病毒载体的文库感染cas9阳性细胞的第一培养物110,所述文库包含至少3个向导RNA(gRNA),用于增强所述细胞的基因组内的DNA的靶标区域的转录,对所述细胞进行测序以获得所述gRNA中的每个的读段计数120,对读段计数超过背景阈值的每个DNA靶标区域的相应gRNA数量进行求和(Σ)(Σ=n)以及对读段计数超过所述背景阈值的所有靶标区域中的gRNA总数进行求和(Σ)(Σ=N)130,用所述病毒载体的文库感染cas9阳性细胞的第二培养物140,将所述第二培养物的所述细胞分类为具有指定表型或不具有指定表型150,选择具有所述指定表型的细胞并且对所选细胞进行测序以获得所述gRNA中的每个的选择后读段计数160,对选择后读段计数超过所述背景阈值的所选细胞中的每个DNA靶标区域的相应gRNA数量进行求和(Σ)(Σ=n’),以及对读段计数超过所述阈值的所选细胞的所有靶标区域中的gRNA总数进行求和(Σ)(Σ=N’)170,对于DNA的靶标区域,根据以下公式来计算在所选细胞中偶然观察到靶标区域的n’个gRNA的概率
其中
在一个实施方案中,可以根据CRISPR技术来感染第一培养物。在一个实施方案中,用病毒载体的文库(例如图2;201)感染cas9阳性细胞的第一培养物并且用病毒载体的文库(例如图2;205)感染cas9阳性细胞的第二培养物)包括使用CRISPR gRNA文库。CRISPR gRNA文库可以是敲除文库,包括例如包括一个或多个靶向基因组中的每个基因的gRNA(例如,sgRNA)的全基因组gRNA敲除文库,其中基因组可以是任何类型的基因组。在一些实施方案中,gRNA文库可以包括合并文库。合并文库的非限制性实例包括基因组规模的CRISPR敲除(GeCKO)文库。参见例如Shalem O等人(2014)Science 343:84-7和Sanjana NE等人(2014)Nat.Methods 11:783-4。文库中的gRNA可以靶向DNA中的任何数量的靶标区域(例如基因)。例如,gRNA可以靶向约50个或更多个基因、约100个或更多个基因、约200个或更多个基因、约300个或更多个基因、约400个或更多个基因、约500个或更多个基因、约1000个或更多个基因、约2000个或更多个基因、约3000个或更多个基因、约4000个或更多个基因、约5000个或更多个基因、约10000个或更多个基因、或约20000个或更多个基因。在一些文库中,可以选择gRNA来靶向特定信号传导通路中的基因。gRNA文库可以使用范围更广的感染复数(MOI)进行施用。在一些方面,较低的MOI可以用于促进感染,以获得每个细胞一个gRNA。
在一个实施方案中,cas阳性细胞可以包含用于切割DNA的靶标区域的cas蛋白或用于调控转录(例如增强或抑制转录)的cas蛋白。用于切割DNA的靶标区域的cas蛋白可以包含RNA结合结构域和核酸酶结构域。用于调控转录的Cas蛋白是失活的,以使其不再具有核酸酶活性。失活的Cas(例如dCas-9)可以与转录活化物或转录阻遏物融合。因此,在一个实施方案中公开了包含具有野生型活性的cas-9或失活的cas-9的cas-9阳性细胞。失活的cas-9可以例如与至少一个转录活化结构域融合。一个或多个gRNA可以结合至所关注的基因的转录起始位点上游的靶标序列,而不是切割DNA,dCas-9和转录调节物可以在活化所关注的基因的转录或抑制所关注的基因的转录中发挥作用。如果dCas-9与一种或多种转录活化物融合,则所述一种或多种转录活化物会将转录因子募集到所关注的基因的转录起始位点,从而活化或上调转录。如果dCas-9与转录阻遏物融合,那么转录将被抑制、下调或抑制。
在一个实施方案中,DNA的靶标区域可以是基因。在一个实施方案中,DNA的靶标区域可以是基因的启动子区域或调控元件区域。在一个实施方案中,DNA的靶标区域调控下游基因或蛋白质。在一个实施方案中,调控下游基因或蛋白质包括下游基因或蛋白质的活化或抑制。
在一些实施方案中,细胞包含选择性标记系统。例如,如本文的方法中所公开,第一培养物的cas-9阳性细胞可以被修饰为含有一个或多个选择性标记。选择性标记系统可以涉及与选择性标记融合或连接的一个或多个标记蛋白,所述选择性标记仅在一个或多个蛋白质受到调控时被活化。“一个或多个标记蛋白”可以是任何可以受到调控的蛋白质,其中受调控意指标记蛋白改变形状、与一个或多个其他蛋白质或核酸结合、活性发生变化或表达水平发生变化。一个或多个标记蛋白可以由一个或多个gRNA靶向的基因调控,因此如果所述基因被Cas9切割,则所述一个或多个标记蛋白不受调控,从而导致选择性标记不被活化。因此,可以选择无活化的选择性标记的细胞作为包含调控一种或多种标记蛋白的基因的那些细胞,所述标记蛋白与细胞中的选择性标记融合或连接。例如,Tau可以与CFP融合或连接。另一种Tau蛋白可以与YFP融合或连接。在tau蛋白聚集时,到达CFP的光会以蓝光的形式发射,从而激发YFP,而YFP又会发射黄光。如果tau蛋白未聚集,则CFP发射出的蓝光不能激发另一种tau蛋白的YFP,因此不会发射黄光。因此,如果调控tau的基因被一个或多个gRNA靶向并因此被切割,则不会发射黄光。最后,该基因可以被鉴定为调控Tau(例如导致Tau聚集)的基因。在CRISPRa中,如果gRNA与靶标区域结合,则会活化下游基因,因此可以选择选择性标记过表达或过量的细胞。例如,使用上述Tau蛋白,与无gRNA的细胞相比,黄光的量增加可以表示调控Tau的基因。已知通路(特别是疾病通路)中的存在任何数量的蛋白质可以用作标记蛋白,以鉴定调控该标记蛋白的基因,并且最终可被发现与特定疾病有关。
在一个实施方案中,可以对细胞集进行分类和选择(例如,通过表型)(例如,图2;206),可以在初始感染后进行一段时间,例如,在感染后1、2、3、4、5、6、7、8、9、10、11、12、13、14、15天等等。在一个实施方案中,可以使用多种类型的筛选/选择机制来利用包含在该细胞集中的选择性标记。在一个实施方案中,选择机制包括以下一者或多者:将第二细胞群暴露于药物,或者将所述第二细胞群暴露于识别蛋白质活性或表达水平的物质。在一个实施方案中,选择具有指定表型的细胞包括根据一个或多个选择性标记对细胞进行分选。在一个实施方案中,细胞的活力可以用于选择。
在一个实施方案中,将第二培养物的细胞分类为具有指定表型或不具有指定表型包括鉴定细胞中存在或不存在指定表型。将第二培养物的细胞分类为具有指定表型或不具有指定表型可以包括将选择机制应用于第二细胞培养物。在一个实施方案中,一旦鉴定出表型,即可选择具有指定表型(或不具有指定表型)的细胞。表型是任何可观察的可以在测定法中测量的特征或功能效应,诸如细胞生长、增殖、形态、酶功能、信号转导、表达模式、下游表达模式、报告基因活化、激素释放、生长因子释放、神经递质释放、配体结合、细胞凋亡和产物形成的变化。在一个实施方案中,指定表型可以是荧光或细胞存活。
可以修饰细胞以传递可以直接选择的表型,例如通过标记的基因组整合或通过未整合到基因组中的胞内标记的存在。如本文所用,“标记”最通常是指当存在于细胞中(例如,被表达)时导致使细胞可视化或将细胞鉴定为含有该标记的属性或表型的生物学特征或性状。许多标记类型是常用的,并且可以是例如可视标记(诸如显色,例如lacZ互补(β-半乳糖苷酶)或荧光,例如绿色荧光蛋白(GFP)或GFP融合蛋白的表达)、RFP、BFP、荧光素酶、β-半乳糖苷酶、增强型绿色荧光蛋白(eGFP)、青色荧光蛋白(CFP)、黄色荧光蛋白(YFP)、增强型黄色荧光蛋白(eYFP)、蓝色荧光蛋白(BFP)、增强型蓝色荧光蛋白(eBFP)、DsRed、ZsGreen、MmGFP、mPlum、mCherry、tdTomato、mStrawberry、J-Red、mOrange、mKO、mCitrine、Venus、YPet、Emerald、CyPet、Cerulean、T-Sapphire和碱性磷酸酶、表型标记(生长速率、细胞形态、集落颜色或集落形态、温度敏感性)、营养缺陷标记(生长要求)、抗生素敏感性和抗性、分子标志物(诸如可通过抗原敏感性区分的生物分子(例如,血型抗原和组织相容性标志物)、细胞表面标志物(例如H2KK)、酶标志物和核酸标志物,例如限制性片段长度多态性(RFLP)、单核苷酸多态性(SNP)和各种其他可扩增的遗传多态性)。因此,例如,所述一种或多种选择性标记可以是可检测的酶,诸如β-半乳糖苷酶或萤光素酶
“选择性标记”或“筛选标记”或“正向选择标记”是指当存在于细胞中时,产生允许从不表达选择性标记性状的其他细胞中选择或分离那些细胞的属性或表型的标记。许多基因可以用作选择性标记,例如编码耐药性或营养缺陷型拯救的基因是熟知的。例如,卡那霉素(新霉素)抗性可以用作选择吸纳有携带编码细菌卡那霉素抗性的基因(例如,酶新霉素磷酸转移酶II)的质粒的细菌的性状。当培养物用新霉素或类似的抗生素处理时,未转染的细胞最终会死亡。该细胞集可以用于药物筛选以鉴定赋予耐药性的基因。细胞可以用所关注的药物处理,富集的gRNA与突变时赋予耐药性的基因相关。对病毒或细菌病原体的抗性筛选可以用于鉴定防止感染或病原体复制的基因。与耐药性筛选一样,病原体暴露后的存活提供了强有力的选择。在癌症中,负向选择CRISPR筛选可以鉴定特定癌症亚型中的“癌基因成瘾”,这可以为分子靶向疗法提供基础。对于发育研究,在人和小鼠多能细胞中进行筛选可以精确定位多能性或分化为不同细胞类型所需的基因。
在一个实施方案中,细胞包含选择性标记系统。选择性标记系统可以涉及荧光蛋白FRET生物传感器。例如,Tau可以与CFP融合或连接。另一种Tau蛋白可以与YFP融合或连接。在tau蛋白聚集时,到达CFP的光会以蓝光的形式发射,从而激发YFP,而YFP又会发射黄光。如果tau蛋白未聚集,则CFP发射出的蓝光不能激发另一种tau蛋白的YFP,因此不会发射黄光。因此,如果调控tau的基因被一个或多个gRNA靶向并因此被切割,则不会发射黄光。最后,该基因可以被鉴定为调控Tau(例如导致Tau聚集)的基因。在CRISPRa中,如果gRNA与靶标区域结合,则会活化下游基因,因此可以选择选择性标记过表达或过量的细胞。例如,使用上述Tau蛋白FRET生物传感器,与无gRNA的细胞相比,黄光的量增加可以表示调控Tau的基因。已知通路(特别是疾病通路)中的存在任何数量的蛋白质可以用作标记蛋白,以鉴定调控该标记蛋白的基因,并且最终可被发现与特定疾病通路有关。
在一个实施方案中,可以对第一培养物的细胞和第二培养物的所选细胞进行测序(例如,图2;202和207)。可以在感染后的不同时间对第一培养物的细胞和第二培养物的所选细胞进行测序。可以在感染后的不同时间对第一培养物的细胞和第二培养物的所选细胞进行测序,以在选择前产生读段计数(第一培养物的细胞)并且在选择后产生读段计数(第二培养物的所选细胞)。例如,可以在感染后3天对第一培养物的细胞进行测序,并且可以在感染后10天对第二培养物的所选细胞进行测序。可以使用任何可用的测序技术(诸如NGS)对细胞进行测序。可以对细胞的核酸进行测序以产生序列数据。序列数据可以包括读段计数。序列数据可以包括文库的一个或多个gRNA的读段计数。对细胞进行测序可以产生可以存储在数据结构中的序列数据。数据结构可包含一个或多个核酸序列和/或样品标识符。
测序产生的读段计数受到CRISPR筛选分析中存在的传统偏差的影响,包括重复的频繁缺失、gRNA敲除效率的差异以及读段计数分布的差异。在根据负二项方法、log
读段计数可以归一化。例如,可以对来自不同样品的读段计数进行中位数归一化,以调整文库大小和读段计数分布的影响。在一个实施方案中,假设N个CRISPR/Cas9基因敲除筛选实验对一组M个gRNA进行,并且实验j中的gRNA i的读段计数为x
其中S是对于j=1至M,
其中x^i是gRNA i的读段计数的几何平均值:
或者,每个gRNA和每个基因的读段计数可以使用每百万计数、总数或大小因子归一化来进行归一化。参见Anders,S.和Huber,W.(2010)Differential expressionanalysis for sequence count data.Genome Biol.,11,R106,该文献以引用的方式整体并入本文。
在一个实施方案中,所公开的方法使用序列数据来确定选择后读段计数超过背景阈值的所选细胞中的每个DNA靶标区域的相应的gRNA数(n’)的总和(Σ)(例如图2;203,208)。所述方法还可以包括确定读段计数超过背景阈值的所选细胞的所有靶标区域中的gRNA的总数(N')(例如图2;204、209)。
因此,所公开的方法可以在所选细胞的序列中鉴定来自文库的gRNA的正出现。可以分析包括读段计数的序列数据以确定每个DNA的靶标区域(例如基因)的选择后“存在之和”或n'。“存在之和”,或每个DNA的靶标区域存在的gRNA的相应的数量,可以通过将每个gRNA的单个读段计数与背景阈值进行比较来确定。确定读段计数超过背景阈值的每个DNA靶标区域的gRNA的相应的数量可以由计算设备执行。背景阈值可以是足以降低序列数据中的背景噪声的任何值。例如,30可以用作背景阈值。因此,“存在之和”表示存在的gRNA数量(读段计数超过背景阈值),与读段计数表示的存在的gRNA的数量形成对比。
在一个实施方案中,步骤可以重复任何次数,所述步骤包括:用所述病毒载体的文库感染cas9阳性细胞的第二培养物;将所述第二培养物的所述细胞分类为具有指定表型或不具有指定表型;选择具有指定表型的细胞并且对所述选择的细胞进行测序以获得所述gRNA中的每个的选择后读段计数;对选择后读段计数超过所述背景阈值的所选细胞中的每个DNA靶标区域的相应gRNA数量进行求和(Σ),其中Σ=n’;以及对读段计数超过所述阈值的所选细胞的所有靶标区域中的gRNA总数进行求和(Σ),其中Σ=N’。
在一个实施方案中,所公开的方法还包括根据在所选细胞中偶然观察到基因的n'个或更多个gRNA的概率来鉴定靶标区域是正向选择的(例如图2;211)。在一些实施方案中,鉴定靶标区域是正向选择的包括确定在所选细胞中偶然观察到所关注的序列的n’个或更多个gRNA的概率满足阈值。
在一些实施方案中,所公开的方法还包括根据在所选细胞中偶然观察到基因的n'个或更多个gRNA的概率来将靶标区域鉴定为第二基因的修饰物。在一些实施方案中,所公开的方法还包括根据在所选细胞中偶然观察到基因的n'个或更多个gRNA的概率来将靶标区域鉴定为治疗靶标。在一些实施方案中,所公开的方法还包括根据在所选细胞中偶然观察到基因的n'个或更多个gRNA的概率来鉴定与指定表型相关的靶标区域。在一些实施方案中,所公开的方法还包括根据在所选细胞中偶然观察到基因的n'个或更多个gRNA的概率来将靶标区域鉴定为表现出保护作用。因此,所公开的方法可以帮助鉴定涉及疾病通路、涉及一个或多个其他基因/蛋白质的调控和/或另外与表型相关的靶标区域(例如,基因)。如果候选基因的基因表达调控导致所选的表型发生变化,则DNA的区域(例如,候选基因)可能与所选的表型“相关”。
在一个实施方案中,所公开的方法还包括确定每个gRNA的富集分数。在一些实施方案中,确定每个gRNA的富集分数包括评估N/N’。可以确定富集分数超过阈值。所述阈值可以相对于其他靶标区域的其他富集分数。gRNA偶然存在的具有高富集分数和低概率的靶标区域可以用于将靶标区域鉴定为与表型相关。
在一个示例性实施方案中,所述方法和系统可以在如图3所示和下文所述的计算机301上实施。类似地,所述方法和系统可以利用一个或多个计算机在一个或多个位置执行一种或多种功能。图3是展示用于执行方法的示例性操作环境的方框图。该示例性操作环境仅为操作环境的实例,并非旨在对操作环境结构的使用范围或功能提出任何限制。也不应将操作环境解释为对示例性操作环境中示出的任一部件或其组合有任何依赖性或要求。
本方法和系统可以用许多其他通用或专用计算系统环境或配置操作。可以适于和所述系统和方法一起使用的计算系统、环境和/或配置的实例包括但不限于个人计算机、服务器计算机、膝上型计算机设备和多处理器系统。另外的实例包括机顶盒、可编程消费性电子产品、网络PC、小型计算机、大型计算机、包括上述任何系统或设备的分布式计算环境等。
所述方法和系统的处理可以由软件部件执行。所述系统和方法可在由一个或多个计算机或其他设备执行的计算机可执行指令(例如程序模块)的一般背景下进行描述。通常,程序模块包括执行特定任务或实现特定抽象数据类型的计算机代码、例程、程序、对象、部件、数据结构等。还可在根据网格的分布式计算环境中实践所述方法,其中任务由通过通信网络连接的远程处理设备执行。在分布式计算环境中,程序模块可以位于包括存储器存储设备的本地和远程计算机存储介质中。
此外,所述系统和方法可以通过呈计算机301形式的计算设备来实施。计算机301的部件可以包括但不限于一个或多个处理器303、系统存储器312以及将包括所述一个或多个处理器303的各个系统部件耦合到系统存储器312的系统总线313。该系统可以利用并行计算。
系统总线313代表几种可能类型的总线结构中的一种或多种,包括使用各种总线结构中的任一种的存储器总线或存储器控制器、外围总线、加速图形端口或本地总线。总线313和本说明书中指定的所有总线也可以通过有线或无线网络连接来实施,并且每个子系统,包括一个或多个处理器303、大容量存储设备304、操作系统305、软件306、数据307、网络适配器308、系统存储器312、输入/输出接口310、显示适配器309、显示设备311和人机接口302,可以包含在物理上分开的位置处的一个或多个远程计算设备314a、b、c内,所述远程计算设备通过这种形式的总线连接,实际上实现了完全分布式系统。
计算机301通常包括各种计算机可读介质。示例性可读介质可以是计算机301可存取的任何可用介质,并且包括,例如并且不意味着限制,易失性和非易失性介质、可移动和不可移动介质。系统存储器312包括易失性存储器形式的计算机可读介质,例如随机存取存储器(RAM),和/或非易失性存储器形式的计算机可读介质,例如只读存储器(ROM)。系统存储器312通常包含数据诸如数据307和/或程序模块诸如操作系统305和软件306,其可由一个或多个处理器303立即存取和/或当前由其操作。
在另一个实施方案中,计算机301还可以包括其他可移动/不可移动、易失性/非易失性计算机存储介质。举例而言,图3示出了大容量存储设备304,其可以为计算机301提供计算机代码、计算机可读指令、数据结构、程序模块和其他数据的非易失性存储。例如并且不意味着限制,大容量存储设备304可以是硬盘、可移动磁盘、可移动光盘、磁带盒或其他磁存储设备、闪存卡、CD-ROM、数字通用盘(DVD)或其他光存储器、随机存取存储器(RAM)、只读存储器(ROM)、和/或电可擦可编程只读存储器(EEPROM)。
任选地,任何数量的程序模块均可存储在大容量存储设备304上,包括例如操作系统305和软件306。操作系统305和软件306中的每一个(或其某种组合)均可包括编程元件和软件306。数据307也可以存储在大容量存储设备304上。数据307可以存储在任何一个或多个数据库中。此类数据库的实例包括
在另一个实施方案中,用户可以经由输入设备(未示出)将命令和信息输入到计算机301中。此类输入设备的实例包括但不限于键盘、点击设备(例如“鼠标”)、麦克风、操纵杆、扫描仪、触觉输入设备(例如手套)和/或其他身体覆盖物等等。这些和其他输入设备可以经由人机接口302连接到一个或多个处理器303,所述人机接口联接到系统总线313,但可以通过其他接口和总线结构诸如并行端口、游戏端口、IEEE 1394端口(也称为Firewire端口)、串行端口或通用串行总线(USB)连接。
在又一个实施方案中,显示设备311也可以经由接口诸如显示适配器309连接到系统总线313。可以设想,计算机301可具有多于一个显示适配器309,并且计算机301可具有多于一个显示设备311。例如,显示设备可以是监视器、LCD(液晶显示器)或投影仪。除显示设备311之外,其他输出外围设备可包括诸如扬声器(未示出)和打印机(未示出)的部件,这些部件可经由输入/输出接口310连接到计算机301。所述方法的任何步骤和/或结果可以任何形式输出到输出设备。此类输出可以是任何形式的视觉再现,包括但不限于文本、图形、动画、音频和/或触觉等。显示器311和计算机301可以是一个设备的一部分或者是独立设备。
计算机301可以使用与一个或多个远程计算设备314a、b、c的逻辑连接在联网环境中操作。例如,远程计算设备可以是个人计算机、便携式计算机、智能电话、服务器、路由器、网络计算机、对等设备或其他公用网络节点等。计算机301与远程计算设备314a、b、c之间的逻辑连接可以经由网络315进行,例如局域网(LAN)和/或通用广域网(WAN)。此类网络连接可以通过网络适配器308进行。网络适配器308可以在有线和无线环境中实施。在一个实施方案中,系统存储器312可以存储使得所述一个或多个远程计算设备314a、b、c可经由网络315访问的一个或多个对象。因此,计算机301可以用作基于云的对象存储器。在另一个实施方案中,一个或多个远程计算设备314a、b、c中的一个或多个可以存储使得计算机301和/或所述一个或多个远程计算设备314a、b、c中的另一个可访问的一个或多个对象。因此,所述一个或多个远程计算设备314a、b、c也可以用作基于云的对象存储器。
出于说明的目的,本文以离散方框形式说明了应用程序和其他可执行程序组件例如操作系统305,但是应认识到,此类程序和组件在不同时间驻留在计算设备301的不同存储部件中,并且由计算机的一个或多个处理器303执行。在一个实施方案中,软件306和/或数据307的至少一部分可存储在计算设备301、远程计算设备314a、b、c和/或它们的组合中的一个或多个上和/或在其上执行。因此,软件306和/或数据307可以在云计算环境中操作,据此可以通过网络315(例如,互联网)执行对软件306和/或数据307的访问。此外,在一个实施方案中,数据307可在计算设备301、远程计算设备314a、b、c和/或它们的组合中的一个或多个上同步。
软件306的实现可以存储在某种形式的计算机可读介质上或通过某种形式的计算机可读介质传输。所述方法中的任一种方法均可由计算机可读介质上包括的计算机可读指令来执行。计算机可读介质可以是可由计算机存取的任何可用介质。举例而言并且不意味着限制,计算机可读介质可包括“计算机存储介质”和“通信介质”。“计算机存储介质”包括以用于存储诸如计算机可读指令、数据结构、程序模块或其他数据等信息的任何方法或技术实现的易失性和非易失性、可移动和不可移动介质。示例性计算机存储介质包括但不限于RAM、ROM、EEPROM、闪存或其他存储器技术、CD-ROM、数字通用盘(DVD)或其他光存储器、磁带盒、磁带、磁盘存储器或其他磁存储设备,或可用于存储所需信息并且可由计算机存取的任何其他介质。
软件306可以被构造为执行本文公开的方法的一些或所有步骤。在一个实施方案中,软件306可以被构造为根据对于DNA的多个靶标区域中的每个,在用包含至少3个向导RNA(gRNA)的文库的载体感染后,对第一细胞群进行的测序,确定读段计数超过背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n),根据对于所述DNA的多个靶标区域中的每个的所述相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第一细胞群中的gRNA的总数(N),根据对于所述DNA的多个靶标区域中的每个,在用包含所述至少3个gRNA的文库的载体感染后,对第二细胞群进行的测序,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n’),根据对于所述DNA的多个靶标区域中的每个的所述相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第二细胞群中的gRNA的总数(N’),对于所述多个靶标区域中的每个靶标区域,根据n、N、n’和N’来确定在所选细胞中偶然观察到所述靶标区域的n’个gRNA的概率,对于包含所关注的序列的靶标区域,根据在所选细胞中偶然观察到所述靶标区域的n’个gRNA的概率来确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率,以及根据在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率来鉴定所关注的序列是正向选择的。
根据对于所述DNA的多个靶标区域中的每个的相应gRNA数量,确定读段计数超过背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于第一细胞群中的gRNA的总数(N)可以包括对读段计数超过背景阈值的第一细胞群中存在的每个gRNA进行计数。
根据对于所述DNA的多个靶标区域中的每个的相应gRNA数量确定读段计数超过背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于第二细胞群中的gRNA的总数(N’)可以包括对读段计数超过背景阈值的第二细胞群的所选细胞中存在的每个gRNA进行计数。
对于所述多个靶标区域中的每个靶标区域,根据n、N、n’和N’来确定在所选细胞中偶然观察到靶标区域的n’个向导的概率可以包括评估
对于包含所关注的序列的靶标区域,根据在所选细胞中偶然观察到所述靶标区域的n’个向导的概率来确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率可以包括评估
根据在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率来鉴定所关注的序列是正向选择的可以包括确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率满足阈值。
软件306还可以被构造为确定每个gRNA的富集分数。确定每个gRNA的富集分数可以包括评估N/N'。
软件306可以被构造为存储以数据307存在的gRNA的读段计数和数量。图4展示了代表数据307的实施方案的示例性数据结构410。数据结构410可以包括一个或多个表、数组等等。数据结构410可以包括多个行和多个列。“基因#”列可以含有唯一基因或DNA的靶标区域的标识符。“每个基因的gRNA”列含有可以与基因或DNA的靶标区域结合的独特gRNA的标识符。“gRNA读段计数(选择前)”列含有读段计数的值。“n”列含有来源于“gRNA读段计数(选择前)”列的每个DNA的靶标区域(例如,基因)存在的gRNA的数量。“gRNA读段计数(选择后)”列含有读段计数的值。“n’”列含有来源于“gRNA读段计数(选择后)”列的每个DNA的靶标区域(例如,基因)存在的gRNA的数量。
出于说明的目的,图4的数据结构410显示出包括存储为数据307的人工值。对于基因1,gRNA A、B和C存在于文库中,并且可以与基因1结合。在测序后,gRNA A、B和C存在于细胞中,读段计数分别为100、200和10。背景阈值30可以应用于读段计数,并且它可以由软件306确定来自文库的gRNA的正出现次数为2(n
如图4的数据结构410所示,gRNA的总数或N为12。N来源于每个基因的n值2+3+3+4(n
“基因#”列含有唯一基因或DNA的靶标区域的标识符。“每个基因的gRNA”列含有可以与基因或DNA的靶标区域结合的独特gRNA的标识符。“gRNA读段计数(选择后)”列含有读段计数的值。“n’”列含有来源于“gRNA读段计数(选择后)”列的每个DNA的靶标区域(例如,基因)存在的gRNA的数量。如图4的数据结构410所示,对于基因1,gRNA A、B和C存在于文库中,并且可以与基因1结合。选择后,在测序后,gRNA A、B和C存在(或不存在)于细胞中,读段计数分别为0、50和0。背景阈值30可以应用于读段计数,并且它可以由软件306确定来自文库的gRNA的正出现次数为1(n
读段计数超过背景阈值的所选细胞中存在的所有靶标区域中的gRNA总数或N'可以通过软件306来确定。如图4的数据结构410所示,gRNA的总数或N’为4。N’来源于每个基因的n值1+3+0+0(n’
在一个实施方案中,数据307还可以被构造为存储软件306的一个或多个结果。图5显示了由软件306产生的示例性结果数据结构510,例如,使用数据结构410作为输入。数据结构510可以包括一个或多个表、数组等等。出于说明的目的,图5显示了人工数据和人工结果。给定文库大小、每个基因的向导数和每个实验中的正向导总数,可以计算正式统计p值,以正向观察实验重复中的向导数。数据结构510表示63,950个gRNA(N)的初始文库。数据结构510表示,在选择后,实验1的细胞群中保留了4,946个gRNA(N')(独特gRNA的数量,而不是gRNA的数量)并且实验2的细胞群中保留了13,606个gRNA(N')(独特gRNA的数量,而不是gRNA的数量)。如数据结构510中所示,靶标区域1具有三(3)个能够与靶标区域1的至少一部分结合的gRNA。对于实验1,假设在选择后细胞群中保留4,946个gRNA,3个gRNA中有3个偶然存在于选择后细胞群中的概率为0.000462378。对于实验2,假设在选择后细胞群中保留13,606个gRNA,3个gRNA中有3个偶然存在于选择后细胞群中的概率为0.009629。数据结构510表示3个gRNA中的2个、3个gRNA中的1个和3个gRNA中的0个偶然存在于选择后细胞群中的概率的结果。
如数据结构510所示,靶标区域2具有四(4)个能够与靶标区域2的至少一部分结合的gRNA。数据结构510表示4个gRNA中的4个、4个gRNA中的3个、4个gRNA中的2个、4个gRNA中的1个和4个gRNA中的0个偶然存在于选择后细胞群中的概率的结果。根据数据结构510中所示的结果,软件306可以确定概率低于阈值(例如,足够小)。例如,对于靶标区域2,实验1,4个gRNA中的4个偶然存在的概率为3.57411E-05,这表示4个gRNA中的4个很可能不仅仅是偶然存在的。
实施例
A.实施例1.开发全基因组CRISPR/Cas9筛选平台以鉴定Tau聚集的遗传修饰物
为了鉴定改变异常tau蛋白聚集过程的基因和通路,开发了一个用于使用CRISPR核酸酶(CRISPRn)sgRNA文库进行全基因组筛选的平台,所述筛选用于鉴定调控细胞被tau疾病相关蛋白质聚集体“接种”的潜力的基因(即当暴露于tau原纤维化蛋白质来源时,断裂时会导致细胞更容易形成tau聚集体的基因)。该筛选采用tau生物传感器人细胞系,该细胞系由稳定表达tau四重复序列结构域tau_4RD的HEK293T细胞组成,tau_4RD包含与CFP或YFP融合的P301S致病性突变的tau微管结合结构域(MBD)。也就是说,HEK293T细胞系含有两个稳定表达与荧光蛋白CFP或荧光蛋白YFP融合的疾病相关蛋白变体的转基因:tau4RD-CFP/tau4RD-YFP(TCY),其中tau重复序列结构域(4RD)包含P301S致病性突变。
在这些生物传感器细胞系中,tau-CFP/tau-YFP蛋白聚集产生FRET信号,这是荧光能量从供体CFP转移到受体YFP的结果。FRET阳性细胞含有tau聚集体,可以通过流式细胞术分选和分离。在基线处,未刺激的细胞以稳定、可溶性状态表达报告基因,FRET信号最小。在刺激(例如,种子颗粒的脂质体转染)时,报告蛋白形成聚集体,产生FRET信号。可以通过FACS来分离含有聚集体的细胞。稳定繁殖的含有聚集体的细胞系Agg[+]可以通过Agg[-]细胞系的克隆连续稀释来分离。
对该tau生物传感器细胞系进行若干修饰,使其可用于遗传筛选。首先,通过慢病毒载体引入表达Cas9的转基因(SpCas9)来修饰这些tau生物传感器细胞。用杀稻瘟素来选择表达Cas9的克隆转基因细胞系,并且通过克隆连续稀释分离以获得单细胞衍生的克隆。通过qRT-PCR来评估克隆的Cas9表达水平,并且通过数字PCR来评估DNA切割活性。
具体而言,在转导编码针对两个所选的靶基因的gRNA的慢病毒后3天和7天,通过数字PCR来评估Cas9突变效率。切割效率受到低表达克隆中的Cas9水平的限制。需要具有足够Cas9表达水平的克隆来实现最大活性。若干具有较低Cas9表达的衍生克隆无法有效切割靶标序列,而具有较高表达的克隆(包括用于筛选的克隆)能够在培养三天后在基因PERK和SNCA中的靶标序列处产生突变,效率为大约80%。在gRNA转导后3天已经观察到有效切割,在7天后仅略有改善。克隆7B10-C3被选为用于后续文库筛选的高性能克隆。
然后,开发了使细胞对tau接种活性敏感的试剂和方法。Tau细胞间增殖可能是含有聚集体的细胞分泌的tau聚集活性的结果。为了研究tau聚集的细胞增殖,获得tau-YFP细胞系的亚克隆,该细胞系由稳定表达tau重复序列结构域tau_4RD的HEK293T细胞组成,tau_4RD包含与YFP融合的P301S致病性突变的tau微管结合结构域(MBD)。
通过用重组原纤维化tau与lipofectamine试剂混合处理这些tau-YFP细胞来获得tau-YFP蛋白稳定处于聚集状态(Agg[+])的细胞,从而使这些细胞稳定表达的tau-YFP蛋白的聚集接种。然后将“接种”的细胞连续稀释以获得单细胞衍生的克隆。然后扩展这些克隆以鉴定克隆细胞系,其中tau-YFP聚集体随时间推移在所有细胞中稳定持续生长和多次传代。将这些tau-YFP_Agg[+]克隆之一用于通过收集已经在汇合的tau-YFP_Agg[+]细胞上放置四天的培养基来产生条件培养基。然后将条件培养基(CM)以3:1的CM:新鲜培养基的比率施用于初始生物传感器tau-CFP/Tau-YFP细胞,以在这些受体细胞的一小部分中诱导tau聚集。未使用lipofectamine。不使用lipofectamine是为了进行尽可能符合生理学的测定法,而不使用lipofectamine诱导受体细胞强制/增加tau聚集。如通过使用流式细胞术所测量,条件培养基在大约0.1%的细胞中持续诱导FRET,所述流式细胞术评估产生FRET信号作为聚集的度量的细胞百分比。
B.实施例2.全基因组CRISPR/Cas9筛选以鉴定Tau聚集的遗传修饰物
为了揭示作为FRET(+)细胞中富集的sgRNA的tau聚集的修饰基因,用两个人全基因组CRISPR sgRNA文库(GeCKO A和GeCKO B)来转导无聚集体的表达Cas9的tau-CFP/tau-YFP生物传感器细胞(Agg[–])(图7),使用慢病毒递送方法在每个靶基因处引入敲除突变。每个CRISPR sgRNA文库靶向5'组成型外显子以进行功能性敲除,每个基因平均覆盖约3个sgRNA(两个文库组合中每个基因总共6个gRNA)。每个文库的读段计数分布(即,文库中每个gRNA的呈现)是正常和相似的。sgRNA被设计为通过避免与脱靶基因组序列具有两个或更少的错配的sgRNA来避免脱靶效应。这些文库涵盖19,050个人类基因和1864个miRNA,以及1000个非靶向对照sgRNA。文库以<0.3的感染复数(MOI)转导,覆盖率>300个细胞/sgRNA。Tau生物传感器细胞在嘌呤霉素选择下生长,以选择每个细胞整合和表达独特sgRNA的细胞。嘌呤霉素选择在转导后24小时开始,浓度为1μg/mL。在初级筛选中使用五个独立的筛选重复。
在转导后第3天和第6天的细胞传代时收集完整的转导细胞群的样品。在第6天传代后,细胞在条件培养基中生长以使它们对接种活性敏感。在第10天,使用荧光辅助细胞分选(FACS)来特异性分离FRET[+]细胞的亚群。筛选由五个重复实验组成。整合sgRNA构建体的DNA分离和PCR扩增允许在每个时间点通过下一代测序(NGS)对sgRNA库进行表征。
与较早时间点第3天和第6天的sgRNA库相比,NGS数据的统计分析能够鉴定在五个实验的第10天FRET[+]亚群中富集的sgRNA。鉴定潜在tau修饰物的第一策略是使用DNA测序在每个样品中使用DESeq算法产生sgRNA读段计数,以发现第10天比第3天丰度更高或第10天比第6天丰度更高,但第6天丰度不比第3天高的sgRNA(倍数变化(fc)≥1.5和负二项式检验p<0.01)。Fc≥1.5表示比率(第10天计数的平均值)/(第3天或第6天计数的平均值)≥1.5。P<0.01表示第10天和第3天或第6天计数之间无统计学差异的概率<0.01。DESeq算法是一种用于“序列计数数据的差异表达分析”的广泛使用的算法。参见例如Anders等人(2010)Genome Biology 11:R106,该文献以引用的方式并入本文。
具体而言,在每个文库中使用两个比较来鉴定显著的sgRNA:第10天与第3天和第10天与第6天。对于这四个比较中的每个,使用DESeq算法,并且被视为显著的截止阈值是倍数变化≥1.5以及负二项式检验p<0.01。一旦在每个文库的这些比较中的每个中鉴定出显著的向导,如果基因满足以下两个标准之一,则该基因被认为具有显著性:(1)至少两个对应于该基因的sgRNA被视为在一个比较中具有显著性(第10天与第3天或第10天与第6天);(2)至少一个sgRNA在两个比较中都具有显著性(第10天与第3天和第10天与第6天)。使用该算法,从第一文库中鉴定出五个基因具有显著性,从第二文库中鉴定出四个基因具有显著性。参见表1。
表1.使用策略#1鉴定的基因。
然而,该第一策略要求每个实验组内的一定读段计数同质性水平可能过于严格。对于相同的sgRNA,很多因素都可以在每个实验组中的样品(第3天、第6天或第10天样品)之间产生读段计数差异,诸如筛选文库中的初始病毒计数、感染或基因编辑效率以及基因编辑后的相对生长速率。因此,第二策略还根据第10天(选择后)每个样品中的每个基因的向导的正出现(读段计数>30)而不是精确读段计数来使用。
选择前CRISPR实验重复四次。如图6所示,对于选择前实验“实验1”中的基因“G1”,gRNA“g1”、“g2”和“g3”存在于细胞群中,读段计数分别为121、1000和302。对于基因“G2”,gRNA“g4”、“g5”、“g6”和“g7”存在于细胞群中,读段计数分别为443、2012、534和150。这种读段计数数据是针对基因“G1”至基因“G21,000”而产生的。对于每个基因,确定“存在之和”或n。“存在之和”,或每个DNA的靶标区域存在的gRNA的相应的数量,通过将每个gRNA的单个读段计数与背景阈值进行比较来确定。在这种情况下,将30用作背景阈值。因此,“存在之和”表示定性存在的gRNA数量,与读段计数表示的存在的gRNA的数量形成对比。对应于基因G1的gRNA的存在之和为3,因为gRNA g1、g2和g3的读段计数均超过背景阈值30。对应于基因G2的gRNA的存在之和为4,因为gRNA g4、g5、g6和g7的读段计数均超过背景阈值30。读段计数超过背景阈值的细胞群中存在的所有靶标区域中的gRNA总数或N表示为59,010。因此,在用于感染细胞群的大约64,000个不同的gRNA的原始文库中,只有大约59,000个不同的gRNA的存在数量超过背景阈值。
具有表型选择的CRISPR实验重复四次。然而,在测序之前,使用荧光技术(例如,FRET荧光)根据表型对细胞群中的细胞进行分选。如果Cas9/CRISPR切割细胞的靶标区域(例如,基因)并且细胞不发荧光,则该基因被成功敲除。如果细胞发荧光,则该基因未被敲除。然后可以对发荧光的细胞进行测序,不发荧光的细胞则不进行测序。所选细胞表示表现出特定表型/标记的细胞。
如图6所示,对于选择后实验“实验1”中的基因“G1”,gRNA“g1”、“g2”和“g3”存在(或不存在)于细胞群中,读段计数分别为0、8和12。对于基因“G2”,gRNA“g4”、“g5”、“g6”和“g7”存在(或不存在)于细胞群中,读段计数分别为4、25、4和150。这种读段计数数据是针对基因“G1”至基因“G21,000”而产生的。对于每个基因,确定“存在之和”或n’。“存在之和”,或每个DNA的靶标区域存在的gRNA的相应的数量,通过将每个gRNA的单个读段计数与背景阈值进行比较来确定。在这种情况下,将30用作背景阈值。因此,“存在之和”表示存在的gRNA数量,与读段计数表示的存在的gRNA的数量形成对比。对应于基因G1的gRNA的存在之和为0,因为gRNA g1、g2和g3的读段计数均不超过背景阈值30。对应于基因G2的gRNA的存在之和为1,因为仅gRNA g7的读段计数超过背景阈值30。读段计数超过背景阈值的细胞群中存在的所有靶标区域中的gRNA总数或N’表示为4,320。因此,在用于感染细胞群的大约64,000个不同的gRNA的原始文库中,只有大约4,320个不同的gRNA的存在数量超过背景阈值。
给定文库大小、每个基因的向导数和选择后样品中的正向导总数,计算正式统计p值,以正向观察选择后样品中的向导数。一旦考虑读段计数,则根据公式
一旦确定偶然观察到所选细胞(选择后)中的靶标区域(例如,基因)的n’个向导的概率,则根据公式
一旦确定偶然观察到所选细胞(选择后)中的靶标区域的n′个或更多个gRNA的概率,则在靶标区域水平上确定平均富集gRNA。与选择前择相比,选择后基因的读段计数的总体富集被用作鉴定正基因的附加参数。平均富集以富集分数表示。富集分数通过评估N/N'来确定。如图6所示,富集分数为59010/4320或13.66。
偶然观察到所选细胞(选择后)中的靶标区域的n′个或更多个gRNA的概率可以用于评估靶标区域是否为正向选择的。富集分数可以另外用于评估靶标区域是否为正向选择的。观察到所选细胞中的靶标区域的n′个或更多个gRNA的概率显著低于偶然观察到所选细胞(选择后)中的靶标区域的n′个或更多个gRNA的概率的靶标区域可以被鉴定为正向选择的靶标区域。另外,具有大于阈值的富集分数的靶标区域可以表示正向选择的靶标区域。
因此,该第二策略表示一种新型的、更灵敏的CRISPR正向选择分析方法。CRISPR正向选择的目标是使用DNA测序来鉴定sgRNA扰动与表型相关的基因。为了降低噪声背景,在这些实验中通常使用同一基因的多个sgRNA以及实验重复。然而,目前常用的统计分析方法要求同一基因的sgRNA之间以及技术重复之间具有一定程度的同质性/一致性,效果不佳。这是因为由于很多可能的原因(例如,不同的感染或基因编辑效率、筛选文库中的初始病毒计数以及其他具有相同表型的sgRNA的存在),这些方法无法处理同一基因的sgRNA和重复之间的巨大差异。相比之下,本实施例中所示的方法对较大差异具有稳健性。它根据单个实验中的每个基因的向导的正出现,而不是每个sgRNA的精确读段计数。给定文库大小、每个基因的sgRNA数和每个实验中的正sgRNA总数,计算正式统计p值,以正向观察实验重复中的sgRNA数。表型选择前后的相对sgRNA序列读取富集也被用作参数。该方法的性能优于最新的广泛使用的方法,包括DESeq、MAGECK等。具体而言,该方法包括以下步骤:
(1)对于每个实验,鉴定具有正向表型的细胞中的任何存在的向导。
(2)在基因水平上,计算每个实验中存在向导的随机概率(也称为p值)。在多个实验中存在的总体概率通过Fisher组合概率检验来计算(参考文献:Fisher,R.A.;Fisher,R.A(1948)."Questions and answers#14".The American Statistician)。也就是说,首先使用来自多个实验的p值来计算检验统计量
(3)在基因水平上计算向导的平均富集:富集分数=选择后的相对丰度/选择前的相对丰度。相对丰度=一个基因的向导的读段计数/所有向导的读段计数。
(4)选择显著小于存在的随机概率以及大于一定富集分数的基因。
C.实施例3
CRISPR/Cas9活化和失活诱变被用于使用CRISPRn sgRNA文库(hGeCKO-A和hGeCKO-B)(靶向编码外显子以进行功能敲除)来筛选Tau和α-突触核蛋白原纤维化和增殖的遗传修饰物。图7显示了样品标识符、实验编号、测序前的时间,并且鉴定了用于实验的文库(Gecko A或Gecko B)。
Gecko A文库由大约63,950个gRNA组成。在63,950个gRNA中,56,116个gRNA靶向18,874个基因,其中大部分基因被3个gRNA靶向。在63,950个gRNA中,6,834个gRNA靶向1,795个微RNA,其中大部分微RNA被4个gRNA靶向。
Gecko B文库由大约56,869个gRNA组成。在56,869个gRNA中,55,869个gRNA靶向18,834个基因,其中大部分基因被3个gRNA靶向。在56,869个gRNA中,没有gRNA靶向微RNA。在1,000个gRNA中没有靶标。
在第3天和第6天,未表现出表型。在第10天,样品呈表型阳性。
图8显示了来自Gecko A文库感染的样品的DNA读段计数。每个条柱表示病毒感染的样品。每个样品在第3天(d03)、第6天(d06)或第10天(d10)进行测序,并且每个样品表示gRNA的测序读数,而不是全基因组。
图9显示了来自Gecko A文库感染的样品的读段计数归一化到中位数的情况。通过用gRNA读段计数除以每个样品中的读段计数的总和并除乘以所有样品中的读段计数的总和的中位数来进行归一化。底部柱状图是定性的,表示读段计数大于阈值30;第10天是选择后。鉴于读段计数的总和相似,第3天和第6天的样品具有多个gRNA,而第10天的样品具有更少的gRNA。
图10显示了在给定文库大小、每个基因的gRNA数和每个实验中的正gRNA总数的情况下计算的正式统计p值,以正向观察实验重复中的大量gRNA。显示了Gecko A文库感染并且在第10天测序的五个样品的p值。
实施方案
实施方案1.一种方法,其包括:
(A)用病毒载体的文库感染cas9阳性细胞的第一培养物,所述文库包含至少3个向导RNA(gRNA),用于切割所述细胞的基因组内的DNA的靶标区域;对所述细胞进行测序以获得所述gRNA中的每个的读段计数;
对读段计数超过背景阈值的每个DNA靶标区域的相应gRNA数量进行求和(Σ),其中Σ=n;以及对读段计数超过所述背景阈值的所有靶标区域中的gRNA总数进行求和(Σ),其中Σ=N;
(B)用所述病毒载体的文库感染cas9阳性细胞的第二培养物;
将所述第二培养物的所述细胞分类为具有指定表型或不具有所述指定表型;选择具有所述指定表型的细胞并且对所选细胞进行测序以获得所述gRNA中的每个的选择后读段计数;
对选择后读段计数超过所述背景阈值的所选细胞中的每个DNA靶标区域的相应gRNA数量进行求和(Σ),其中Σ=n’;以及
对读段计数超过所述阈值的所选细胞的所有靶标区域中的gRNA总数进行求和(Σ),其中Σ=N’;(C)对于DNA的靶标区域,根据公式
实施方案2.一种方法,其包括:
(A)用病毒载体的文库感染cas9阳性细胞的第一培养物,所述文库包含至少3个向导RNA(gRNA),用于增强所述细胞的基因组内的DNA的靶标区域的转录;
对所述细胞进行测序以获得所述gRNA中的每个的读段计数;
对读段计数超过背景阈值的每个DNA靶标区域的相应gRNA数量进行求和(Σ),其中Σ=n;以及
对读段计数超过所述背景阈值的所有靶标区域中的gRNA总数进行求和(Σ),其中Σ=N;
(B)用所述病毒载体的文库感染cas9阳性细胞的第二培养物;
将所述第二培养物的所述细胞分类为具有指定表型或不具有所述指定表型;
选择具有所述指定表型的细胞并且对所选细胞进行测序以获得所述gRNA中的每个的选择后读段计数;
对选择后读段计数超过所述背景阈值的所选细胞中的每个DNA靶标区域的相应gRNA数量进行求和(Σ),其中Σ=n’;以及
对读段计数超过所述阈值的所选细胞的所有靶标区域中的gRNA总数进行求和(Σ),其中Σ=N’;
(C)对于DNA的靶标区域,根据公式
对于包含基因的DNA的靶标区域,根据公式
实施方案3.如前述实施方案中任一项所述的方法,其中所述第一培养物根据CRISPR技术感染。
实施方案4.如前述实施方案中任一项所述的方法,其中所述第一培养物的所述cas-9阳性细胞被修饰为含有一个或多个选择性标记。
实施方案5.如前述实施方案中任一项所述的方法,其中所述指定表型是荧光。
实施方案6.如前述实施方案中任一项所述的方法,其中所述指定表型是细胞存活。
实施方案7.如实施方案4所述的方法,其中所述一个或多个选择性标记包括荧光标记。
实施方案8.如实施方案7所述的方法,其中所述荧光标记是FRET生物传感器的一部分。
实施方案9.如实施方案4所述的方法,其中所述一个或多个选择性标记是可检测的酶。
实施方案10.如实施方案9所述的方法,其中所述可检测的酶是β-半乳糖苷酶。
实施方案11.如实施方案9所述的方法,其中所述可检测的酶是荧光素酶。
实施方案12.如前述实施方案中任一项所述的方法,其中靶标区域包含基因。
实施方案13.如前述实施方案中任一项所述的方法,其中将所述第二培养物的细胞分类为具有所述指定表型或不具有所述指定表型包括将选择机制应用于所述第二细胞培养物。
实施方案14.如实施方案13所述的方法,其中所述选择机制包括以下一者或多者:将第二细胞群暴露于药物,或者将所述第二细胞群暴露于识别蛋白质活性或表达水平的物质。
实施方案15.如实施方案4所述的方法,其中选择具有所述指定表型的细胞包括根据所述一个或多个选择性标记对所述细胞进行分选。
实施方案16.如前述实施方案中任一项所述的方法,其还包括根据在所选细胞中偶然观察到所述基因的n′个或更多个gRNA的概率来鉴定所述靶标区域是正向选择的。
实施方案17.如实施方案16所述的方法,其中鉴定所述靶标区域是正向选择的包括确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率满足阈值。
实施方案18.如前述实施方案中任一项所述的方法,其还包括确定每个gRNA的富集分数。
实施方案19.如实施方案18所述的方法,其中确定每个gRNA的所述富集分数包括评估N/N’。
实施方案20.如实施方案2-19中任一项所述的方法,其中所述cas-9阳性细胞包含失活的cas-9。
实施方案21.如实施方案20所述的方法,其中所述失活的cas-9与至少一个转录活化结构域融合。
实施方案22.如前述实施方案中任一项所述的方法,其中DNA的所述靶标区域调控下游基因或蛋白质。
实施方案23.如实施方案22所述的方法,其中调控下游基因或蛋白质包括所述下游基因或蛋白质的活化或抑制。
实施方案24.如前述实施方案中任一项所述的方法,其还包括根据在所选细胞中偶然观察到所述基因的n′个或更多个gRNA的概率来将所述靶标区域鉴定为第二基因的修饰物。
实施方案25.如前述实施方案中任一项所述的方法,其还包括根据在所选细胞中偶然观察到所述基因的n′个或更多个gRNA的概率来将所述靶标区域鉴定为治疗靶标。
实施方案26.如前述实施方案中任一项所述的方法,其还包括根据在所选细胞中偶然观察到所述基因的n′个或更多个gRNA的概率来将所述靶标区域鉴定为与所述指定表型相关。
实施方案27.如前述实施方案中任一项所述的方法,其还包括根据在所选细胞中偶然观察到所述基因的n′个或更多个gRNA的概率来将所述靶标区域鉴定为表现出保护作用。
实施方案28.一种方法,其包括:根据对于DNA的多个靶标区域中的每个,在用包含至少3个向导RNA(gRNA)的文库的载体感染后,对第一细胞群进行的测序,确定读段计数超过背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n);根据对于所述DNA的多个靶标区域中的每个的所述相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第一细胞群中的gRNA的总数(N);根据对于所述DNA的多个靶标区域中的每个,在用包含所述至少3个gRNA的文库的载体感染后,对第二细胞群进行的测序,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n’);
根据对于所述DNA的多个靶标区域中的每个的所述相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第二细胞群中的gRNA的总数(N’);对于所述多个靶标区域中的每个靶标区域,根据n、N、n’和N’来确定在所选细胞中偶然观察到所述靶标区域的n’个gRNA的概率;对于包含所关注的序列的靶标区域,根据在所选细胞中偶然观察到所述靶标区域的n’个gRNA的概率来确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率;以及根据在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率来鉴定所关注的序列是正向选择的。
实施方案29.如实施方案28所述的方法,其中所述第一细胞群和所述第二细胞群包含cas-9阳性细胞。
实施方案30.如实施方案29所述的方法,其中根据对于DNA的多个靶标区域中的每个,在用包含至少3个gRNA的文库的载体感染后,对第一细胞群进行的测序,确定读段计数超过背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n)包括:用所述病毒载体的文库感染所述第一细胞群;对所述第一细胞群的细胞进行测序以获得所述gRNA中的每个的读段计数;以及对于所述DNA的多个靶标区域中的每个,对读段计数超过所述背景阈值的每个gRNA进行计数。
实施方案31.如实施方案30所述的方法,其中根据对于所述DNA的多个靶标区域中的每个的相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第一细胞群中的gRNA的总数(N)包括对读段计数超过所述背景阈值的所述第一细胞群中存在的每个gRNA进行计数。
实施方案32.如实施方案31所述的方法,其中根据对于所述DNA的多个靶标区域中的每个,在用包含所述至少3个gRNA的文库的载体感染后,对第二细胞群进行的测序,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n’)包括:将所述第二细胞群的细胞分类为具有指定表型或不具有所述指定表型;
选择具有所述指定表型的细胞;对所选细胞进行测序以获得所述gRNA中的每个的读段计数;以及对于所述DNA的多个靶标区域中的每个,对读段计数超过所述背景阈值的每个gRNA进行计数。
实施方案33.如实施方案28-32中任一项所述的方法,其中根据对于所述DNA的多个靶标区域中的每个的相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第二细胞群中的gRNA的总数(N’)包括对读段计数超过所述背景阈值的所述第二细胞群的所选细胞中存在的每个gRNA进行计数。
实施方案34.如实施方案28-33中任一项所述的方法,其中对于所述多个靶标区域中的每个靶标区域,根据n、N、n’和N’来确定在所选细胞中偶然观察到靶标区域的n’个gRNA的概率包括评估
实施方案35.如实施方案28-34中任一项所述的方法,其中对于包含所关注的序列的靶标区域,根据在所选细胞中偶然观察到所述靶标区域的n’个gRNA的概率来确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率包括评估
实施方案36.如实施方案28-35中任一项所述的方法,其中根据在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率来鉴定所关注的序列是正向选择的包括确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率满足阈值。
实施方案37.如实施方案28-36中任一项所述的方法,其还包括确定每个gRNA的富集分数。
实施方案38.如实施方案37所述的方法,其中确定每个gRNA的富集分数包括评估N/N’。
实施方案39.如实施方案28-38中任一项所述的方法,其中所述cas-9阳性细胞包含失活的cas-9。
实施方案40.如实施方案39所述的方法,其中所述失活的cas-9与至少一个转录活化结构域融合。
实施方案41.一种装置,其包括:一个或多个处理器;和存储处理器可执行指令的存储器,当由所述一个或多个处理器执行时,所述处理器可执行指令使得所述装置:(A)接收第一读段计数数据,其中所述第一读段计数数据通过以下方式产生:用病毒载体的文库感染cas9阳性细胞的第一培养物,所述文库包含至少3个向导RNA(gRNA),用于切割所述细胞的基因组内的DNA的靶标区域,以及对所述细胞进行测序以获得所述gRNA中的每个的读段计数;根据所述第一读段计数数据,对读段计数超过背景阈值的每个DNA靶标区域的相应gRNA数量进行求和(Σ),其中Σ=n;以及根据所述第一读段计数数据,对读段计数超过所述背景阈值的所有靶标区域中的gRNA总数进行求和(Σ),其中Σ=N;(B)接收第二读段计数数据,其中所述第二读段计数数据通过以下方式产生:用所述病毒载体的文库感染cas9阳性细胞的第二培养物,将所述第二培养物的所述细胞分类为具有指定表型或不具有所述指定表型,以及选择具有所述指定表型的细胞并且对所选细胞进行测序以获得所述gRNA中的每个的选择后读段计数;根据所述第二读段计数数据,对选择后读段计数超过所述背景阈值的所选细胞中的每个DNA靶标区域的相应gRNA数量进行求和(Σ),其中Σ=n’;以及根据所述第二读段计数数据,对读段计数超过所述阈值的所选细胞的所有靶标区域中的gRNA总数进行求和(Σ),其中Σ=N’;(C)对于DNA的靶标区域,根据公式
实施方案42.如实施方案41所述的装置,其中所述第一培养物根据CRISPR技术感染。
实施方案43.如实施方案41-42中任一项所述的装置,其中所述第一培养物的所述cas-9阳性细胞被修饰为含有一个或多个选择性标记。
实施方案44.如实施方案41-43中任一项所述的装置,其中靶标区域包含基因。
实施方案45.如实施方案41-44中任一项所述的装置,其中将所述第二培养物的细胞分类为具有所述指定表型或不具有所述指定表型包括将选择机制应用于所述第二细胞培养物。
实施方案46.如实施方案45所述的装置,其中所述选择机制包括以下一者或多者:将第二细胞群暴露于药物,或者将所述第二细胞群暴露于识别蛋白质活性或表达水平的物质。
实施方案47.如实施方案41-46中任一项所述的装置,其中选择具有所述指定表型的细胞包括根据所述一个或多个选择性标记对所述细胞进行分选。
实施方案48.如实施方案41-47中任一项所述的装置,其还包括根据在所选细胞中偶然观察到所述基因的n′个或更多个gRNA的概率来鉴定所述靶标区域是正向选择的。
实施方案49.如实施方案48所述的装置,其中鉴定所述靶标区域是正向选择的包括确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率满足阈值。
实施方案50.如实施方案41-49中任一项所述的装置,其还包括确定每个gRNA的富集分数。
实施方案51.如实施方案50所述的装置,其中确定每个gRNA的所述富集分数包括评估N/N’。
实施方案52.如实施方案41-51中任一项所述的装置,其中所述cas-9阳性细胞包含失活的cas-9。
实施方案53.如实施方案52所述的装置,其中所述失活的cas-9与至少一个转录活化结构域融合。
实施方案54.一种用于确定偶然观察到一个或多个向导RNA(gRNA)的概率的非暂时性计算机可读介质,所述非暂时性计算机可读介质存储处理器可执行指令,所述处理器可执行指令在由一个或多个处理器执行时,使得所述一个或多个处理器:(A)接收第一读段计数数据,其中所述第一读段计数数据通过以下方式产生:用病毒载体的文库感染cas9阳性细胞的第一培养物,所述文库包含至少3个向导RNA(gRNA),用于切割所述细胞的基因组内的DNA的靶标区域,以及对所述细胞进行测序以获得所述gRNA中的每个的读段计数;根据所述第一读段计数数据,对读段计数超过背景阈值的每个DNA靶标区域的相应gRNA数量进行求和(Σ),其中Σ=n;以及根据所述第一读段计数数据,对读段计数超过所述背景阈值的所有靶标区域中的gRNA总数进行求和(Σ),其中Σ=N;(B)接收第二读段计数数据,其中所述第二读段计数数据通过以下方式产生:用所述病毒载体的文库感染cas9阳性细胞的第二培养物,将所述第二培养物的所述细胞分类为具有指定表型或不具有所述指定表型,以及选择具有所述指定表型的细胞并且对所选细胞进行测序以获得所述gRNA中的每个的选择后读段计数;根据所述第二读段计数数据,对选择后读段计数超过所述背景阈值的所选细胞中的每个DNA靶标区域的相应gRNA数量进行求和(Σ),其中Σ=n’;以及根据所述第二读段计数数据,对读段计数超过所述阈值的所选细胞的所有靶标区域中的gRNA总数进行求和(Σ),其中Σ=N’;(C)对于DNA的靶标区域,根据公式
实施方案55.如实施方案54所述的非暂时性计算机可读介质,其中所述第一培养物根据CRISPR技术感染。
实施方案56.如实施方案54-55中任一项所述的非暂时性计算机可读介质,其中所述第一培养物的所述cas-9阳性细胞被修饰为含有一个或多个选择性标记。
实施方案57.如实施方案54-56中任一项所述的非暂时性计算机可读介质,其中靶标区域包含基因。
实施方案58.如实施方案54-57中任一项所述的非暂时性计算机可读介质,其中将所述第二培养物的细胞分类为具有所述指定表型或不具有所述指定表型包括将选择机制应用于所述第二细胞培养物。
实施方案59.如实施方案58所述的非暂时性计算机可读介质,其中所述选择机制包括以下一者或多者:将第二细胞群暴露于药物,或者将所述第二细胞群暴露于识别蛋白质活性或表达水平的物质。
实施方案60.如实施方案54-59中任一项所述的非暂时性计算机可读介质,其中选择具有所述指定表型的细胞包括根据所述一个或多个选择性标记对所述细胞进行分选。
实施方案61.如实施方案54-60中任一项所述的非暂时性计算机可读介质,其还包括根据在所选细胞中偶然观察到所述基因的n′个或更多个gRNA的概率来鉴定所述靶标区域是正向选择的。
实施方案62.如实施方案61所述的非暂时性计算机可读介质,其中鉴定所述靶标区域是正向选择的包括确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率满足阈值。
实施方案63.如实施方案54-62中任一项所述的非暂时性计算机可读介质,其还包括确定每个gRNA的富集分数。
实施方案64.如实施方案63所述的非暂时性计算机可读介质,其中确定每个gRNA的所述富集分数包括评估N/N’。
实施方案65.如实施方案54-64中任一项所述的非暂时性计算机可读介质,其中所述cas-9阳性细胞包含失活的cas-9。
实施方案66.如实施方案65所述的非暂时性计算机可读介质,其中所述失活的cas-9与至少一个转录活化结构域融合。
实施方案67.一种装置,其包括:一个或多个处理器;和存储处理器可执行指令的存储器,当由所述一个或多个处理器执行时,所述处理器可执行指令使得所述装置:根据对于DNA的多个靶标区域中的每个,在用包含至少3个向导RNA(gRNA)的文库的载体感染后,对第一细胞群进行的测序,确定读段计数超过背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n);根据对于所述DNA的多个靶标区域中的每个的所述相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第一细胞群中的gRNA的总数(N);根据对于所述DNA的多个靶标区域中的每个,在用包含所述至少3个gRNA的文库的载体感染后,对第二细胞群进行的测序,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n’);根据对于所述DNA的多个靶标区域中的每个的所述相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第二细胞群中的gRNA的总数(N’);对于所述多个靶标区域中的每个靶标区域,根据n、N、n’和N’来确定在所选细胞中偶然观察到所述靶标区域的n’个gRNA的概率;对于包含所关注的序列的靶标区域,根据在所选细胞中偶然观察到所述靶标区域的n’个gRNA的概率来确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率;以及根据在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率来鉴定所关注的序列是正向选择的。
实施方案68.如实施方案67所述的装置,其中所述第一细胞群和所述第二细胞群包含cas-9阳性细胞。
实施方案69.如实施方案67-68中任一项所述的装置,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置根据对于DNA的多个靶标区域中的每个,在用包含至少3个gRNA的文库的载体感染后,对第一细胞群进行的测序,确定读段计数超过背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n),使得所述装置:接收第一读段计数数据,所述第一读段计数数据通过以下方式产生:用所述病毒载体的文库感染所述第一细胞群,以及对所述第一细胞群的细胞进行测序以获得所述gRNA中的每个的读段计数;以及根据所述第一读段计数数据,对于所述DNA的多个靶标区域中的每个,对读段计数超过所述背景阈值的每个gRNA进行计数。
实施方案70.如实施方案69所述的装置,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置根据对于所述DNA的多个靶标区域中的每个的相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第一细胞群中的gRNA的总数(N)使得所述装置对读段计数超过所述背景阈值的所述第一细胞群中存在的每个gRNA进行计数。
实施方案71.如实施方案67-70中任一项所述的装置,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置根据对于所述DNA的多个靶标区域中的每个,在用包含所述至少3个gRNA的文库的载体感染后,对第二细胞群进行的测序,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n’),使得所述装置:接收第二读段计数数据,所述第二读段计数数据通过以下方式产生:将所述第二细胞群的细胞分类为具有指定表型或不具有所述指定表型;选择具有所述指定表型的细胞;对所选细胞进行测序以获得所述gRNA中的每个的读段计数;以及根据所述第二读段计数数据,对于所述DNA的多个靶标区域中的每个,对读段计数超过所述背景阈值的每个gRNA进行计数。
实施方案72.如实施方案67-71中任一项所述的装置,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置根据对于所述DNA的多个靶标区域中的每个的相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第二细胞群中的gRNA的总数(N’)使得所述装置对读段计数超过所述背景阈值的所述第二细胞群的所选细胞中存在的每个gRNA进行计数。
实施方案73.如实施方案67-72中任一项所述的装置,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置对于所述多个靶标区域中的每个靶标区域,根据n、N、n’和N’来确定在所选细胞中偶然观察到靶标区域的n’个gRNA的概率使得所述装置评估
实施方案74.如实施方案67-73中任一项所述的装置,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置对于包含所关注的序列的靶标区域,根据在所选细胞中偶然观察到所述靶标区域的n’个gRNA的概率来确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率使得所述装置评估
实施方案75.如实施方案67-74中任一项所述的装置,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置根据在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率来鉴定所关注的序列是正向选择的使得所述装置评估确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率满足阈值。
实施方案76.如实施方案67-76中任一项所述的装置,其还包括处理器可执行指令,所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置确定每个gRNA的富集分数。
实施方案77.如实施方案76所述的装置,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置确定每个gRNA的富集分数,所述处理器可执行指令使得所述装置评估N/N'。
实施方案78.如实施方案67-77中任一项所述的装置,其中所述cas-9阳性细胞包含失活的cas-9。
实施方案79.如实施方案78所述的装置,其中所述失活的cas-9与至少一个转录活化结构域融合。
实施方案80.一种用于确定偶然观察到一个或多个向导RNA(gRNA)的概率的非暂时性计算机可读介质,所述非暂时性计算机可读介质存储处理器可执行指令,所述处理器可执行指令在由一个或多个处理器执行时,使得所述一个或多个处理器:根据对于DNA的多个靶标区域中的每个,在用包含至少3个向导RNA(gRNA)的文库的载体感染后,对第一细胞群进行的测序,确定读段计数超过背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n);根据对于所述DNA的多个靶标区域中的每个的所述相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第一细胞群中的gRNA的总数(N);根据对于所述DNA的多个靶标区域中的每个,在用包含所述至少3个gRNA的文库的载体感染后,对第二细胞群进行的测序,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n’);根据对于所述DNA的多个靶标区域中的每个的所述相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第二细胞群中的gRNA的总数(N’);对于所述多个靶标区域中的每个靶标区域,根据n、N、n’和N’来确定在所选细胞中偶然观察到所述靶标区域的n’个gRNA的概率;对于包含所关注的序列的靶标区域,根据在所选细胞中偶然观察到所述靶标区域的n’个gRNA的概率来确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率;以及
根据在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率来鉴定所关注的序列是正向选择的。
实施方案81.如实施方案80所述的非暂时性计算机可读介质,其中所述第一细胞群和所述第二细胞群包含cas-9阳性细胞。
实施方案82.如实施方案80-81中任一项所述的非暂时性计算机可读介质,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置根据对于DNA的多个靶标区域中的每个,在用包含至少3个gRNA的文库的载体感染后,对第一细胞群进行的测序,确定读段计数超过背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n),使得所述一个或多个处理器:接收第一读段计数数据,所述第一读段计数数据通过以下方式产生:用所述病毒载体的文库感染所述第一细胞群,以及对所述第一细胞群的细胞进行测序以获得所述gRNA中的每个的读段计数;以及根据所述第一读段计数数据,对于所述DNA的多个靶标区域中的每个,对读段计数超过所述背景阈值的每个gRNA进行计数。
实施方案83.如实施方案80-82中任一项所述的非暂时性计算机可读介质,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置根据对于所述DNA的多个靶标区域中的每个的相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第一细胞群中的gRNA的总数(N)使得所述一个或多个处理器对读段计数超过所述背景阈值的所述第一细胞群中存在的每个gRNA进行计数。
实施方案84.如实施方案80-83中任一项所述的非暂时性计算机可读介质,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置根据对于所述DNA的多个靶标区域中的每个,在用包含所述至少3个gRNA的文库的载体感染后,对第二细胞群进行的测序,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的每个所存在的相应gRNA数量(n’),使得所述一个或多个处理器:接收第二读段计数数据,所述第二读段计数数据通过以下方式产生:将所述第二细胞群的细胞分类为具有指定表型或不具有所述指定表型;选择具有所述指定表型的细胞;对所选细胞进行测序以获得所述gRNA中的每个的读段计数;以及根据所述第二读段计数数据,对于所述DNA的多个靶标区域中的每个,对读段计数超过所述背景阈值的每个gRNA进行计数。实施方案85.如实施方案80-84中任一项所述的非暂时性计算机可读介质,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置根据对于所述DNA的多个靶标区域中的每个的相应gRNA数量,确定读段计数超过所述背景阈值的所述DNA的多个靶标区域中的所有靶标区域中存在于所述第二细胞群中的gRNA的总数(N)使得所述一个或多个处理器对读段计数超过所述背景阈值的所述第二细胞群的所选细胞中存在的每个gRNA进行计数。
实施方案86.如实施方案80-85中任一项所述的非暂时性计算机可读介质,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置对于所述多个靶标区域中的每个靶标区域,根据n、N、n’和N’来确定在所选细胞中偶然观察到靶标区域的n’个gRNA的概率使得所述一个或多个处理器评估
实施方案87.如实施方案80-86中任一项所述的非暂时性计算机可读介质,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置对于包含所关注的序列的靶标区域,根据在所选细胞中偶然观察到所述靶标区域的n’个gRNA的概率来确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率使得所述一个或多个处理器评估
实施方案88.如实施方案80-87中任一项所述的非暂时性计算机可读介质,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置根据在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率来鉴定所关注的序列是正向选择的使得所述一个或多个处理器评估确定在所选细胞中偶然观察到所关注的序列的n′个或更多个gRNA的概率满足阈值。
实施方案89.如实施方案80-88中任一项所述的非暂时性计算机可读介质,其还包括处理器可执行指令,所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述一个或多个处理器确定每个gRNA的富集分数。
实施方案90.如实施方案89所述的非暂时性计算机可读介质,其中所述处理器可执行指令在由所述一个或多个处理器执行时,使得所述装置确定每个gRNA的富集分数包括使得所述一个或多个处理器评估N/N'。
实施方案91.如实施方案80-90中任一项所述的非暂时性计算机可读介质,其中所述cas-9阳性细胞包含失活的cas-9。
实施方案92.如实施方案91所述的非暂时性计算机可读介质,其中所述失活的cas-9与至少一个转录活化结构域融合。
本领域的技术人员将认识到或能够使用不超过常规实验来确定本文所描述的方法和组合物的具体实施方案的许多等同物。此类等同物旨在被以下权利要求所涵盖。
- 用于CRISPR选择的方法和系统
- 一种适用于CRISPR/Cas9系统的基因组DNA片段编辑精准度的分析方法及应用