一种组织粘性可吸收材料及其制备方法与应用
文献发布时间:2024-04-18 20:01:23
技术领域
本发明涉及一种组织粘性可吸收材料,特别是涉及一种组织粘性可吸收材料及其制备方法与应用。
背景技术
每年数以千万计的人遭受各种各样的组织创伤,包括皮肤割伤、慢性伤口(如糖尿病溃疡)及手术切口等。缝合和钉合是应对创伤的传统手术方式,但会造成组织额外损伤且不能有效防止体液和气体渗漏。组织粘合材料可以在伤口部位进行密封,在防止渗漏的同时还可提供机械支撑和止血作用,为手术干预创伤提供了有效的方案。
目前,现有的组织粘合材料有着明显的化学特性缺点,在临床上的应用仍然有限。如纤维蛋白类粘合剂虽然被认为是金标准,但其粘性低,吻合口易发生渗漏;氰基丙烯酸酯类粘合剂具有很强的稳定性,但被认为有毒性,会引起明显的炎症反应,且需要额外的固化过程,临床使用不便。理想的组织粘合材料应具有生物相容性、生物可降解性、无毒性且能与组织产生坚固的粘连。
发明内容
本发明要解决的技术问题是提供一种组织粘性可吸收材料及其制备方法,该组织粘性可吸收材料具有生物相容性、生物可降解性、无毒性,还具有较好的组织粘性,无需额外固化过程即可直接粘连创伤组织,可广泛应用于组织创面的粘合,如皮肤、肺、胃、肠、胰腺等创面的粘合与愈合。
为解决上述技术问题,本发明的技术方案是:
一种组织粘性可吸收材料,其分子结构为多臂分子链结构,其结构单元由乙交酯、丙交酯、己内酯、β-丙内酯、环氧乙烷或环氧丙烷中的其中一种或多种单体聚合而成。
进一步地,本发明所述的一种组织粘性可吸收材料,其分子链由反应型基团封端,反应型基团为N-羟基丁二酰亚胺-NHS、醛基-CHO、异氰酸酯–NCO或巯基-SH中的其中一种。
进一步地,本发明所述的一种组织粘性可吸收材料,其多臂分子链结构是由引发剂引发聚合而成,引发剂为乙二醇、丙三醇、季戊四醇、赤藓糖醇、双季戊四醇或三季戊四醇中的其中一种。
进一步地,本发明所述的一种组织粘性可吸收材料,其多臂分子链结构中的单臂分子结构链段为聚丙交酯链段、聚乙交脂链段、聚己内酯链段、聚乙二醇链段、聚(丙交酯-co-乙交酯)链段、聚(丙交酯-co-己内酯)链段、聚(丙交酯-co-乙二醇)链段、聚(乙交酯-co-己内酯)链段、聚(乙交酯-co-乙二醇)链段、聚(乙二醇-co-己内酯)链段、聚(丙交酯-co-乙交酯-co-己内酯)链段、聚(丙交酯-co-乙交酯-co-乙二醇)链段、聚(丙交酯-co-己内酯-co-乙二醇)链段、聚(乙交酯-co-己内酯-co-乙二醇)链段、聚(丙交酯-co-乙交酯-co-己内酯-co-乙二醇)链段,聚(乙交酯-co-己内酯-co-丙二醇)链段或聚(丙交酯-co-β-丙内酯-co-丙二醇)链段中的其一种,由乙交酯、丙交酯、己内酯、β-丙内酯、环氧乙烷、环氧丙烷中的任意两种以上的单体共聚而成。
进一步地,本发明所述的一种组织粘性可吸收材料,其数均分子量为1000-200000g/mol,更进一步地,其数均分子量为2000-100000g/mol。
上述组织粘性可吸收材料的制备方法包括以下步骤:
S1.当引发剂为丙三醇、双季戊四醇、三季戊四醇时,按照1:(36.5-59.02)的摩尔比将单体加入引发剂中,室温至160℃下共混10-20分钟得到共混物;
当引发剂为乙二醇、季戊四醇、赤藓糖醇时,按照1mmol:(1-8)mmol:(60-120)mL的比例将引发剂、氢化钾溶解于四氢呋喃中,室温搅拌反应4小时得到引发剂溶液,将环氧乙烷或环氧丙烷加入引发剂溶液中,室温搅拌反应24小时,然后加入其他单体,80℃下蒸干溶剂后共混10-20分钟得到共混物,引发剂与单体的摩尔比为1:(28.6-95.49);
S2.按照0.0045:1的重量比将辛酸亚锡加入步骤S1得到的共混物中,100-140℃下反应20-24小时得到粗制预产物;
S3.按照1g:(1-2)mL的比例将步骤S2得到的粗制预产物溶解于二氯甲烷中,然后用乙醚0-5℃下洗涤纯化粗制预产物3-5次,室温真空干燥12-24小时除去溶剂得到预产物一,所述预产物一为分子链段的端基为羟基的聚合物;
S4.将步骤S3得到的预产物一溶解于二氯甲烷中,0-5℃下加入丁二酸酐和4-二甲氨基吡啶,15-30℃下搅拌反应20-30小时得到预产物二,预产物一、二氯甲烷、丁二酸酐、4-二甲氨基吡啶的比例为(0.2-0.5)g:1mL:(4-35)mmol:(1.2-12)mmol;
S5.将步骤S4得到的预产物二用乙醚0-5℃下洗涤纯化3-5次,室温真空干燥12-24小时除去溶剂得到预产物三,所述预产物三为分子链段的端基为羧基的聚合物;
S6.将步骤S5得到的预产物三溶解于二氯甲烷中,0-5℃下加入N-羟基丁二酰亚胺、4-二甲氨基吡啶和N,N'-二异丙基碳二亚胺,15-30℃下搅拌反应20-30小时得到预产物四,预产物三、二氯甲烷、N-羟基丁二酰亚胺、4-二甲氨基吡啶、N,N'-二异丙基碳二亚胺的比例为(0.2-0.5)g:1mL:(4-35)mmol:(1.2-12)mmol:(4-35)mmol;
S7.将步骤S6得到的预产物四用乙醚0-5℃下洗涤纯化3-5次,室温真空干燥12-24小时除去溶剂得到分子链段的端基为N-羟基丁二酰亚胺-NHS的组织粘性可吸收材料;
S8.将步骤S3得到的预产物一溶解于二氯甲烷中,加入4-甲酰基苯甲酸和4-二甲氨基吡啶,混合均匀后0-5℃下加入二环己基碳二亚胺,搅拌反应24小时得到预产物五,预产物一、二氯甲烷、4-甲酰基苯甲酸、4-二甲氨基吡啶、二环己基碳二亚胺的比例为(0.2-0.5)g:1mL:(4-35)mmol:(1.2-12)mmol:(4-35)mmol;将预产物五用乙醚0-5℃下洗涤纯化3-5次,室温真空干燥12-24小时除去溶剂得到分子链段的端基为醛基-CHO的组织粘性可吸收材料;
S9.将步骤S3得到的预产物一溶解于甲苯中得到浓度为0.2-0.5g/mL的预产物一甲苯溶液,将预产物一甲苯溶液加入浓度为0.2-0.5g/mL的六亚甲基二异氰酸酯甲苯溶液中,预产物一与六亚甲基二异氰酸酯的摩尔比为1:(4-30),搅拌均匀后加入辛酸亚锡,辛酸亚锡的重量为预产物一甲苯溶液与六亚甲基二异氰酸酯甲苯溶液重量之和的0.45%,60℃下搅拌反应2小时得到预产物六;将预产物六用乙醚0-5℃下洗涤纯化3-5次,室温真空干燥12-24小时除去溶剂得到分子链段的端基为异氰酸酯-NCO的组织粘性可吸收材料;
S10.将步骤S3得到的预产物一、二硫代二丙酸和4-二甲氨基吡啶溶解于二氯甲烷中,混合均匀后0-5℃下加入N,N'-二异丙基碳二亚胺,搅拌反应8小时后得到反应物一,将反应物一用0-5℃的甲醇洗涤三次得到固体粉末,预产物一、二氯甲烷、4-二甲氨基吡啶、N,N'-二异丙基碳二亚胺、甲醇的比例为(0.2-0.5)g:1mL:(1.2-12)mmol:(4-35)mmol:(10-15)mL,预产物一、二硫代二丙酸的摩尔比为1:(4-30);
S11.将步骤S10得到的固体粉末、二硫苏糖醇溶于二氯甲烷中,搅拌反应8小时后得到反应物二,将反应物二用0-5℃的甲醇洗涤三次得到分子链段的端基为巯基-SH的组织粘性可吸收材料,固体粉末、二氯甲烷、甲醇的比例为(0.5-1)g:1mL:(10-15)mL,固体粉末、二硫苏糖醇的摩尔比为1:(2-10)。
进一步地,本发明所述步骤S1中,单体为乙交酯、丙交酯、己内酯、β-丙内酯、环氧乙烷或环氧丙烷中的其中一种或多种。
本发明还提供了上述组织粘性可吸收材料在制备组织粘合材料中的应用。
与现有技术相比,本发明具有以下有益效果:
(1)本发明通过具有多臂羟基的引发剂引发环氧基团开环聚合获得聚合物材料,经化学改性获得活性基团封端,活性基团中的N-羟基丁二酰亚胺、醛基能与人体组织中蛋白质的氨基和巯基发生反应产生粘结,异氰酸酯能与人体组织中蛋白质的氨基和羟基发生反应产生粘结,巯基能与人体组织中蛋白质的氨基发生反应产生粘结,因此本发明制得的组织粘性可吸收材料的组织粘性较好且临床使用方便,将材料直接涂抹至组织创面部位,按压后即可粘合。
(2)本发明所使用的原料皆为生物相容性好的材料,且原料易得,成本低廉,适合工业化生产,制得的组织粘性可吸收材料具有生物相容性、生物可降解性、无毒性。
附图说明
此处所说明的附图用来提供对本发明的进一步理解,构成本发明的一部分,并不构成对本发明的不当限定,在附图中:
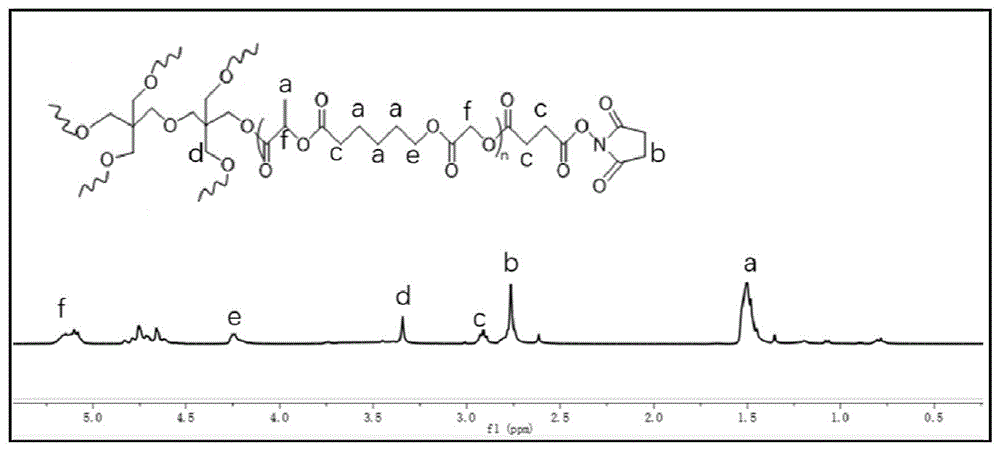
图1为本发明实施例1制得的组织粘性可吸收材料的分子结构示意图及核磁共振氢谱图;
图2为本发明实施例2制得的组织粘性可吸收材料的分子结构示意图及核磁共振氢谱图;
图3为本发明实施例3制得的组织粘性可吸收材料的分子结构示意图及核磁共振氢谱图;
图4为本发明实施例4制得的组织粘性可吸收材料的分子结构示意图及核磁共振氢谱图;
图5为本发明实施例5制得的组织粘性可吸收材料的分子结构示意图及核磁共振氢谱图;
图6为本发明实施例6制得的组织粘性可吸收材料的分子结构示意图及核磁共振氢谱图;
图7为本发明实验例1的抗破裂压力测试图;
图8为本发明实验例1中内皮细胞置于含实施例2组织粘性可吸收材料培养皿培养24小时后的显微照片;
图9为本发明实验例3的体外降解测试图。
具体实施方式
下面将结合具体实施例来详细说明本发明,在此本发明的示意性实施例及其说明用来解释本发明,但并不作为对本发明的限定。
实施例1
利用双季戊四醇作为引发剂引发丙交酯、乙交脂、己内酯开环聚合,通过化学改性获得端基为N-羟基丁二酰亚胺-NHS,数均分子量为6000g/mol的组织粘性可吸收材料,其步骤如下:
S1.将13.79mmol丙交酯、13.79mmol乙交脂和13.79mmol己内酯加入1mmol双季戊四醇中,160℃下共混10分钟得到共混物;
S2.按照0.0045:1的重量比将辛酸亚锡加入步骤S1得到的共混物中,130℃下反应20小时得到粗制预产物;
S3.将步骤S2得到的粗制预产物溶解于10mL二氯甲烷中,然后用乙醚0℃下洗涤纯化粗制预产物3次,室温真空干燥12小时除去溶剂得到预产物一,预产物一为包括双季戊四醇段、聚(丙交酯-co-乙交脂-co-己内酯)链的端基为羟基的聚合物;
S4.将步骤S3得到的预产物一溶解于二氯甲烷中,0℃下加入丁二酸酐和4-二甲氨基吡啶,25℃下搅拌反应24小时得到预产物二,预产物一、二氯甲烷、丁二酸酐、4-二甲氨基吡啶的比例为0.2g:1mL:8mmol:2.7mmol;
S5.将步骤S4得到的预产物二用乙醚0℃下洗涤纯化3次,室温真空干燥12小时除去溶剂得到预产物三,预产物三为包括双季戊四醇段、聚(丙交酯-co-乙交脂-co-己内酯)链的端基为羧基的聚合物;
S6.将步骤S5得到的预产物三溶解于二氯甲烷中,0℃下加入N-羟基丁二酰亚胺、4-二甲氨基吡啶和N,N'-二异丙基碳二亚胺,25℃下搅拌反应24小时得到预产物四,预产物三、二氯甲烷、N-羟基丁二酰亚胺、4-二甲氨基吡啶、N,N'-二异丙基碳二亚胺的比例为0.2g:1mL:9mmol:2.7mmol:9mmol;
S7.将步骤S6得到的预产物四用乙醚0℃下洗涤纯化3次,室温真空干燥12小时除去溶剂得到数均分子量为6000g/mol,包括双季戊四醇段、聚(丙交酯-co-乙交脂-co-己内酯)链的端基为N-羟基丁二酰亚胺-NHS的组织粘性可吸收材料,其分子结构示意图及核磁共振氢谱图如图1所示。
实施例2
利用丙三醇作为引发剂引发丙交酯、己内酯开环聚合,通过化学改性获得端基为N-羟基丁二酰亚胺-NHS,数均分子量为8000g/mol的组织粘性可吸收材料,其步骤如下:
S1.将29.51mmol丙交酯、29.51mmol己内酯加入1mmol丙三醇中,110℃下共混20分钟得到共混物;
S2.按照0.0045:1的重量比将辛酸亚锡加入步骤S1得到的共混物中,110℃下反应20小时得到粗制预产物;
S3.将步骤S2得到的粗制预产物溶解于10mL二氯甲烷中,然后用乙醚0℃下洗涤纯化粗制预产物4次,室温真空干燥24小时除去溶剂得到预产物一,预产物一为包括丙三醇段、聚(丙交酯-co-己内酯)链的端基为羟基的聚合物;
S4.将步骤S3得到的预产物一溶解于二氯甲烷中,0℃下加入丁二酸酐和4-二甲氨基吡啶,25℃下搅拌反应24小时得到预产物二,预产物一、二氯甲烷、丁二酸酐、4-二甲氨基吡啶的比例为0.5g:1mL:4mmol:1.4mmol;
S5.将步骤S4得到的预产物二用乙醚0℃下洗涤纯化4次,室温真空干燥24小时除去溶剂得到预产物三,预产物三为包括丙三醇段、聚(丙交酯-co-己内酯)链的端基为羧基的聚合物;
S6.将步骤S5得到的预产物三溶解于二氯甲烷中,0℃下加入N-羟基丁二酰亚胺、4-二甲氨基吡啶和N,N'-二异丙基碳二亚胺,25℃下搅拌反应24小时得到预产物四,预产物三、二氯甲烷、N-羟基丁二酰亚胺、4-二甲氨基吡啶、N,N'-二异丙基碳二亚胺的比例为0.2g:1mL:4.5mmol:1.4mmol:4.5mmol;
S7.将步骤S6得到的预产物四用乙醚0℃下洗涤纯化3次,室温真空干燥24小时除去溶剂得到数均分子量为8000g/mol,包括丙三醇段、聚(丙交酯-co-己内酯)链的端基为N-羟基丁二酰亚胺-NHS的组织粘性可吸收材料,其分子结构示意图及核磁共振氢谱图如图2所示。
实施例3
利用季戊四醇作为引发剂引发丙交酯、己内酯、环氧乙烷开环聚合,通过化学改性获得端基为N-羟基丁二酰亚胺-NHS,数均分子量为10000g/mol的组织粘性可吸收材料,其步骤如下:
S1.将1mmol季戊四醇、3.6mmol氢化钾溶解于120mL四氢呋喃中,室温搅拌反应4小时得到引发剂溶液,将31.83mmol环氧乙烷加入引发剂溶液中,室温搅拌反应24小时,然后加入31.83mmol丙交酯和31.83mmol己内酯,80℃下蒸干溶剂后共混20分钟得到共混物;
S2.按照0.0045:1的重量比将辛酸亚锡加入步骤S1得到的共混物中,100℃下反应22小时得到粗制预产物;
S3.将步骤S2得到的粗制预产物溶解于10mL二氯甲烷中,然后用乙醚0℃下洗涤纯化粗制预产物5次,室温真空干燥24小时除去溶剂得到预产物一,预产物一为包括季戊四醇段、聚(丙交酯-co-乙二醇-co-己内酯)链的端基为羟基的聚合物;
S4.将步骤S3得到的预产物一溶解于二氯甲烷中,0℃下加入丁二酸酐和4-二甲氨基吡啶,25℃下搅拌反应24小时得到预产物二,预产物一、二氯甲烷、丁二酸酐、4-二甲氨基吡啶的比例为0.3g:1mL:5.3mmol:1.8mmol;
S5.将步骤S4得到的预产物二用乙醚0℃下洗涤纯化5次,室温真空干燥24小时除去溶剂得到预产物三,预产物三为包括季戊四醇段、聚(丙交酯-co-乙二醇-co-己内酯)链的端基为羧基的聚合物;
S6.将步骤S5得到的预产物三溶解于二氯甲烷中,0℃下加入N-羟基丁二酰亚胺、4-二甲氨基吡啶和N,N'-二异丙基碳二亚胺,25℃下搅拌反应24小时得到预产物四,预产物三、二氯甲烷、N-羟基丁二酰亚胺、4-二甲氨基吡啶、N,N'-二异丙基碳二亚胺的比例为0.3g:1mL:6mmol:1.8mmol:6mmol;
S7.将步骤S6得到的预产物四用乙醚5℃下洗涤纯化5次,室温真空干燥24小时除去溶剂得到数均分子量为10000g/mol,包括季戊四醇段、聚(丙交酯-co-乙二醇-co-己内酯)链的端基为N-羟基丁二酰亚胺-NHS的组织粘性可吸收材料,其分子结构示意图及核磁共振氢谱图如图3所示。
实施例4
利用赤藓糖醇作为引发剂引发环氧乙烷、丙交酯、己内酯开环聚合,通过化学改性获得端基为异氰酸酯-NCO,数均分子量为40000g/mol的组织粘性可吸收材料,其步骤如下:
S1.将0.5mmol季戊四醇、1.8mmol氢化钾溶解于120mL四氢呋喃中,室温搅拌反应4小时得到引发剂溶液,将9mmol环氧乙烷加入引发剂溶液中,室温搅拌反应24小时,然后加入14mmol丙交酯和14mmol己内酯,80℃下蒸干溶剂后共混20分钟得到共混物;
S2.按照0.0045:1的重量比将辛酸亚锡加入步骤S1得到的共混物中,120℃下反应21小时得到粗制预产物;
S3.将步骤S2得到的粗制预产物溶解于10mL二氯甲烷中,然后用乙醚0℃下洗涤纯化粗制预产物3次,室温真空干燥24小时除去溶剂得到预产物一,预产物一为包括赤藓糖醇段、聚(乙二醇-co-丙交酯-co-己内酯)链的端基为羟基的聚合物;
S4.将步骤S3得到的预产物一溶解于甲苯中得到浓度为0.5g/mL的预产物一甲苯溶液,将预产物一甲苯溶液加入浓度为0.5g/mL的六亚甲基二异氰酸酯甲苯溶液中,预产物一与六亚甲基二异氰酸酯的摩尔比为1:30,搅拌均匀后加入辛酸亚锡,辛酸亚锡的重量为预产物一甲苯溶液与六亚甲基二异氰酸酯甲苯溶液重量之和的0.45%,60℃下搅拌反应2小时得到预产物六;
S5.将步骤S4得到的预产物六用乙醚5℃下洗涤纯化3次,室温真空干燥18小时除去溶剂得到数均分子量为40000g/mol,包括赤藓糖醇段、聚(乙二醇-co-丙交酯-co-己内酯)链的端基为异氰酸酯-NCO的组织粘性可吸收材料,其分子结构示意图及核磁共振氢谱图如图4所示。
实施例5
利用乙二醇作为引发剂引发环氧丙烷、丙交酯、己内酯开环聚合,通过化学改性获得端基为醛基-CHO,数均分子量为2000g/mol的组织粘性可吸收材料,其步骤如下:
S1.将1mmol乙二醇、2mmol氢化钾溶解于60mL四氢呋喃中,室温搅拌反应4小时得到引发剂溶液,将21mmol环氧丙烷加入引发剂溶液中,室温搅拌反应24小时,然后加入3.8mmol丙交酯和3.8mmol己内酯,80℃下蒸干溶剂后共混15分钟得到共混物;
S2.按照0.0045:1的重量比将辛酸亚锡加入步骤S1得到的共混物中,110℃下反应20小时得到粗制预产物;
S3.将步骤S2得到的粗制预产物溶解于10mL二氯甲烷中,然后用乙醚0℃下洗涤纯化粗制预产物3次,室温真空干燥12小时除去溶剂得到预产物一,预产物一为包括乙二醇段、聚(乙二醇-co-丙交酯-co-己内酯)链的端基为羟基的聚合物;
S4.将步骤S3得到的预产物一溶解于二氯甲烷中,加入4-甲酰基苯甲酸和4-二甲氨基吡啶,混合均匀后5℃下加入二环己基碳二亚胺,搅拌反应24小时得到预产物五,预产物一、二氯甲烷、4-甲酰基苯甲酸、4-二甲氨基吡啶、二环己基碳二亚胺的比例为0.5g:1mL:10mmol:2mmol:20mmol;
S5.将步骤S4得到的预产物五用乙醚5℃下洗涤纯化3次,室温真空干燥24小时除去溶剂得到数均分子量为2000g/mol,包括乙二醇段、聚(丙交酯-co-己内酯)链的端基为醛基-CHO的组织粘性可吸收材料,其分子结构示意图及核磁共振氢谱图如图5所示。
实施例6
利用三季戊四醇作为引发剂引发β-丙内酯开环聚合,通过化学改性获得端基为巯基-SH,数均分子量为3000g/mol的组织粘性可吸收材料,其步骤如下:
S1.将36.5mmolβ-丙内酯加入1mmol三季戊四醇中,120℃下共混20分钟得到共混物;
S2.按照0.0045:1的重量比将辛酸亚锡加入步骤S1得到的共混物中,130℃下反应24小时得到粗制预产物;
S3.将步骤S2得到的粗制预产物溶解于10mL二氯甲烷中,然后用乙醚0℃下洗涤纯化粗制预产物3次,室温真空干燥12小时除去溶剂得到预产物一,预产物一为包括三季戊四醇段、聚丙内酯链的端基为羟基的聚合物;
S4.将步骤S3得到的预产物一、二硫代二丙酸和4-二甲氨基吡啶溶解于二氯甲烷中,混合均匀后2℃下加入N,N'-二异丙基碳二亚胺,搅拌反应8小时后得到反应物一,将反应物一用2℃的甲醇洗涤三次得到固体粉末,预产物一、二氯甲烷、4-二甲氨基吡啶、N,N'-二异丙基碳二亚胺、甲醇的比例为0.2g:1mL:1.2mmol:4mmol:10mL,预产物一、二硫代二丙酸的摩尔比为1:10;
S5.将步骤S4得到的固体粉末、二硫苏糖醇溶于二氯甲烷中,搅拌反应8小时后得到反应物二,将反应物二用0℃的甲醇洗涤三次得到数均分子量为3000g/mol,包括三季戊四醇段、聚丙内酯链的端基为醛基-CHO的组织粘性可吸收材料,其分子结构示意图及核磁共振氢谱图如图6所示,固体粉末、二氯甲烷、甲醇的比例为0.5g:1mL:10mL,固体粉末、二硫苏糖醇的摩尔比为1:10。
实验例1:
抗破裂压力测试:将实施例1-6制得的组织粘性可吸收材料应用于猪小肠漏液封堵实验。根据外科密封胶的标准测试方法(ASTM F2392-04),将上述实施例进行抗破裂压力测试:使用25G针在猪小肠上刺一个孔,涂抹100μL实施例对孔洞进行密封,如图1所示。将小肠两端夹紧,以1mL/min的恒定速率向小肠的上部注入蒸馏水施加压力。在整个实验过程中监测膜内压力,抗破裂压力被定义为材料破损和小肠开始泄漏的压力,实施例1-6的抗破裂压力数据如表1所示。由表1的实验数据可见,本发明提供的组织粘性生物材料能够有效粘接组织创伤部位,从而有效防止损伤组织部位气体泄漏及血液渗漏。
表1
细胞相容性测试:将实施例1-6制得的组织粘性可吸收材料放入48孔板中,加入100μL的内皮细胞(密度为5000细胞/mL),100μL含8%CCK-8试剂的新鲜无血清培养基,并在5%二氧化碳,95%空气和95%湿度气氛中37℃下培养24小时,以不含实施例的48孔板中的细胞作为阳性对照组。24小时培养后,实施例1-6的内皮细胞存活率如表1所示。由表1的实验数据可见,所有实施例均不会对细胞增值产生负面影响,表明实施例1-6均有良好的细胞相容性。
实验例2:
生物相容性测试:将实施例1-6制得的组织粘性可吸收材料进行生物相容性测试,测试根据相关国标进行,实施例1-6的生物相容性测试结果如表2所示。由表2可见,本发明提供的组织粘性生物材料具有良好的生物无毒性、无过敏及无刺激性。
表2
实验例3:
体外降解测试:分别取20mg实施例1-4制得的组织粘性可吸收材料所形成的薄膜浸没入磷酸盐缓冲盐水(PBS)溶液中,设置溶液的pH值为7.4,温度为37℃,并使用台式轨道摇床以150rpm的速度摇动溶液,定期取出薄膜,干燥后称取薄膜质量,计算出质量保持率。实施案例1-4样品的质量保持率随时间的变化如图9所示。由图9可见,本发明制得的组织粘性可吸收材料在磷酸盐缓冲盐水(PBS)溶液中具有良好的降解性能,其降解周期根据设计分子量的变化而变化:高分子量的实施例降解周期长,反之,低分子量的实施例降解周期短。
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
- 电子设备供电电路及电子设备
- 网口POE供电电路、供电电路、监控系统及供电方法
- 电子设备端口类型检测方法、装置、检测电路及电子设备
- 电子设备的供电电路、供电方法和一种电子设备
- 电子设备的供电电路、供电方法和一种电子设备