α1,2-岩藻糖基转移酶突变体及表达其的重组大肠杆菌和应用
文献发布时间:2024-04-18 20:01:23
技术领域
本发明涉及生物技术领域,尤其是涉及一种α1,2-岩藻糖基转移酶突变体及表达其的重组大肠杆菌和应用。
背景技术
母乳低聚糖(Human milk oligosaccharides,HMOs)是母乳中一类独特的游离型低聚糖,在母乳中的含量仅次于水、乳糖和脂肪,是母乳中第三大固体成分,被认为是天然母乳和人工配方奶粉的重要区别。因为它们几乎是母乳喂养婴儿的益生元的唯一来源,对新生儿的生长有许多重要的健康影响。例如,它们可以通过塑造肠道微生物群,促进双歧杆菌增殖,发挥独特的健康效应,包括调节肠上皮细胞应答、抗粘附抗菌抗病毒,抵抗致病性微生物感染、预防坏死性小肠结肠炎,调节免疫反应、改善大脑发育和认知等。因此,近年来HMOs引起了广泛的商业关注。其中,一些HMOs组分,如LNT(乳糖-N-四糖)、LNnT(乳糖-N-新四糖)、2'-FL(2'-岩藻糖基乳糖)、3'-FL(3'-岩藻糖基乳糖)、3'-SL(3'-唾液酸乳糖)和6'-SL(6'-唾液酸乳糖)已被已经被美国FDA批准为“公认的安全(GRAS)”的食品,并允许商业化生产和添加到婴儿配方奶粉中。
2'-岩藻糖基乳糖(2'-FL)是母乳中含量最丰富、最早商业化的一个HMO结构,也是当下所有HMOs中最受关注的组分,目前已获得美国、欧盟、澳大利亚和亚太地区多个监管机构的批准,主要应用于婴儿配方奶粉、营养食品和食品添加剂。目前,已经报导了四种方法,包括母乳分离、化学合成、酶促合成和微生物发酵,被用于2'-FL的合成研究。前三种传统的生产方法存在原料或底物来源有限、价格昂贵,且生产工艺复杂,不够经济环保等因素,目前均不适用于2'-FL的规模化生产。随着合成生物学和代谢工程技术的快速发展,使得通过微生物构建细胞工厂日益易于操作,促进了微生物合成2'-FL的各种策略的发展,这已被证明是一种很有前途并被广泛采用的方法。
大肠杆菌因其快速生长、遗传背景清晰易改造,且易于扩大培养,已成为HMOs合成的首选微生物宿主。目前通过多种代谢工程策略,包括模块化途径优化与构建、辅因子供应优化和底盘微生物修饰等等,大肠杆菌已被开发为2'-FL的主要生产者,但目前相对较低的产量和高生产成本仍然是2'-FL工业化生产中亟待改善的问题。
一般来说,对于2'-FL的微生物合成,需要考虑三个关键要素:供体GDP-L-岩藻糖、受体乳糖和α1,2-岩藻糖基转移酶(FT)。α1,2-FT能将L-岩藻糖基从供体转移到受体的半乳糖基上,催化合成2'-FL。供体GDP-L-岩藻糖的主流生产方法是从头合成,主要以糖酵解的重要中间体6-磷酸果糖为起点,经过五个酶促级联步骤(ManA[甘露糖-6-磷酸异构酶],ManB[磷酸甘露糖变位酶],ManC[α-D-甘露糖1-磷酸鸟嘌呤基转移酶],Gmd[GDP-甘露糖4,6-脱氢酶]和WcaG[GDP-L-岩藻糖合成酶])催化合成,参见图1。通常利用高拷贝数质粒过表达这五个基因,以及阻断竞争性代谢途径的关键酶(WcaJ)来强化GDP-L-岩藻糖的供应水平。乳糖是一种常见碳源,价格相对低廉,因此通常直接外源添加到反应体系中,作为底物使用。为了减少乳糖的降解损失,通常需要阻断底盘宿主的乳糖分解代谢途径。在2'-FL的微生物合成途径中,α1,2-FT是关键限速步骤之一。此前,有多个研究小组已经成功从不同细菌中克隆并鉴定了几种具有良好催化活性的α1,2-FTs,如幽门螺杆菌来源的FutC、大肠杆菌O126来源的WbgL、脆弱拟杆菌来源的WcfB、螺杆菌11S02629-2来源的BKHT、生脂固氮螺菌来源的SAMT。尽管如此,这些基因在大肠杆菌中活性表达水平通常很低,导致催化性能低效,而且均来源于病原微生物,对食品生产安全性具有潜在的威胁,一定程度上限制了它们在2'-FL合成中的应用,同时也为在不同国家申请2'-FL的生产和应用埋下了潜在的障碍。另外,这些α1,2-FTs还能产生3-FL和/或DFL等副产物,导致底物特异性较低,这为下游的纯化过程带来了新的挑战。因此,寻找高活性且高特异性的α1,2-FT基因对于2'-FL的高效生产具有重大意义,最好是来自于人类胃肠道中的安全微生物。
到目前为止,利用高拷贝质粒对参与从头合成途径的基因进行过表达是提高2'-FL产量的常用措施,绝大部分全细胞合成2'-FL的方法都依赖于所携带的质粒来表达目的基因。重组质粒对于单基因的克隆和短期表达是有利的,但在利用细菌宿主细胞合成次级代谢产物时一般需要对多个途径基因进行共表达,这种情况下使用质粒表达系统往往存在一些不可避免的稳定性缺陷。一方面,质粒在细胞增殖过程中具有结构和分离不稳定性,这可能导致目的基因的丢失或表达失活;其次,为了在细胞中维持表达质粒的遗传,通常需要添加选择标记,如抗生素抗性基因,这种选择系统导致生产成本的增加,同时由于抗生素的使用在食品安全生产上受法律限制,导致下游的纯化提取工艺成本又增加;另外,高拷贝质粒复制时产生的代谢负担和某些基因的强烈过表达可能导致生长速度降低,导致胞内代谢通量和能量分布的失衡,进而影响目标产物的合成。因此,开发安全稳定、环境友好、经济高效的2'-FL合成方法成为了必然的趋势。
有鉴于此,特提出本发明。
发明内容
本发明的目的在于提供一种α1,2-岩藻糖基转移酶突变体及表达其的重组大肠杆菌和应用,该突变体具有较高的岩藻糖基化活性和较高的底物特异性,不会形成副产物DFL和3-FL,以缓解上述中的至少一个技术问题。
为解决上述技术问题,本发明特采用如下技术方案:
根据本发明的一个方面,提供了一种α1,2-岩藻糖基转移酶突变体,其具有如下氨基酸序列的片段:由SEQ ID NO.1所示氨基酸序列经如下一种突变或几种突变的组合后得到的序列:D66G、F101Y、L128I、L136I、V138I、Q205M、W224Y和V232I。
根据本发明的另一个方面,还提供了一种生物材料,所述生物材料选自(ⅰ)~(ⅲ)中任一项:
(i)多核苷酸,含有编码上述的α1,2-岩藻糖基转移酶突变体的片段;
(ⅱ)载体,所述载体携带(ⅰ)中多核苷酸;
(ⅲ)细胞,所述细胞携带(ⅰ)中多核苷酸,或含有(ⅱ)中载体,或表达上述的α1,2-岩藻糖基转移酶突变体。
根据本发明的另一个方面,还提供了一种重组大肠杆菌,所述重组大肠杆菌的染色体基因组中含有至少一个拷贝的糖基转移酶基因JAGAPm,每个所述糖基转移酶基因JAGAPm分别独立的编码上述的α1,2-岩藻糖基转移酶突变体。
根据本发明的另一个方面,还提供了上述的重组大肠杆菌的制备方法,该制备方法包括通过至少一种CRISPR/Cas基因编辑系统,清除、下调和/或敲入大肠杆菌基因组染色体中的基因和/或启动子区域。
根据本发明的另一个方面,还提供了一种2'-岩藻糖基乳糖的制备方法,所述制备方法包括由表达上述的α1,2-岩藻糖基转移酶突变体的细胞发酵得到。
根据本发明的另一个方面,还提供了上述的α1,2-岩藻糖基转移酶突变体,或上述的生物材料,或上述的重组大肠杆菌,或上述的重组大肠杆菌的制备方法,或上述的制备方法在制备母乳低聚糖中的应用。
与现有技术相比,本发明具有如下有益效果:
本发明对一个普雷沃氏菌来源的α1,2-岩藻糖基转移酶(JAGAP)进行多轮定点突变,最终获得了一株具有较高岩藻糖基化活性和较高底物特异性的α1,2-岩藻糖基转移酶突变体,能够高效实现将底物乳糖转化为2'-FL,而且不会形成副产物DFL和3-FL。
本发明通将α1,2-岩藻糖基转移酶突变体插入到大肠杆菌染色体上进行整合表达,以及在一些优选的实施方式中将从头合成的相关途径基因也插入到大肠杆菌染色体上进行整合表达,所获得的重组菌株相较于携带质粒表达的重组菌株,具有良好的遗传稳定性和生长状况。
本发明利用上述重组大肠杆菌在5L发酵罐中以甘油为碳源,乳糖为底物发酵生产2'-FL的产量可达到106.42g/L,单位生产效率可达到1.34g/L/h,该发酵水平高于现有技术水平,且在发酵过程中无需添加任何抗生素。因此,本发明的原材料廉价易得,无危害人体健康的添加剂,对后续产物提纯无不利影响,能够达到商业应用的要求。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
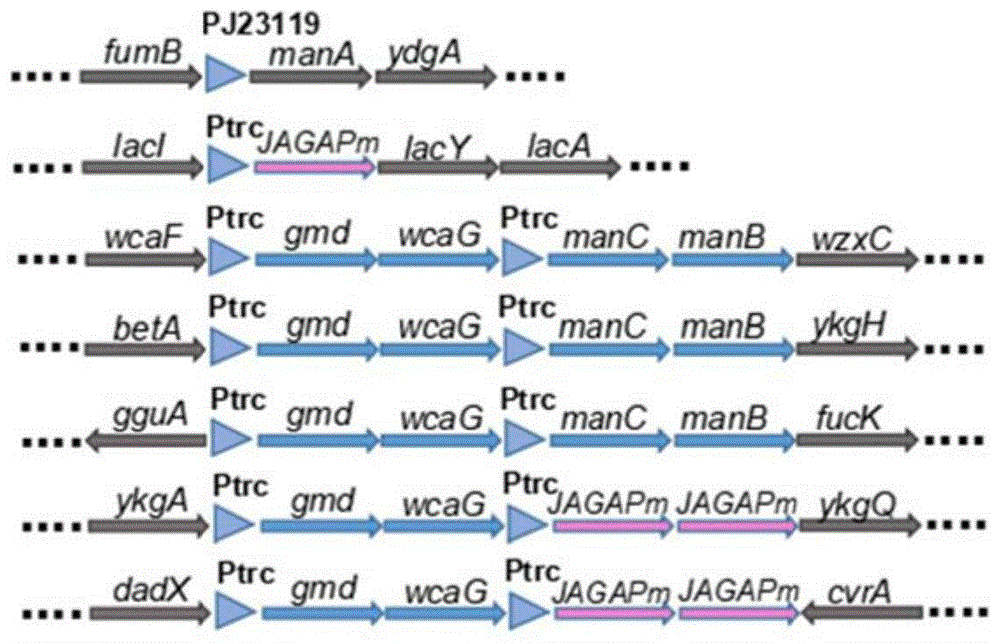
图1为实施例3制备的重组大肠杆菌TK-C010中重组基因的示意图;
图2为实施例5中示差折光检测重组菌TK-R009的检测结果;
图3为实施例7扩大培养时,发酵罐中各物质含量随时间的变化。
具体实施方式
下面将结合实施例对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
发明人从素食爱好者的粪便中分离得到一株普雷沃氏菌,该菌为革兰阴性厌氧菌,是一种经典的肠道和呼吸系统共生细菌。本发明通过对普雷沃氏菌来源的α1,2-FT(JAGAP,SEQ ID NO.1)进行多轮定点突变,得到一株具有较高岩藻糖基化活性和较高底物特异性的α1,2-岩藻糖基转移酶突变体,该突变体包括具有如下氨基酸序列的片段:由SEQID NO.1所示氨基酸序列经如下一种突变或几种突变的组合后得到的序列:
D66G,即SEQ ID NO.1所示氨基酸序列的第66位由D(天冬氨酸)突变成G(甘氨酸);
F101Y,即SEQ ID NO.1所示氨基酸序列的第101位由F(苯丙氨酸)突变成Y(酪氨酸);
L128I,即SEQ ID NO.1所示氨基酸序列的第128位由L(亮氨酸)突变成I(异亮氨酸);
L136I,即SEQ ID NO.1所示氨基酸序列的第136位由L(亮氨酸)突变成I(异亮氨酸);
V138I,即SEQ ID NO.1所示氨基酸序列的第138位由V(缬氨酸)突变成I(异亮氨酸);
Q205M,即SEQ ID NO.1所示氨基酸序列的第205位由Q(谷氨酰胺)突变成M(蛋氨酸);
W224Y,即SEQ ID NO.1所示氨基酸序列的第224位由W(色氨酸)突变成Y(酪氨酸);
和,V232I,即SEQ ID NO.1所示氨基酸序列的第232位由V(缬氨酸)突变成I(异亮氨酸)。
可选的实施方式中,α1,2-岩藻糖基转移酶突变体含有由SEQ ID NO.1所示氨基酸序列经JAGAPm1~10任意一方式突变后得到的序列:
JAGAPm1:突变位点为D66G;
JAGAPm2:突变位点为L128I;
JAGAPm3:突变位点为Q205M;
JAGAPm4:突变位点为W224Y;
JAGAPm5:突变位点为V232I;
JAGAPm6:突变位点为D66G和L128I;
JAGAPm7:突变位点为Q205M和W224Y;
JAGAPm8:突变位点为Q205M和V232I;
JAGAPm9:突变位点为D66G、L128I、Q205M和V232I;
JAGAPm10:突变位点为D66G、L128I、Q205M和W224Y。
SEQ ID NO.1:
MVYVELTDRLGNNLFQIAAAASLASYNNTNYAAYISDFILPNGQKLETSINAYKNNILRNVSFIKDMPTDIIKYEQDGFNYSPLPYHDRIYLKGYFQSEKFFNKELVRNLFSIDEDTYKYINNKYGYLLKKEVISLHVRRGDYVYRPRRQRLCSLTYYKNAIRYFGKDKLFLILSDDIEWCKAHFKGKNFIYSDQESAIVDLYLQTLCTHNIISNSTFSWWGAWLNNNKNKVVIAPKQWFGKHLSHYKTEDMIPETWIRLNNPRSLSIILRAFKYDVLDFFDRTRGINKFRG。
可选的实施方式中,所述α1,2-岩藻糖基转移酶突变体的氨基酸序列如SEQ IDNO.2所示。
SEQ ID NO.2:
MVYVELTDRLGNNLFQIAAAASLASYNNTNYAAYISDFILPNGQKLETSINAYKNNILRNVSFIKGMPTDIIKYEQDGFNYSPLPYHDRIYLKGYFQSEKFFNKELVRNLFSIDEDTYKYINNKYGYILKKEVISLHVRRGDYVYRPRRQRLCSLTYYKNAIRYFGKDKLFLILSDDIEWCKAHFKGKNFIYSDQESAIVDLYLMTLCTHNIISNSTFSWWGAWLNNNKNKIVIAPKQWFGKHLSHYKTEDMIPETWIRLNNPRSLSIILRAFKYDVLDFFDRTRGINKFRG。
根据本发明的另一个方面,还提供了一种生物材料,所述生物材料选自(ⅰ)~(ⅲ)中任一项:
(i)多核苷酸,含有编码上述的α1,2-岩藻糖基转移酶突变体的片段。本文中“多核苷酸”指的是任何长度的核苷酸的聚合物形式,多核苷酸包括核糖核苷酸和/或脱氧核糖核苷酸。多核苷酸的实例包括但不限于单链、双链或多链DNA或RNA、基因组DNA、cDNA、DNA-RNA杂合体或者包含嘌呤和嘧啶碱基或其他天然、化学或生化修饰、非天然或衍生的核苷酸碱基的聚合物。多核苷酸编码上述α1,2-岩藻糖基转移酶突变体,编码可选地为编码正义链或反义链。多核苷酸可以是天然存在的、合成的、重组的或它们的任意组合。术语“多核苷酸”和“核酸”本文中可互换地使用。可选的实施方式中,所述多核苷酸含有核苷酸序列如SEQID NO.4所示的片段;或含有核苷酸序列与SEQ ID NO.4至少具有70%、75%、80%、85%、90%、95%或98%同一性的核苷酸序列的片段。
SEQ ID NO.4:
atggtttatgtggagctgaccgatcgcctgggtaataatctgtttcagattgcagcagcagccagcctggcaagctataataataccaattatgcagcatatattagcgattttattctgccgaatggtcagaaactggaaaccagcattaatgcctacaaaaataacattctgcgtaatgttagctttatcaaaggtatgccgaccgacattattaaatatgaacaggatggttttaactatagtccgctgccgtatcatgatcgtatttatctgaaaggttatttccagagcgaaaaatttttcaacaaagaactggttcgtaatctgtttagcattgatgaagatacctataaatacattaacaataaatatggttatatcctgaaaaaagaagttattagcctgcatgttcgtcgcggtgattatgtttatcgtccgcgtcgtcagcgtctgtgctcactgacatattataaaaatgcaattcgttattttggcaaagataaactgtttctgattctgagtgatgatattgaatggtgtaaagcacattttaaaggcaaaaactttatctatagcgatcaagaaagcgcaattgttgatctgtatctgatgaccctgtgtacccacaatattattagcaatagcacctttagctggtggggtgcatggctgaataacaacaagaataaaattgttattgccccgaaacagtggtttggtaaacatctgagccattataaaaccgaagatatgattccggaaacctggattcgtctgaataatcctcgtagcctgagcatcatcctgcgtgcatttaaatatgatgtgctggatttttttgatcgtacacgtggtattaataaatttcgtggttaa。
本文中“同一性”或类似术语是指,在最佳比对下,待比较序列之间相同核苷酸或氨基酸之百分比。该百分比为纯粹统计学的,且两个序列之间的差异可能为(但不一定)随机分布于待比较序列之整个长度上。两个序列之比较通常藉由在最佳比对之后,相对于片段或“比较窗口”以鉴别对应序列之局部区。
(ⅱ)载体,所述载体携带前述(ⅰ)多核苷酸。所述载体是本领域技术人员公知的,包括但不限于:质粒、噬菌粒、柯斯质粒、人工染色体等。可选的实施方式中,载体中包含基因工程中常用的调控元件,例如增强子、启动子、内部核糖体进入位点(IRES)和其他表达控制元件(例如转录终止信号,或者多腺苷酸化信号和多聚U序列等)。
(ⅲ)细胞,所述细胞携带(ⅰ)中多核苷酸,或含有(ⅱ)中载体,或表达上述的α1,2-岩藻糖基转移酶突变体。
可选的实施方式中,所述细胞携带(ⅰ)中多核苷酸,所述多核苷酸可以整合于载体,例如整合于质粒,独立于所述细胞染色体基因组独自复制和/或表达;所述多核苷酸也可以整合于所述细胞的染色体基因组中。
可选的实施方式中,所述细胞含有(ⅱ)中载体,所述载体在所述细胞中复制和/或表达。
可选的实施方式中,所述细胞表达上述的α1,2-岩藻糖基转移酶突变体,所述α1,2-岩藻糖基转移酶突变体可以由整合至所述细胞染色体基因组中的编码基因表达,和/或,独立于所述细胞染色体基因组的载体中的编码基因表达。可选的实施方式中,部分α1,2-岩藻糖基转移酶突变体由整合至所述细胞染色体基因组中的编码基因表达,部分α1,2-岩藻糖基转移酶突变体由独立于所述细胞染色体基因组的载体中的编码基因表达。
本文中细胞可以指单个细胞,也可以指细胞系,或包含细胞的培养物。本文中细胞包括其后代,后代可以由于天然的、偶然的或故意的突变不一定与原代细胞完全相同,在形态学上和/或在基因组DNA上与原代细胞存在差异。所述细胞可以为天然细胞或转化体。本发明不限制提供的上述(ⅲ)细胞的具体种类,包括但不限于真核或原核细胞,更具体的包括但不限于细菌、真菌、植物、昆虫或动物细胞。
可选的实施方式中,本发明提供的生物材料中含有多个拷贝的编码α1,2-岩藻糖基转移酶突变体的基因片段时,各拷贝之间可以相同也可以不同。例如每个拷贝编码上述不同实施方式中,含有不同突变位点的α1,2-岩藻糖基转移酶突变体。
根据本发明的另一个方面,还提供了一种重组大肠杆菌,所述重组大肠杆菌的染色体基因组中含有至少一个拷贝的糖基转移酶基因JAGAPm,所述糖基转移酶基因JAGAPm编码上述的α1,2-岩藻糖基转移酶突变体。
可选的实施方式中,JAGAPm基因在染色体基因组中的拷贝数为1~9个,例如可以为但不限于为1、2、3、4、5、6、7、8或9个拷贝,优选为3个拷贝。可选的实施方式中,所述重组大肠杆菌中含有至少两个JAGAPm基因拷贝时,各拷贝之间可以相同也可以不同。例如每个拷贝编码上述不同实施方式中,含有不同突变位点的α1,2-岩藻糖基转移酶突变体。
可选的实施方式中,所述重组大肠杆菌的染色体基因组中还含有至少一个拷贝的甘露糖-6-磷酸异构酶基因manA;至少一个拷贝的磷酸甘露糖变位酶基因manB;至少一个拷贝的α-D-甘露糖1-磷酸鸟嘌呤基转移酶基因manC;至少一个拷贝的GDP-甘露糖4,6-脱氢酶基因gmd;和,至少一个拷贝的GDP-L-岩藻糖合成酶基因wcaG。通过将2'-FL从头合成的相关途径基因(manA、manC、manB、gmd、wcaG、JAGAPm)插入到大肠杆菌染色体上进行整合表达,以获得不含质粒的稳定型重组菌株;进一步通过对插入基因的拷贝数进行优化,以提高2'-FL生产效价和单位生产率,为后续2'-FL的规模化生产提供更好的基础。
可选的实施方式中,manB基因和manC基因在染色体基因组中的拷贝数分别独立的为1~5个,例如可以为但不限于为1、2、3、4或5个拷贝,优选为3个拷贝。
可选的实施方式中,gmd基因和wcaG基因在染色体基因组中的拷贝数分别独立的为1~7个,例如可以为但不限于为1、2、3、4、5、6或7个拷贝,优选为5个拷贝。
可选的实施方式中,所述重组大肠杆菌的染色体基因组中,manA基因、manB基因、manC基因、gmd基因和wcaG基因的拷贝数分别独立的至少为两个时,每个基因的拷贝的核苷酸序列可以相同也可以不同,例如当所述染色体基因组中含有多个manB基因拷贝时,各拷贝来源于不同株的大肠杆菌,和/或,其中一个或几个拷贝的manB基因经突变,以期获得功能改进的磷酸甘露糖变位酶。
可选的实施方式中,所述重组大肠杆菌的染色体基因组中,至少manB基因、manC基因、gmd基因和wcaG基因的部分拷贝分别独立的来源于E.coli str.K-12substr.MG1655(GenBank:NC_000913.3)。可选的实施方式中,所述重组大肠杆菌的染色体基因组中,manB基因的部分或全部拷贝的核苷酸序列为Gene ID:946574所示序列;可选的实施方式中,所述重组大肠杆菌的染色体基因组中,manC基因的部分或全部拷贝的核苷酸序列为Gene ID:946580所示序列;可选的实施方式中,所述重组大肠杆菌的染色体基因组中,gmd基因的部分或全部拷贝的核苷酸序列为Gene ID:946562所示序列;可选的实施方式中,所述重组大肠杆菌的染色体基因组中,wcaG基因的部分或全部拷贝的核苷酸序列为Gene ID:946563所示序列。
可选的实施方式中,JAGAPm基因、manB基因、manC基因、gmd基因和wcaG基因的整合位点分别独立的选自lacZ、wcaJ、fucI、betA-ykgH、fumB-ydgA、ykgA-ykgQ、dadA-cvrA、yjiPyjiR、ileY ygaQ中的一个或多个。上述A-B表示两个位点之间(A表示betA、fumB、ykgA、dadA、yjiP或ileY,B表示对应的ykgH、ydgA、ykgQ、cvrA、yjiR或ygaQ),具体的为位点A的5’端起始至位点B的3’端结束的任意一位置。可选的实施方式中,上述基因整合至两个位点之间的间隔序列结构,不影响原有两个基因自身的表达。实际操作中要考虑所选序列的编辑效率和靶位点基因的必要性,例如靶位点基因对菌株生长代谢可能是非必需的,根据具体需求,可选的实施方式中会破坏或者取代插入位点相邻的某个基因,即非两个位点之间的间隔序列结构的位置。
可选的实施方式中,manA基因由强启动子调控表达;所述强启动子选自Ba_J54200、BBa_J23100、BBa_J23101和BBa_J23119中的至少一种,优选为BBa_J23119。
可选的实施方式中,manB基因、manC基因、gmd基因和wcaG基因由诱导型启动子调控表达,所述诱导型启动子选自Para、Prha、Ptet、Plac、Ptrc、Ptac和PT7中的至少一种,优选为Ptrc。
可选的实施方式中,所述重组大肠杆菌的染色体基因组中含有至少一个manC-manB,所述manC-manB为以顺反子结构进行组合串联表达的manC基因和manB基因,由顺反子上游启动子控制串联表达。
可选的实施方式中,所述重组大肠杆菌的染色体基因组中含有至少一个gmd-wcaG,所述gmd-wcaG为以顺反子结构进行组合串联表达的gmd基因和wcaG基因,由顺反子上游启动子控制串联表达。
可选的实施方式中,所述重组大肠杆菌的wcaJ基因和lacZ基因被下调或清除,下调或清除wcaJ基因用于阻断竞争性代谢途径的关键酶来强化GDP-L-岩藻糖的供应水平;下调或清除lacZ基因以阻断重组大肠杆菌的乳糖分解代谢途径。
可选的实施方式中,所述重组大肠杆菌的wcaJ基因和lacZ基因被下调或清除;且所述染色体基因组中整合有如下基因:
fumB和ydgA位点之间整合有BBa_J23119启动子调控的manA基因;
lacI和lacY之间整合有Ptrc启动子调控的JAGAPm基因;
wcaF和wzxC位点之间,betA和ykgH位点之间,以及fucI和fucK位点之间分别依次整合有所述gmd-wcaG和所述manC-manB,所述gmd-wcaG和所述manC-manB分别由启动子Ptrc调控;
ykgA和ykgQ位点之间以及dadX和Ptrc位点之间依次整合有所述gmd-wcaG和两个串联的JAGAPm基因;所述gmd-wcaG由启动子Ptrc调控;两个串联的JAGAPm基因由启动子Ptrc调控。
可选的实施方式中,上述重组大肠杆菌的染色体基因组中至少一个JAGAPm基因编码的α1,2-岩藻糖基转移酶突变体具有JAGAPm9的突变方式,即突变位点为D66G、L128I、Q205M和V232I。编码具有JAGAPm9的突变方式的α1,2-岩藻糖基转移酶突变体的JAGAPm基因记为JAGAPm9。可选的实施方式中,上述重组大肠杆菌的染色体基因组中JAGAPm基因编码的α1,2-岩藻糖基转移酶突变体全部按照上述JAGAPm9所示的突变方式突变。
可选的实施方式中,上述重组大肠杆菌的染色体基因组中至少一个JAGAPm9基因编码的α1,2-岩藻糖基转移酶突变体的氨基酸序列如SEQ ID NO.2所示。可选的实施方式中,所述重组大肠杆菌的染色体基因组中的JAGAPm9基因编码的α1,2-岩藻糖基转移酶突变体的氨基酸序列全部如SEQ ID NO.2所示。
可选的实施方式中,上述重组大肠杆菌的染色体基因组中至少一个JAGAPm9基因的核苷酸序列如SEQ ID NO.4所示。可选的实施方式中,所述重组大肠杆菌的染色体基因组中JAGAPm9基因的核苷酸序列全部如SEQ ID NO.4所示。
可选的实施方式中,上述重组大肠杆菌的出发菌株是BL21 Star(DE3)。
根据本发明的另一个方面,还提供了上述重组大肠杆菌的制备方法,包括通过至少一种CRISPR/Cas基因编辑系统,清除、下调和/或敲入大肠杆菌基因组染色体中的基因和/或启动子区域。
CRISPR/Cas基因编辑技术能够高效快速连续地对大肠杆菌基因组进行目标基因的敲除失活以及靶基因或靶序列的整合敲入。可选的实施方式中,使用CRISPR/Cas9基因编辑系统制备上述重组大肠杆菌。
可选的实施方式中,所述制备方法包括先使用CRISPR/Cas基因编辑系统下调或清除大肠杆菌的wcaJ基因和lacZ基因,然后使用CRISPR/Cas基因编辑系统将manA基因的启动子替换为Ba_J54200、BBa_J23100、BBa_J23101和BBa_J23119中的至少一种;以及将manC基因和gmd基因的启动子替换为Para、Prha、Ptet、Plac、Ptrc、Ptac和PT7中的至少一种;然后使用CRISPR/Cas基因编辑系统向经上述步骤改造的大肠杆菌中敲入更多拷贝的manB基因、manC基因、gmd基因和wcaG基因。
根据本发明的另一个方面,还提供了一种2'-岩藻糖基乳糖的制备方法,该制备方法由表达上述的α1,2-岩藻糖基转移酶突变体的细胞发酵得到。
可选的实施方式中,所述制备方法中用于发酵生产2'-岩藻糖基乳糖的细胞中,编码α1,2-岩藻糖基转移酶突变体的基因整合于所述细胞的基因组染色体中。
可选的实施方式中,所述制备方法中用于发酵生产2'-岩藻糖基乳糖的细胞中,编码α1,2-岩藻糖基转移酶突变体的基因整合于独立于所述细胞的基因组染色体的载体中。
可选的实施方式中,所述制备方法中用于发酵生产2'-岩藻糖基乳糖的细胞中,部分拷贝的编码α1,2-岩藻糖基转移酶突变体的基因整合于所述细胞的基因组染色体中,部分拷贝的编码α1,2-岩藻糖基转移酶突变体的基因整合于独立于所述细胞的基因组染色体的载体中。
可选的实施方式中,上述任意一独立于所述细胞的基因组染色体的载体是质粒。
可选的实施方式中,所述表达上述的α1,2-岩藻糖基转移酶突变体的细胞为上述任意一实施方式中的重组大肠杆菌。
本发明提供的2'-岩藻糖基乳糖的制备方法中,对表达上述的α1,2-岩藻糖基转移酶突变体的细胞的发酵和培养条件,本领域技术人员可根据本领域已知的、常规的方式进行选择,例如但不限于培养环境、培养设备、培养体积和培养基本领域技术人员可以根据本领域众所周知的,且如各种一般和更具体的教材、参考文献、工艺手册、商品说明和标准文件等记载的方法来进行,本发明对此不做限制。
可选的实施方式中,所述表达上述的α1,2-岩藻糖基转移酶突变体的细胞为大肠杆菌,碳源选自甘油,发酵底物为乳糖。
可选的实施方式中,所述制备方法包括将上述重组大肠杆菌接种至种子培养基中扩大培养,制备得到种子液;再将种子液接种至发酵培养基中,通过诱导发酵合成2'-岩藻糖基乳糖。
可选的实施方式中,种子培养基为LB液体培养基;发酵培养基为M9液体培养基。
可选的实施方式中,随着发酵过程中乳糖的消耗,在发酵过程中补充乳糖;在一些具体的实施方式中,当发酵液中乳糖低于10g/L时,间歇性补加乳糖至终浓度达到20g/L。可选的实施方式中,发酵过程中发酵体系的pH维持在6.5~7.5。
根据本发明的另一个方面,还提供了上述的α1,2-岩藻糖基转移酶突变体,或上述的生物材料,或上述的重组大肠杆菌,或上述的重组大肠杆菌的制备方法,或上述的制备方法在制备母乳低聚糖中的应用。
下面通过具体的实施例进一步说明本发明,但是,应当理解为,这些实施例仅仅是用于更详细地说明之用,而不应理解为用于以任何形式限制本发明。
在以下实施例中,生产2'-FL的基因工程菌以E.coli BL21 star(DE3)为出发宿主细胞。
在以下实施例中,涉及的培养基如下:
LB培养基配方(g/L):酵母粉5,蛋白胨10,NaCl 10,固体培养基另加1.8%琼脂粉。
M9培养基配方(g/L):甘油10,Na
在以下实施例中,涉及的发酵样品检测条件如下:
(1)仪器:Agilent HPLC system(1,100Series);示差检测器(Agilent);氨基柱NH
(2)试剂:FL标准品,购自Carbosynth,纯度90%;乳糖标准品,购自国药,纯度98.0%,流动相:70%乙腈;
(3)色谱条件:柱温25℃,示差检测器35℃,流速1.1mL/mL,进样量20μl。
在以下实施例中,涉及的引物如表1所示:
表1序列信息
/>
实施例1重组质粒的构建
具体步骤如下:
(1)根据糖基转移酶JAGAP的氨基酸序列信息,进行密码子优化,获得该α1,2-FT基因的核苷酸序列(SEQ ID NO.3所示),并交由南京金斯瑞生物公司合成。
SEQ ID NO.3:
atggtttatgtggagctgaccgatcgcctgggtaataatctgtttcagattgcagcagctgccagtctggcaagctataacaatacgaattatgcagcctatattagcgattttattctgccgaatggtcagaaactggaaacaagcatcaatgcatataaaaataacattctgcgtaatgttagttttattaaagatatgccgaccgatattatcaaatatgaacaggatggttttaactatagcccgctgccgtatcatgatcgtatttatctgaaaggttatttccagagcgaaaagttctttaataaagaactggtgcgtaatctgtttagcattgatgaagatacctataaatacattaataacaaatatggttatctgctgaaaaaagaagttatcagcctgcatgttcgtcgcggtgattatgtttatcgtccgcgtcgtcagcgtctgtgcagcctgacctattacaaaaatgcaattcgttattttggtaaagataaactatttctgattctgagcgatgatattgaatggtgtaaagcacattttaaaggtaaaaattttatctattcagaccaagaaagcgcaattgttgatctgtatctgcagaccctgtgtacccacaatattattagcaatagcacctttagctggtggggtgcatggctgaataataataaaaacaaagttgttatcgcaccgaaacagtggtttggcaaacatctgagccattataaaaccgaagatatgattccggaaacctggattcgtctgaataatcctcgtagcctgagcattattctgcgtgcatttaaatatgatgttctggatttttttgatcgtacccgtggtattaacaaatttcgtggctaa。
(2)利用NdeI与AvrII酶切位点设计引物(如表1所示),将α1,2-FT基因序列与商业化载体pETDuet-1进行连接;经过化学转化法转入E.coli DH5α感受态细胞中,涂布于含有浓度为50μg/L氨苄青霉素抗性的LB固体培养基平板上,于37℃培养箱培养12h左右;将长出的单菌落提质粒进行酶切和测序验证,获得正确的重组质粒pET-JAGAP。
(3)参照上述(1)和(2)步骤,根据糖基转移酶JAGAP定点突变的氨基酸序列信息,获得一系列定点突变后的α1,2-FT基因核苷酸序列,并分别构建到商业化载体pETDuet-1上,获得重组质粒pET-JAGAPm1、pET-JAGAPm2.....pET-JAGAPm10(见表2)。
表2 JAGAPm1~10的突变位点
(4)通过设计和合成相应的引物(如表1所示),分别扩增来源于E.coli str.K-12sμbstr.MG1655(GenBank:NC_000913.3)的磷酸甘露糖变位酶基因manB(Gene ID:946574),α-D-甘露糖1-磷酸鸟嘌呤基转移酶基因manC(Gene ID:946580),GDP-甘露糖4,6-脱氢酶基因gmd(Gene ID:946562)和GDP-L-岩藻糖合成酶基因wcaG(Gene ID:946563),获得manC-manB和gmd-wcaG基因片段;利用限制性内切酶NcoI和NotI对manC-manB片段和pRSFDuet-1质粒进行双酶切,酶切后将载体和manC-manB片段连接,连接产物转化至E.coli DH5α感受态细胞,并在卡那霉素板上进行筛选,获得重组质粒pR-CB。然后利用限制性内切酶NdeI与AvrII对gmd-wcaG片段和pR-CB质粒进行双酶切,酶切后将载体和gmd-wcaG片段进行连接,连接产物转化至E.coli DH5α感受态细胞,并在卡那霉素板上进行筛选,获得重组质粒pR-CBGW。
(5)同理,分别将JAGAPm9、manC-manB和gmd-wcaG基因片段克隆到载体pTrc99a上NcoI与HindIII酶切位点之间,获得重组质粒pTrc-JAGAPm9、pTrc-CB和pTrc-GW。
实施例2基因敲除
本实施例利用CRISPR/Cas9基因编辑技术能够高效快速连续地对大肠杆菌基因组进行目标基因的敲除失活。详细步骤如下阐述:
本实施例以wcaJ基因为例,详细阐述基因敲除的步骤,其他基因的敲除与此相同。
(1)引物设计:根据NCBI收录的大肠杆菌BL21(DE3)基因组序列信息(GenBank:CP001509.3)设计并合成待敲除基因wcaJ的鉴定引物和缺失引物(如表1)。
(2)目的基因的鉴定:利用鉴定引物对wcaJ-DF/DR和大肠杆菌BL21(DE3)基因组为模板,PCR克隆目标基因wcaJ,PCR扩增条带大小符合预期,且核苷酸测序结果与NCBI中GenBank:CP001509.3序列一致。
(3)sgRNA序列的设计:利用http://chopchop.cbu.uib.no/数据库,设计和筛选基因wcaJ敲除所需的sgRNA,即PAM位点及其上游20bp核苷酸序列,引物wcaJ-N20-F/R如表1中所示。
(4)同源臂的制备:利用引物对wcaJ-UP-F/R和大肠杆菌BL21(DE3)基因组为模板,PCR扩增目标基因wcaJ的上游同源臂序列,获得wcaJ-UP片段;利用引物对wcaJ-DO-F/R和大肠杆菌BL21(DE3)基因组为模板,PCR扩增目标基因wcaJ的下游同源臂序列,获得wcaJ-DO片段。
(5)Donor片段的制备:利用引物对wcaJ-UP-F和wcaJ-DO-R,以wcaJ-UP和wcaJ-DO片段为混合模板,通过重叠PCR将上下游同源臂融合,获得wcaJ-Donor片段。
(6)引导质粒gRNA-wcaJ-N20的制备:
①wcaJ-N20 oligo dsDNA的制备:利用互补引物对wcaJ-N20-F/R退火自搭形成oligo dsDNA双链,反应体系设置:引物wcaJ-N20-F/R各10μL;T4 ligase buffer 5μL;去离子水25μL,总体积50μL。退火反应程序:94℃煮2min,然后放置室温自然降温,待降到16℃,孵育10min。最后,将退火后产物稀释200倍,冻存于-80℃保存备用。
②pEcgRNA的线性化:利用限制性内切酶BsaI对引导质粒pEcgRNA进行酶切处理,酶切产物切胶回收,纯化备用。
③酶切后将线性化pEcgRNA和wcaJ-N20 oligo dsDNA片段按1:3的摩尔比进行混合,加入T4DNA连接酶后在22℃下连接2~5h,连接产物转化至大肠杆菌DH5α感受态细胞,在链霉素板上进行筛选,获得重组质粒pEcgRNA-wcaJ-N20。
(7)制备大肠杆菌BL21(DE3)的电转化感受态细胞并将质粒pEcCas转入其中,获得重组菌BL21(DE3)/pEcCas。随后转接含有pEcCas质粒的重组菌接种于LB液体培养基中,37℃、220rpm培养,当OD(600)值达到0.2左右时,加入终浓度为20mM的L-阿拉伯糖诱导重组酶表达,继续培养至OD(600)为0.6~0.8时,低温低速离心,收集菌体,制备大肠杆菌重组菌的电转感受态细胞。
(8)电转化:将100ng引导质粒pEcgRNA-wcaJ-N20和400ng Donor片段依次加入到含pEcCas质粒的重组菌感受态细胞中,电击复苏后涂布于含有壮观霉素(50μg/ml)和卡那霉素(50μg/ml),通过鉴定引物对,PCR扩增和测序确认获得目的基因被删除的突变菌株。
(9)编辑质粒的消除:鉴定成功的突变菌接种于含有卡那霉素(50μg/ml)培养基中,同时加入20mM鼠李糖,诱导引导质粒pEcgRNA-wcaJ-N20发生丢失,37℃、220rpm培养16~24h后,于含有10g/L蔗糖的固体平板上划线分离单菌,获得pEcCas质粒丢失的突变菌株E.coli BL21(DE3)△wcaJ(TK-C001)。
(10)重复上述1)~9)步骤,对基因组上lacZ基因进行敲除,最终获得双敲除菌株E.coli BL21(DE3)△wcaJ△lacZ(TK-C002)。
实施例3:基因整合
本实施例利用CRISPR/Cas9基因编辑技术能够高效快速连续地对大肠杆菌基因组进行靶基因或靶序列的整合敲入。所述操作步骤与实施例2中基因敲除部分操作一致,只是在Donor片段的设计和构建上略有不同,其中间融合了待敲入的目的基因序列。
(一)靶基因启动子的替换
本实施例以manA基因启动子的替换为例,详细阐述操作步骤,其他基因启动子的编辑与此相同。
(1)引物设计:根据NCBI收录的大肠杆菌BL21(DE3)基因组序列信息,设计并合成manA基因的引物(如表1)。
(2)目的基因的鉴定:利用鉴定引物对manA-DF/DR和大肠杆菌BL21(DE3)基因组为模板,PCR克隆目标基因manA及其启动子部分的序列,PCR扩增条带大小符合预期,且核苷酸测序结果与NCBI中GenBank:CP001509.3序列一致。
(3)sgRNA序列的设计:利用http://chopchop.cbu.uib.no/数据库,设计和筛选manA基因编辑所需的sgRNA,即PAM位点及其上游20bp核苷酸序列,如引物manA-N20-PAM所示(如表1)。
(4)同源臂的制备:为了在基因组上manA基因5’端上游敲入启动子BBa_J23119,我们将BBa_J23119启动子序列设计到上游同源臂的3’端和下游同源臂的5’端引物中,如引物manA-UP-R和manA-DO-F所示;利用引物对manA-UP-F、manA-UP-R和大肠杆菌BL21(DE3)基因组为模板,PCR扩增目标基因manA启动子的上游同源臂序列,获得manA-UP片段;利用引物对manA-DO-F、manA-DO-R和大肠杆菌BL21(DE3)基因组为模板,PCR扩增目标基因manA的下游同源臂序列,获得manA-DW片段。
(5)Donor片段的制备:利用引物对manA-UP-F和manA-DO-R,以manA-UP和manA-DW片段为混合模板,通过overPCR将上下游同源臂融合,获得含有目标启动子序列的整合片段manA-Donor-BBa_J23119。
(6)引导质粒gRNA-manA-N20的制备:参照实施例2中步骤(6);
(7)参照实施例2中步骤(7),制备电转化感受态细胞TK-C002/pEcCas;
(8)电转化:参照实施例2中步骤(8),将引导质粒pEcgRNA-manA-N20和Donor片段电转化到感受态细胞TK-C002/pEcCas中;复苏后涂布于含有壮观霉素(50μg/ml)和卡那霉素(50μg/ml),通过鉴定引物对,PCR扩增和测序确认获得manA基因启动子被编辑的突变菌株。
(9)编辑质粒的消除:鉴定成功的突变菌接种于含有卡那霉素(50μg/ml)培养基中,同时加入20mM鼠李糖,诱导引导质粒pEcgRNA-manA-N20发生丢失,37℃、220rpm培养16~24h后,于含有10g/L蔗糖的固体平板上划线分离单菌,从而消除pEcCas质粒,最终获得菌株E.coli BL21(DE3)△wcaJ△lacZ,manA::PJ23119-manA(TK-C003)。
(10)重复上述1)~9)步骤,在基因组上gmd基因5’端的上游敲入启动子Ptrc序列,最终获得菌株E.coli BL21(DE3)△wcaJ△lacZ,manA::PJ23119-manA,gmd::Ptrc-gmd(TK-C004)。
(11)重复上述1)~9)步骤,在基因组上manC基因5’端的上游敲入启动子Ptrc序列,最终获得菌株E.coli BL21(DE3)△wcaJ△lacZ,manA::PJ23119-manA,gmd::Ptrc-gmd,manC::Ptrc-manC(TK-C005)。
(二)靶基因的敲入
本实施例以α1,2-FT基因JAGAPm9的敲入为例,详细阐述操作步骤,其他基因的敲入与此相同。
(1)引物设计:根据菌株TK-C005基因组上现存的乳糖操纵子序列信息,设计并合成基因JAGAPm9敲入所需要的引物(如表1)。
(2)sgRNA序列的设计:利用http://chopchop.cbu.uib.no/数据库,设计和筛选所需的sgRNA,即PAM位点及其上游20bp核苷酸序列,如引物lac-N20-PAM所示(如表1)。
(3)JAGAPm9基因整合型Donor的制备:以菌株TK-C005基因组为模版,利用引物对lac-UP-F/R和lac-DW-F/R分别扩增上下游同源臂序列,获得DNA片段lac-UP和lac-DW;以重组质粒pTrc-JAGAPm9为模版,利用引物对trc-JAGAPm9-F/R进行PCR扩增,获得JAGAPm9基因表达盒;以片段lac-UP、lac-DW和JAGAPm9基因表达盒为混合模版,利用引物lac-UP-F和lac-DW-R进行overlap PCR扩增,将三个片段进行连接,获得JAGAPm9基因整合型Donor片段:Plac::Ptrc-JAGAPm9。
(4)引导质粒gRNA-Plac-N20的制备:参照实施例2中步骤(6);
(5)参照实施例2中步骤(7),制备电转化感受态细胞TK-C005/pEcCas;
(6)电转化:参照实施例2中步骤(8),将引导质粒pEcgRNA-Plac-N20和Plac::Ptrc-JAGAPm9片段电转化到感受态细胞TK-C005/pEcCas中;复苏后涂布于含有壮观霉素(50μg/ml)和卡那霉素(50μg/ml),通过鉴定引物对,PCR扩增和测序确认获得基因组上lac启动子被JAGAPm9基因表达盒所替换的突变菌株。
(7)编辑质粒的消除:参照实施例2中步骤(9),最终获得菌株E.coli BL21(DE3)△wcaJ△lacZ,manA::PJ23119-manA,gmd::Ptrc-gmd,manC::Ptrc-manC,Plac::Ptrc-JAGAPm9(TK-C006)。
(8)重复上述1)~7)步骤,利用重组质粒pTrc-CB和pTrc-GW为模版,扩增获得Ptrc-manCB片段和Ptrc-gmd-wcaG-rrnB terminater片段,并将二者融合;在菌株TK-C006基因组上betA-ykgH两个基因之间的间隔序列处敲入Ptrc-manCB-Ptrc-gmd-wcaG-rrnBterminater,最终获得菌株E.coli BL21(DE3)△wcaJ△lacZ,manA::PJ23119-manA,gmd::Ptrc-gmd,manC::Ptrc-manC,Plac::Ptrc-JAGAPm9,betA-ykgH::Ptrc-manCB-Ptrc-gmd-wcaG-rrnB terminater(TK-C007);
(9)重复上述1)~7)步骤,构建新的融合片段,将整合位点调整为fucI-fucK两个基因之间的间隔序列处,fucI基因被插入取代,并将Ptrc-manCB-Ptrc-gmd-wcaG-rrnBterminater敲入菌株TK-C007基因组上fucI-fucK两个基因之间,最终获得菌株E.coliBL21(DE3)△wcaJ△lacZ,manA::PJ23119-manA,gmd::Ptrc-gmd,manC::Ptrc-manC,Plac::Ptrc-JAGAPm9,betA-ykgH::Ptrc-manCB-Ptrc-gmd-wcaG-rrnB terminater,fucI-fucK::Ptrc-manCB-Ptrc-gmd-wcaG-rrnB terminater(TK-C008);
(10)重复上述1)~7)步骤,构建新的融合片段,将整合位点调整为ykgA-ykgQ两个基因之间的间隔序列处,并将Ptrc-gmd-wcaG-Ptrc-JAGAPm9-rrnB terminater敲入菌株TK-C008基因组上ykgA-ykgQ两个基因之间,最终获得菌株E.coli BL21(DE3)△wcaJ△lacZ,manA::PJ23119-manA,gmd::Ptrc-gmd,manC::Ptrc-manC,Plac::Ptrc-JAGAPm9,betA-ykgH::Ptrc-manCB-Ptrc-gmd-wcaG-rrnB terminater,fucI-fucK::Ptrc-manCB-Ptrc-gmd-wcaG-rrnB terminater,ykgA-ykgQ::Ptrc-gmd-wcaG-Ptrc-JAGAPm9-rrnBterminater(TK-C009);
(11)重复上述1)~7)步骤,构建新的融合片段,将整合位点调整为dadX-cvrA两个基因之间的间隔序列处,并将Ptrc-gmd-wcaG-Ptrc-JAGAPm9-rrnB terminater敲入菌株TK-C009基因组上dadX-cvrA两个基因之间,最终获得菌株E.coli BL21(DE3)△wcaJ△lacZ,manA::PJ23119-manA,gmd::Ptrc-gmd,manC::Ptrc-manC,Plac::Ptrc-JAGAPm9,betA-ykgH::Ptrc-manCB-Ptrc-gmd-wcaG-rrnB terminater,fucI-fucK::Ptrc-manCB-Ptrc-gmd-wcaG-rrnB terminater,ykgA-ykgQ::Ptrc-gmd-wcaG-Ptrc-JAGAPm9-rrnBterminater,dadX-cvrA::Ptrc-gmd-wcaG-Ptrc-JAGAPm9-rrnB terminater(TK-C010)。TK-C010中重组基因的示意图如图1所示。
实施例4含质粒版重组菌株的构建
(1)活化编辑后的TK-C002菌株,制备电转化感受态细胞,分别转化双质粒pR-CBGW和pET-JAGAP,pR-CBGW和pET-JAGAPm1、pET-JAGAPm2.....pET-JAGAPm10。在含有卡那霉素(50μg/ml)和氨苄青霉素(100μg/ml)的双抗平板上筛选正确的单克隆,获得携带2'-FL从头合成途径的重组菌株,命名为TK-R001、TK-R002.....TK-R011。
(2)活化编辑后的TK-C003菌株,制备电转化感受态细胞,转化双质粒pR-CBGW和pET-JAGAPm9。在含有卡那霉素(50μg/ml)和氨苄青霉素(100μg/ml)的双抗平板上筛选正确的单克隆,获得重组菌株TK-R012。
(3)活化编辑后的TK-C004菌株,制备电转化感受态细胞,转化双质粒pR-CBGW和pET-JAGAPm9。在含有卡那霉素(50μg/ml)和氨苄青霉素(100μg/ml)的双抗平板上筛选正确的单克隆,获得重组菌株TK-R013。
(4)活化编辑后的TK-C005菌株,制备电转化感受态细胞,转化双质粒pR-CBGW和pET-JAGAPm9。在含有卡那霉素(50μg/ml)和氨苄青霉素(100μg/ml)的双抗平板上筛选正确的单克隆,获得重组菌株TK-R014。
(5)活化编辑后的TK-C005菌株,制备电转化感受态细胞,转化质粒pET-JAGAPm9。在含有氨苄青霉素(100μg/ml)的抗性平板上筛选正确的单克隆,获得重组菌株TK-R015。
(6)活化编辑后的TK-C006菌株,制备电转化感受态细胞,转化双质粒pR-CBGW和pET-JAGAPm9。在含有卡那霉素(50μg/ml)和氨苄青霉素(100μg/ml)的双抗平板上筛选正确的单克隆,获得重组菌株TK-R016。
(7)活化编辑后的TK-C006菌株,制备电转化感受态细胞,转化质粒pET-JAGAPm9。在含有氨苄青霉素(100μg/ml)的抗性平板上筛选正确的单克隆,获得重组菌株TK-R017。
实施例5整合版(无质粒)重组菌株的构建
具体构建方法详见实施例3中步骤二所述,最后获得整合版重组菌株TK-C006、TK-C007、TK-C008、TK-C009、TK-C010。
实施例6摇瓶发酵生产2'-FL
具体步骤如下:
(1)将实施例4和5制备得到的重组大肠杆菌菌株在LB固体培养基上划线,37℃过夜培养,得到单菌落。
(2)挑取单菌落接种于装有20mL LB液体培养基的100mL锥形瓶中,于37℃、220r/min培养12h,得到种子液。
(3)以2%接种量取种子液转接至装有50mL发酵培养基的250mL锥形瓶中,于33~37℃、220rpm条件下培养至OD600=0.8~1.0时,加入0.5mM IPTG和15g/L乳糖,并转入28℃、250rpm条件下继续振荡培养诱导2'-FL合成。
(4)诱导发酵72h后,取样1ml,检测生物量、残留乳糖和产物水平,结果如图2和表3所示。重组大肠杆菌中随着各途径基因拷贝数的逐步增加,不断升高。最终质粒版重组菌株TK-R016在摇瓶发酵合成2'-FL产量最高达到7.06g/L;整合版重组菌株TK-C010在摇瓶发酵合成2'-FL产量最高达到8.72g/L;同时,所述重组菌发酵液中未检测到副产物DFL和3-FL的形成。
表3
实施例7重组菌株在5L发酵罐条件下发酵生产2'-FL
具体步骤如下:
(1)将重组菌株TK-R016和TK-C010分别在LB固体培养基上划线,37℃过夜培养,得到单菌落。
(2)挑取单菌落接种于装有50mL LB液体培养基的250mL锥形瓶中,于37℃、220r/min培养12h,制备得到种子液。
(3)按2%(v/v)的接种量转接种子液到装有150mL发酵培养基的500mL锥形瓶中,于37℃、220r/min培养6~7h,得到二级种子液。
(4)按照5%(v/v)的接种量将二级种子液转入装有2.3L M9发酵培养基的5L发酵罐中,于通气量1VVM,初始转速200rpm,34℃条件下培养至OD600=10,一次性添加终浓度为0.50mM的异丙基-β-D-硫代半乳糖苷(IPTG)诱导目的基因表达,同时降低温度至28℃;诱导培养后2h,开始添加终浓度为30g/L的乳糖;此后,当发酵液中乳糖低于10g/L时,间歇性补加乳糖至终浓度达到20g/L。直到发酵结束前24h,停止添加乳糖;发酵过程中以28%氨水维持pH=7.0±0.5,以80%(m/v)甘油碳源为补料维持发酵进行;同时,通过与转速偶联设置,控制发酵过程中溶氧水平=30~40%。
(5)诱导80h后停止发酵,取样检测。结果证实,利用5L罐成功实现了重组大肠杆菌的高密度发酵,重组菌株TK-C010在5L发酵罐中发酵合成2'-FL产量最高达到106.42g/L,单位生产效率达到1.34g/L/h。发酵罐中各物质含量随时间的变化如图3所示。
实施例8重组菌株传代稳定性
具体步骤如下:
(1)将重组大肠杆菌菌株TK-R016和TK-C010在无抗生素的LB固体培养基上划线培养,获得单菌落。
(2)挑取平板上的单菌落在新的LB固体培养基上划线培养,再次得到单菌落。
(3)以此方式推行,得到五代单菌落,分别命名为TK-R016-G1、TK-R016-G2、TK-R016-G3、TK-R016-G4、TK-R016-G5;TK-C010-G1、TK-C010-G2、TK-C010-G3、TK-C010-G4、TK-C010-G5。
(4)将五代单菌落分别接种于装有20mL LB液体培养基的100mL锥形瓶中,于37℃、220r/min培养12h,得到种子液。
(5)以2%接种量取种子液转接至装有50mL发酵培养基的250mL锥形瓶中,于35℃、220rpm条件下培养至OD600=0.8~1.0时,加入0.5mM IPTG和15g/L乳糖,并转入28℃、250rpm条件下继续振荡培养诱导产物合成。
(6)诱导发酵72h后,取样检测,结果如表4所示。从表4结果可以看出,基因组中整合有目的基因的重组菌株传代稳定性好,5代后依据具有良好的发酵能力。
表4
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 用于新生儿疾病筛查的智能化采血设备
- 一种用于新生儿疾病筛查采血装置