一种食管癌新辅助放化疗疗效评估的生物标记
文献发布时间:2023-06-19 09:29:07
技术领域
本发明涉及生物技术领域,特别是涉及一种食管癌新辅助放化疗疗效评估的生物标记。
背景技术
食管癌多数患者晚期诊断预后差,一旦发生转移,5年生存率将降至4%。对于局部晚期ESCC,术前新放化疗(nCRT)是治疗的主要组成部分。然而,病人在手术后常有疾病复发。同时,nCRT的预后也不同,nCRT 反应的患者生存时间更长,而无反应的患者预后更差。重要的是,nCRT可能增加术后并发症,临床参数(TNM分级、肿瘤位置)不能预测nCRT的反应。因此,通过识别新的生物标记物来预测nCRT反应,将有助于ESCC的管理。此外,它将帮助临床医生停止非有效治疗,从而避免过度治疗这些无反应的病人。
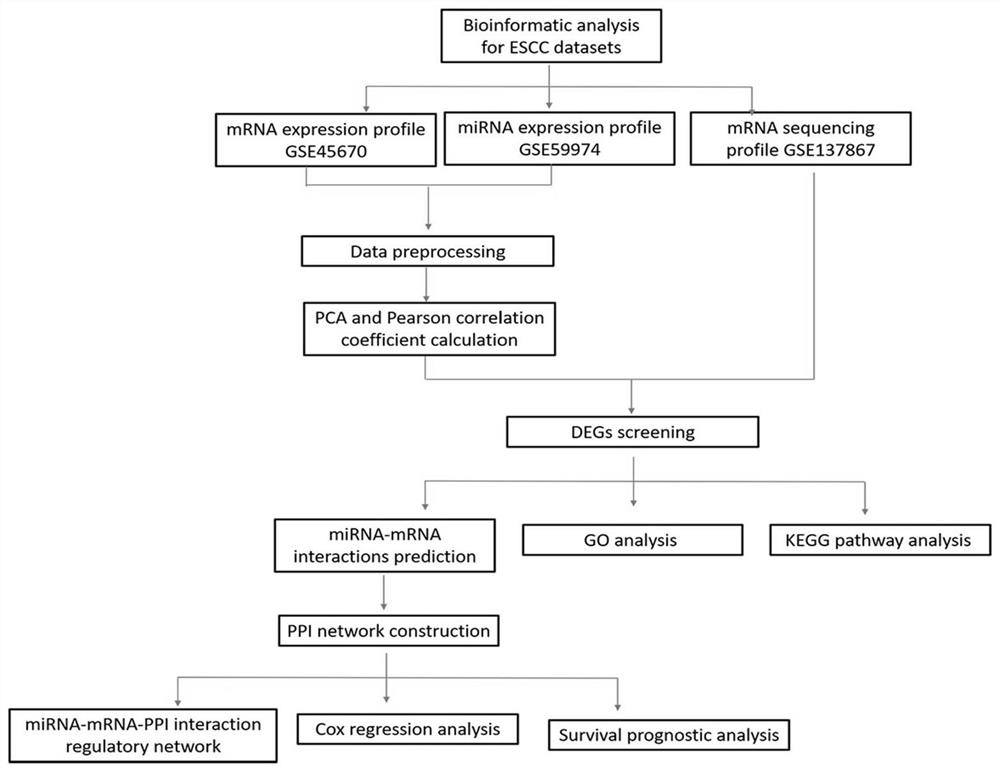
近年来,基因芯片分析和RNA测序技术的发展为探索基因表达变异与临床预后的关系提供了有利的信息。本研究在RNA测序和生物信息学分析的基础上,对ESCC中nCRT应答组和非应答组的差异表达基因(DEGs) 或差异表达miRNA进行了鉴定。此外,我们还构建了蛋白质相互作用(PPI)网络和miRNA mRNA网络,以探索与疾病进展相关的候选基因。Cox回归分析探讨ESCC患者生存时间与候选基因或miRNAs的相关性。ESCC 数据集的生物信息学分析示意图如图1所示。本发明探讨了ESCC的转录组表达模式,识别具有预后价值的新的生物标记物可以更好地了解ESCC的进展。
发明内容
本发明的技术方案如下:
MMP12蛋白作为生物标记物在制备食管癌新辅助放化疗疗效评估试剂、试剂盒或检测装置中的应用。
本发明提供了一种食管癌新辅助放化疗疗效评估的生物标记。在本发明中,术前新放化疗(nCRT)是治疗局部晚期食管癌的标准方法。然而,对于许多患者,nCRT的结果是不同的。在整合PPI网络和miRNA mRNA 网络后,我们筛选出8个与疾病进展相关的候选基因,包括肿瘤坏死因子、AKR1C1、AKR1C2、ICAM1、GPR68、 GNB4、SERPINE1和MMP12。此外,MMP12的异常表达水平与食管鳞癌的TNM分期、淋巴结转移、生存时间显著相关(P<0.05),本发明通过微阵列数据分析确定了预测食管鳞癌患者nCRT反应的新生物标记物,MMP12 可能是ESCC患者有用的肿瘤生物标志物和治疗靶点。
检测MMP12蛋白表达的产品在食管癌新辅助放化疗疗效评估中的应用。
一种食管癌新辅助放化疗疗效检测试剂盒,包括与MMP12蛋白特异性结合的检测物。
所述检测物优选自与MTHFD2蛋白特异性结合的抗体或者与MTHFD2编码基因特异性结合的引物或探针。
本发明的有益效果是:术前新放化疗(nCRT)是治疗局部晚期食管癌的标准方法。然而,对于许多患者, nCRT的结果是不同的。本发明旨在探讨预测食管鳞状细胞癌(ESCC)nCRT反应的潜在生物标记物。微阵列数据集(登录号GSE45670和GSE59974)从基因表达综合数据库(GEO)下载,包括ESCC nCRT应答者样本和非应答者样本。同时,对4例ESCC患者在nCRT前后的癌组织进行mRNA表达谱分析。筛选ESCC应答者和非应答者的差异表达基因(DEGs)和miRNAs。对这些DEGs进行功能富集分析,然后构建蛋白-蛋白相互作用 (PPI)网络和miRNA mRNA调控网络。最后,进行单变量生存分析,以确定在ESCC发展中具有预后价值的候选生物标记物。我们从ESCC应答组中鉴定了大量DEGs和差异表达的miRNAs。GO和KEGG分析表明,这些失调基因主要涉及生物过程和途径,包括“刺激反应”、“细胞对有机物的反应”、“信号转导调节”、“糖尿病并发症中的年龄-年龄信号途径”和“类固醇激素生物合成”。在整合PPI网络和miRNA mRNA网络后,我们筛选出8个与疾病进展相关的候选基因,包括肿瘤坏死因子、AKR1C1、AKR1C2、ICAM1、GPR68、GNB4、 SERPINE1和MMP12。单因素cox回归分析显示,失调miRNAs(hsa-miR-34b-3p、hsa-miR-127-5p、hsa-miR- 144-3p、hsa-miR-486-5p等)与ESCC患者生存时间无显著相关性。此外,MMP12的异常表达水平与食管鳞癌的TNM分期、淋巴结转移、生存时间显著相关(P<0.05),本发明通过微阵列数据分析确定了预测食管鳞癌患者nCRT反应的新生物标记物,MMP12可能是ESCC患者有用的肿瘤生物标志物和治疗靶点。
附图说明
图1为本发明的ESCC数据集生物信息学分析示意图。
图2为对mRNA表达数据集GSE45670进行主成分分析和相关分析
图2A为基于转录组学的主成分分析(PCA)和相关分析示意图。
图2B为热图显示组织标本的相关分析的示意图。
图3为本发明筛选ESCC-nCRT应答者和非应答者的差异表达基因(DEGs),火山图显示了DEGs的分布, ESCC应答者和ESCC非应答者样本中DEG的双向聚类分析。这些基因指的是两个数据集中的重叠基因失调的示意图。
图4为差异表达的miRNAs的热图
图4A为本发明火山图显示ESCC反应组和非反应组miRNA的差异表达示意图。
图4B为本发明热图显示ESCC反应组和非反应组miRNA的差异表达示意图。
图4C为本发明ESCC应答者与ESCC非应答者差异表达miRNAs的聚类分析示意图。
图5为miRNA基因相互作用的PPI网络分析与预测
图5A为本发明miRNA基因调控网络构建确定候选基因示意图。
图5B为本发明蛋白质-蛋白质相互作用(PPI)网络分析确定候选基因示意图。
图5C为本发明这些差异表达基因的PPI网络分析示意图。
图5D为本发明miRNA的mRNA调控网络分析,以确定与ESCC进展相关的关键基因。红色代表上调,而蓝色代表下调基因示意图。
图6为本发明ESCC候选基因的Kaplan-Meier生存曲线,包括肿瘤坏死因子、AKR1C1、ICAM1、MMP12等示意图。
图7为本发明单因素cox回归分析及ESCC应答者hsa-miR-34b-3p、hsa-miR-127-5p、hsa-miR-144-3p等差异表达mirna的生存分析结果示意图。
注:仅AKR1C1和AKR1C2被下调其余基因被上调。
具体实施方式
为了方便理解本发明的上述技术方案,以下通过具体使用方式上对本发明的技术方案进行详细说明:
下面结合附图对本发明做进一步说明。
实施例1
材料和方法
数据源和数据预处理。
首先,从基因表达综合数据库(GEO)下载基因表达谱GSE45670和miRNA表达谱GSE59974。这两组数据来自同一临床样本,包括17个ESCC无应答样本和11个ESCC应答样本。GSE45670数据集在hg-u133+u2 Affymetrix人类基因组u133+2.0平台上进行检测,GSE45670数据集在Agilent-038169人miRNA病毒v18.0 平台上进行分析。
此外,对南京医科大学附属常州第二人民医院4例nCRT前后的癌组织进行了活检。本研究经医院伦理研究委员会批准。转录组学数据(登录号GSE13786, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137867),并对这些临床标本进行mRNA表达分析。GSE137867被视为验证数据集。从三组数据中筛选出DEGs,重叠基因被认为是与ESCC相关的候选基因。
对这些微阵列数据进行数据标准化。我们从序列矩阵文件中提取基因表达值,并将相应的探针ID转换为基因符号。剔除异常信息后,选取ESCC应答者或非应答者的表达值进行主成分分析(PCA)。
PCA分析和DEGs筛选。
PCA是一种多变量技术,它将多个相关变量线性转换成一组值,而不相关变量表示主成分。它被广泛用于探索高维数据,如基因组和转录组表达数据。在本研究中,我们使用主成分分析法来研究样本在实验组与对照组之间的分布。通过对异常样本的检测和剔除,最终得到了一系列相似度较高的样本。
基于分位数归一化方法对miRNA基质和mRNA表达谱进行了归一化。采用Limma软件包[16]对实验组和正常对照组的DEGs和miRNAs进行筛选,阈值设为P<0.1,折叠变化>1.5(|log2FC|>0.585)。此外,在相同的截断准则下,我们筛选了GSE137867与地学数据集的重叠DEG进行进一步分析。
与ESCC相关的DEGs的功能富集分析。
对DEGs进行了基因本体论(GO)和京都基因与基因组百科全书(KEGG)信号途径富集分析。利用 Fisher精确检验,我们筛选了一组富含DEGs的生物过程和途径。P<0.05为显著性差异,P值越小的色谱柱表明DEGs与通路类型之间的联系越紧密。
miRNA与mRNA相互作用的预测。
通过研究各种数据库(TargetScan、miRTarBase、miRDB和miRanda),我们进一步预测了miRNA的相互作用。对于上调的miRNAs,我们探讨了它们与下调的靶基因的相关性。随后,我们重点研究了下调的miRNAs,并研究了它们与ESCC-nCRT应答器上调的靶基因的相关性。应用cytoscape软件对差异表达miRNAs与mRNAs 的关系进行可视化研究。
监管网络建设。
在线工具串(https://string-db.org网站/)用于分析蛋白质之间的相互作用。交互作用得分>0.7(高置信度)被设定为截止标准。在筛选关系对后,利用Cytoscape分析了PPI网络的拓扑性质(连通度、贴近度和中间度)。得分较高的蛋白质被认为是网络中的枢纽因子,可能是疾病进展的关键候选基因。
此外,我们还利用citoscape软件整合PPI网络和miRNA调控网络,预测参与ESCC进展的候选miRNA。
生存分析。
应用R软件中的生存包建立cox回归模型。基于TCGA数据集进行单变量cox回归分析和生存分析,以确定与ESCC生存相关的关键基因和mirna。
结果
从ESCC数据集中筛选DEG。
首先,对mRNA表达数据集GSE45670进行主成分分析和相关分析。数据预处理结果如图2所示。在 P<0.1和fold change>1.5(|log2FC|>0.585)阈值下,我们从ESCC应答组和非应答组中筛选出1311个 DEGs,其中上调基因672个,下调基因639个。对RNA序列GSE137867采用相同的截止标准,在nCRT前后对标本进行了大量的DEGs筛选。
此外,我们整合了RNA序列剖面与地理数据集之间的重叠DEGs。在这些基因重叠中,有17个基因在两个数据集中均上调,而8个基因在两个数据集中均下调(表1)。对这些DEGs进行了火山图和双向聚类分析,结果如图3所示。此外,通过设置P<0.1和折叠变化>1.5的阈值(log2FC>0.585),也可以鉴定差异表达的 miRNAs。结果显示为散点图和火山图。最后,我们从ESCC应答者组和非应答者组中分别鉴定出44个上调 mirna和15个下调mirna。表2中列出了差异表达的miRNAs,图4显示了热图。
对这些候选基因进行KEGG、GO分析。
GO分析结果显示,这些失调的DEG主要与几个生物学过程有关(表3),如“刺激反应”(计数=18, FDR=0.001617666)、“信号调节”(计数=12,FDR=0.001658521)、“有机物反应”(计数=11, FDR=0.001658521),“细胞对有机物的反应”(计数=11,FDR=0.001658521),“信号转导的调节”(计数=11,FDR=0.002048231)。此外,由这些基因富集的KEGG途径(表4)是“糖尿病并发症中的年龄信号途径” (计数=3,P=5.23e-04)、“Epstein-Barr病毒感染”(计数=3,P=3.91e-03)、“类固醇激素生物合成” (计数=2,P=3.91e-03)、“非洲锥虫病”(计数=2,P=1.63e-03)和“疟疾”(计数=2,P=2.85e-03)。
miRNA基因相互作用的PPI网络分析与预测。
通过预测上调miRNA的靶基因和下调miRNA的靶基因,我们分别为上调miRNA(图5A)和下调miRNA (图5B)构建了两个miRNA靶网络。在调控网络中,有几个分子被鉴定为连接度高于其他基因的枢纽基因,包括PER2(度=6)、PCTP(度=6)、hsa-miR-486-5p(度=6)。
利用在线工具串和Cytoscape软件,我们构建了DEGs的PPI网络(表5,图5C),最终得到肿瘤坏死因子(TNF)、醛固酮还原酶家族1成员C1(AKR1C1)、AKR1C2、细胞间粘附分子1(ICAM1)等8个基因间的 5对相互作用,卵巢癌G蛋白偶联受体1(GPR68)、鸟嘌呤核苷酸结合蛋白亚基β4(GNB4)、纤溶酶原激活物抑制剂1(SERPINE1)和基质金属蛋白酶12(MMP12)。
通过整合PPI网络和miRNA网络,我们最终构建了一个由多个miRNA和基因组成的调控网络。在这些基因中,PER2、PCTP、TNF和hsa-miR-486-5p的相互作用高于其他基因。因此,这些基因被鉴定为与疾病进展相关的hub基因。
在ESCC-nCRT应答者中识别具有预后价值的关键基因。
我们对独立队列患者(数据来源于TCGA数据库)进行生存分析,分析8个基因在ESCC患者中的预后意义。分析信号包括肿瘤坏死因子、AKR1C1、AKR1C2、ICAM1等基因,根据基因表达的中位数将患者分为高表达组和低表达组。比较两组患者的生存率,预测有预后价值的候选基因。结果显示MMP-12高表达组的预后较低表达组差(生存率危险比为1.737,P<0.05)。
对hsa-miR-34b-3p、hsa-miR-127-5p、hsa-miR-144-3p、hsa-miR-486-5p等差异表达的mirna进行单因素cox回归分析和生存分析。。然而,结果显示,任何失调的miRNAs与ESCC患者的预后没有显著相关性 (图7)。
结论
在本发明中,我们试图识别潜在的肿瘤生物标记物来预测ESCC的nCRT反应。在整合RNA测序数据集和地理数据集的重叠基因后,我们确定了两个数据集中均上调的17个基因,以及两个数据集中均下调的8个基因。功能富集分析显示,失调基因主要参与“刺激反应”、“信号转导调节”、“糖尿病并发症中的年龄-年龄信号通路”、“类固醇激素生物合成”。通过对PPI网络和miRNA网络的分析,我们确定了8个与ESCC进展相关的候选基因,如TNF、AKR1C1、AKR1C2、ICAM1、GPR68、GNB4、SERPINE1和MMP12。最后,单变量回归分析显示,MMP12的高表达水平与预后不良显著相关(图4)。
以往的研究已经探索了应答者和非应答者样本之间的基因表达差异,以预测食管癌的nCRT应答。Maher 等人确定了8个在应答组中有统计学差异表达的基因;根据预测模型的分析,5个基因可以在很大比例的食管癌患者中以高准确率(95%)预测nCRT应答。在最近的一篇论文中,研究人员发现应答者和非应答者之间存在9个失调基因;功能富集分析显示,4个基因(miR-422、CDK4、Cyclin D2和E2F3)主要与G1/S检查点发育有关,这些基因可以调节肿瘤对nCRT的敏感性。Els-Visser等系统地评估了22项食管癌研究中基因表达与临床结果的关系,发现基因表达状态、对nCRT的反应和淋巴结转移存在很大的异质性。
本发明显示,有8个基因被筛选为与ESCC进展相关的候选基因,包括肿瘤坏死因子、AKR1C1、ICAM1 等。AKR1C1/C2编码的酶属于aldo/keto还原酶超家族,这些酶在肿瘤细胞的耐药性中起着关键作用,与多环芳烃的代谢有关[29]。AKR1C1和AKR1C2在AKR1C亚家族中具有高度同源性,在7种氨基酸中存在差异。先前的研究表明AKR1C1/C2与EDHB诱导的食管癌细胞增殖抑制有关,这可能通过EDHB(酶的底物)促进食管癌的治疗。
在miRNA的mRNA调控网络中,本发明发现PER2、PCTP、TNF和hsa-miR-486-5p是与疾病进展相关的中枢基因,其连接度高于其他基因。MiRNA已经被证实通过调节癌基因或抗癌基因的水平在癌症发展中起主要作用。在ESCC患者中检测到特殊mirna的异常表达水平,如miR-10b、miR-21、miR-26a上调和miR-125b、 miR-203、miR-205下调。最近的一项研究表明,miR-486-5p在食管癌中表达下调,它可能通过调节细胞迁移而在疾病转移中发挥肿瘤抑制基因的作用。通过185例食管鳞癌组织芯片分析,Ren等人发现66.2%的食管鳞癌组织中miR-486-5p表达降低,miR-486-5p的异常表达与食管癌的预后有关。我们的结果预测下调miR- 486-5p,与靶基因ICAM1相互作用在ESCC的进展中起关键作用。ICAM1或CD54是一种90kda的糖基化跨膜蛋白,在内皮细胞和各种间充质干细胞上高度表达。它在细胞转移、增殖和多种细胞免疫反应中起重要作用。 ICAM1在肝癌和ESCC干细胞中表达失调,通过调控ESCC细胞中的转移相关基因,促进上皮向间质的转化。 ICAM1被认为是lncRNA-ECM参与肿瘤发生和ESCC进展的靶基因。然而,ICAM1和miR-486-5p在ESCC中的作用仍然不清楚,尤其是两者的相互作用。据我们所知,只有最近的研究报道下调miR-486-5p可以通过调节ICAM-1在乳腺癌中的转移介质来抑制肿瘤转移。由此推测,miR-486-5p对ICAM-1的调节可能在ESCC的转移和发展中起重要作用,而抑制ICAM-1可能是ESCC治疗的一种可能策略。
除hsa-miR-486-5p外,还对hsa-miR-34b-3p、hsa-miR-127-5p、hsa-miR-144-3p等几种肿瘤组织中差异表达的mirna进行了生存分析。。然而,任何失调的miRNAs与ESCC患者的预后没有显著相关性。根据文献检索,miR-127在几种癌症类型中被报道下调,包括胃癌、胶质母细胞瘤和肝细胞癌。在ESCC患者中, Gao等人发现miR-127通过调节癌基因Formin-like 3(FMNL3)在肿瘤样本中发挥抑癌作用[46]。另一篇文章报道了血清中miRNAs表达的分析结果,7个血清miRNAs最终被鉴定为ESCC生物标志物,包括miR-127- 3p[47]。至于其他miRNAs,组织或血清miR-144的表达在胃癌中被评估,然后低miR-144的表达预测胃肠道癌的预后不良。此外,miRNA-34b在ESCC的发生发展中起着致癌作用,根据大量中国人群分析,rs4938723/pri-miR-34b/c多态性与ESCC易感性有关。因此,这些miRNAs在ESCC放化疗反应中的潜在作用需要更多的实验研究来证实。
MMP-12蛋白又称人巨噬细胞金属弹性蛋白酶(HME)或巨噬细胞弹性蛋白酶(ME),属于MMPs家族。 MMP12与许多疾病中的弹性蛋白降解和巨噬细胞迁移有关,如慢性阻塞性肺病、皮肤病和癌症。然而,MMP12 在肿瘤中的作用是矛盾的。MMP12的异常表达见于多种癌症类型,包括肝细胞癌、肺癌、结肠癌和鼻咽癌。高表达HME/MMP12 mRNA的结直肠癌患者与正常组相比,生存率明显提高。在胃癌中,Cheng等人也观察到MMP12/HME蛋白的高表达,MMP12过表达的患者表现出更好的生存率。抗肿瘤作用可能与MMP12诱导血管生成抑制素有关,从而抑制肿瘤血管生成。然而,在大多数其他类型的癌症中,上调的MMP12被广泛报道与较低的生存时间有关。据我们所知,MMP12在ESCC中的潜在作用尚不确定。最近的一项研究表明,与正常鳞状上皮相比,MMP12在可切除肿瘤组织中高表达,MMP12的高表达与ESCC中较低的生存时间呈正相关。与之前的研究一致,本发明结果也显示MMP12的过度表达与病理分期和生存率低有关。在miRNA的mRNA调控网络中, MMP12与肿瘤坏死因子相互作用被认为是与疾病进展相关的中枢基因。然而,MMP12和TNFs的相互作用在肿瘤发展中的报道还很少。基于这些发现,本发明揭示MMP12与TNFs相互作用是ESCC进展的主要机制。
表1为从两个mRNA图谱(GSE45670和GSE137867)中鉴定出25个DEGs,其中ESCC应答组与对照组相比,有17个上调基因和8个下调基因示意表。
表1
表2为从miRNA图谱(GSE59974数据集)中鉴定出59个差异表达的miRNA,其中ESCC应答组与对照组相比有44个上调基因和15个下调基因示意表。
表2
表3为与ESCC(前十大生物过程术语)相关的差异表达基因的基因本体分析示意表。
表3
表4为ESCC差异表达基因的KEGG途径分析示意表。
表4
表5为基于连通度评价的ESCC差异表达基因蛋白质相互作用网络分析示意表。
表5
此外,本发明还存在一些局限性。应进行实验验证,以确定候选DEGs或miRNAs在ESCC发展中的确切生物学行为。同时,由于ESCC样本的数量较少,有必要在更大的队列中进一步验证这些基因的预测特性。
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的仅为本发明的优选例,并不用来限制本发明,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定
- 一种食管癌新辅助放化疗疗效评估的生物标记
- 直肠癌术前同期新辅助放化疗疗效评估系统和方法