一种测定生物样品中芬太尼类药物的方法及试剂盒
文献发布时间:2023-06-19 09:38:30
技术领域
本发明属于生物技术领域,具体涉及一种测定生物样品中芬太尼类药物的方法及试剂盒。
背景技术
芬太尼(fentanyl)是一种合成阿片类药物,于1960年由比利时科学家PaulJanseen首次合成,作为镇痛药出售,其镇痛效果为吗啡的80倍。芬太尼类药物为临床主要的麻醉用药,但药物剂量一旦过量,会导致呼吸抑制甚至死亡的危险。由于其强大的镇痛效果,芬太尼类物质被广泛使用,芬太尼非药用以及滥用的现象也随之出现。
芬太尼(及其衍生物)同阿片μ受体结合并具有高亲和力、高脂溶性和强内在活性。这些既是芬太尼的重要药理药效作用特点,但也是导致其致命的不良反应或毒性的主要原因,表现为同时具有强效镇痛效应和高滥用潜力,可快速透过细胞膜进入血脑屏障进入大脑,短时间形成血药高峰,极易形成耐受和药物依赖。芬太尼可通过皮肤、粘膜吸收,因此,此类物质中毒不但发生在滥用者中,而且可发生在无防护措施情况下芬太尼类药物处置或接触的工作人员中。
目前适用于对生物样品中芬太尼及其衍生药物进行初步筛选的分析方法多样,例如:免疫分析法、气相色谱-质谱、液相色谱-质谱(HPLC-MS/MS)方法等。免疫分析是利用抗原(靶标)和抗体的特异性结合来进行测定的一种方法,但不同的免疫分析法对芬太尼类似物的交叉反应具有局限性,且某些交叉反应是未知的;气相色谱-质谱方法不能直接测定非挥发性,极性或热不稳定性的物质,需要对目标物进行衍生化反应,与未来分析化学快速、高效的发展趋势不符。目前液相色谱-质谱方法作为最常用的检测手段,相比较于气相色谱-质谱方法来说,该技术灵敏度更高,更加稳定,而且适用范围更广,但非法和处方芬太尼类物质的液相色谱-质谱检测方法报道较少,导致处方芬太尼类药物的过量流行和非法芬太尼类药物的出现。因此,亟需一种可靠、快速、准确的分析检测方法,并将其应用于解决实际问题。
发明内容
针对现有技术中存在的问题,本发明目的是要提供一种测定生物样品中芬太尼类药物的方法及试剂盒。
为实现上述目的,本发明提供了如下方案:
本发明提供一种测定生物样品中芬太尼类药物的方法,采用HPLC-MS/MS测定生物样品中芬太尼类药物含量,具体包括以下步骤:
(1)样品预处理:将样品振荡离心后通过芬太尼类药物净化萃取管去除提取液中的共萃取杂质;
(2)HPLC-MS/MS检测:液相色谱采用0.1%甲酸-水溶液和0.1%甲酸-乙腈作为流动相;质谱采用电喷雾离子源正离子多反应监测模式检测。
进一步地,所述芬太尼类药物具体包括:乙酰芬太尼、异丁酰芬太尼、丙烯酰芬太尼、奥芬太尼、芬太尼、戊酰芬太尼和呋喃芬太尼。
进一步地,所述净化萃取管由混合净化剂、固相萃取柱、筛板、针筒推杆及滤膜组成。
进一步地,所述样品包括全血、唾液和尿液样品。
进一步地,步骤(1)所述混合净化剂各组分及用量为27mg C18吸附剂、29mg EMR(Bond Elut EMR-Lipid增强型脂质去除剂)、143mg NH
进一步地,所述混合净化剂各组分及用量的确定方式为采用化学计量学技术进行确定。
进一步地,所述化学计量学技术包括Plackett-Burman筛选试验及响应面-中心组合设计。
进一步地,所述滤膜是0.22μm亲水性PTFE微孔滤膜。
进一步地,步骤(2)所述的样品具体的振荡离心方式为:取样品加入2倍样品体积的乙腈振荡提取5min,在离心力15000g下离心10min。
进一步地,步骤(3)所述液相色谱色谱条件为:流动相A:0.1%甲酸水溶液(V/V),流动相B:0.1%甲酸-乙腈(V/V);色谱柱:Waters BEH C18色谱柱(100mm×2.1mm,1.7μm);流速:300μL/min;进样量:10μL。
所述质谱条件为:基于电喷雾离子源采取正离子多反应监测模式;定量检测方式:多反应监测模式;气帘气压力:20psi;碰撞气压力:7psi;去簇电压:120V;离子源电压:4.5kV;离子源温度:500℃。
本发明还提供一种测定生物样品中芬太尼类药物的试剂盒,其特征在于,包括净化萃取管、标准溶液、0.1%甲酸水溶液(V/V)、预装乙腈的离心管、质控品以及配套的一次性耗材。
本发明公开了以下技术效果:
1、本发明设计、研发了一种适用于血浆、唾液、尿液中7种芬太尼类药物快速萃取、处理的试剂盒,其中以一种新型萃取净化管为主体。该净化管原理类似于分散固相萃取技术,将混合净化剂预填充在净化管内,两端以筛板固定,操作动作类似于针筒吸取、排除液体,仅需抽、推两个动作即可在1min内使样品溶液与混合填料层充分接触两次,从而快速、高效完成净化;相比于传统净化方法,操作更加简单、快捷,无需氮吹浓缩、复溶等步骤,非专业技术人员也能操作,避免了因操作人员专业经验缺乏引起的测定结果准确度和精密度低等问题。
2、本发明的样品处理回收率高,稳定性好。本发明利用化学计量学技术对净化剂的吸附效果、回收率进行评价,快速筛选出对试验结果具有显著性影响的种类;同时设计中心组合实验矩阵高效预测、计算最佳配比和用量,相比传统控制变量优化方法可对每个因素的重要程度进行评价,且可在保证结果准确性、推导理论最佳值的情况下大大减少所需实验次数。
3、优化后的净化管可有效降低样品中的基质共萃取杂质,使基质效应控制在可接受范围内;同时,混合净化填料均匀,稳定性好,方法精密度、准确度及灵敏度均令人满意。
4、设计的集约化试剂盒所用试剂均预先填装和分小代包装,样本仅需一台小型离心机配合即可完成整个预处理过程,非常适用于现场取样、净化,后续可与便携式质谱设备或台式质谱衔接,可快速定性、定量分析样品中7种芬太尼类药物,这也是目前传统分析方法难以具备的优势。
附图说明
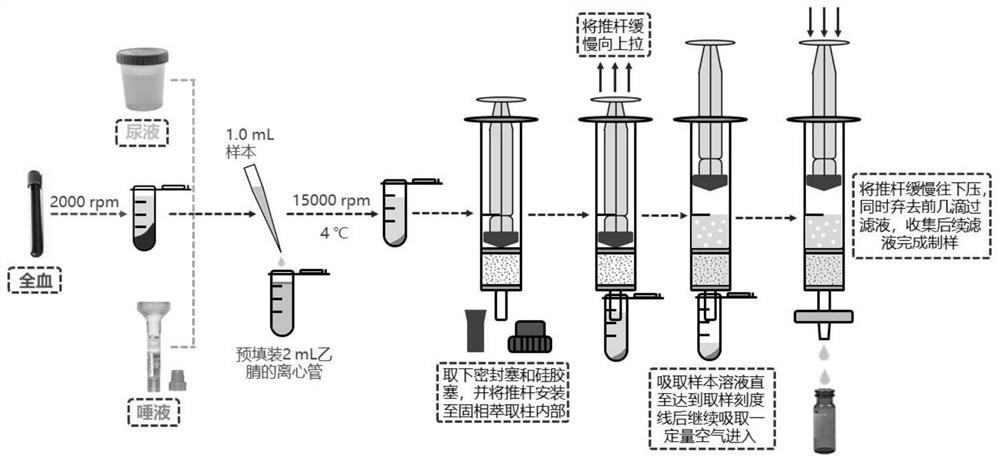
图1是净化装置预处理示意图;
图2是芬太尼类药物标准溶液(5.0ng/mL)MRM谱图;
图3是芬太尼类药物净化管设计示意图;
图4是净化效果对比的总离子流图;图4-A是使用萃取净化管后效果;图4-B是提取后未经净化直接进样;
图5是三种基质中进行三种浓度水平加标实验结果示意图。
具体实施方式
下面结合附图进一步说明本发明的实施例,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为还具体公开了该范围的上限和下限之间的每个中间值。在任何陈述值或陈述范围内的中间值以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。
在不背离本发明的范围或精神的情况下,可对本发明说明书的具体实施方式做多种改进和变化,这对本领域技术人员而言是显而易见的。由本发明的说明书得到的其他实施方式对技术人员而言是显而易见得的。本申请说明书和实施例仅是示例性的。
关于本文中所使用的“包含”、“包括”、“具有”、“含有”等等,均为开放性的用语,即意指包含但不限于。
本发明所涉及到的试剂和材料及实验仪器如下:
A.试剂与材料
乙酰芬太尼、异丁酰芬太尼、丙烯酰芬太尼、奥芬太尼、芬太尼、戊酰芬太尼、呋喃芬太尼标准溶液(100μg/mL,甲醇溶液),Sigma-Aldrich西格玛奥德里奇(上海)贸易有限公司;甲醇(CH
B.实验仪器
LC-20AD液相色谱仪,日本Shimadzu岛津公司;5500型质谱仪,美国Sciex公司;Milli-Q超纯水仪,美国Millipore公司。
实施例1
生物样品中芬太尼类药物含量的测定
1.1标准储备液的配制
芬太尼类药物混合储备标准溶液(10.0μg/mL):准确吸取各标准溶液1.0mL后用乙腈定容至10.0mL,于20℃下密封保存,有效期12个月;
1.2混合标准工作溶液的配制
芬太尼类药物混合标准工作溶液(1.0μg/mL):从各支标准储备溶液各吸取1.0mL后用乙腈定容至10.0mL,于-20℃下密封保存,有效期1个月。
1.3色谱条件
采用Waters BEH C18色谱柱(100mm×2.1mm,1.7μm),或相当者进行液相分离,柱温设置40℃,流速0.3mL/min,进样体积10μL,流动相A:0.1%甲酸水溶液(V/V),流动相B:0.1%甲酸-乙腈(V/V),液相梯度条件见表1。
表1液相梯度洗脱条件
1.4质谱条件
离子源:电喷雾离子源(ESI);正离子模式;定量检测方式:多反应监测模式(MRM),离子源电压4.5kV,源温度500℃,气帘气压力:20psi;碰撞气压力:7psi;芬太尼类标准物质多反应监测质谱条件见表2。
表2芬太尼类标准物质多反应监测质谱条件
1.5.样品提取
全血样品:取3mL以上全血样品转移至离心管中,以2000rpm速度离心5min后得到血浆。准确移取血浆样品1.0mL于离心管中(离心管中预先已装有2mL乙腈),多余血浆样品于-20℃条件下保存用于复测,振荡提取5min后继续以15000rpm,4℃条件离心10min,结束后转移上清液于新的试管中。
唾液、尿液样品:准确移取唾液、尿液样品1.0mL于离心管中(离心管中预先已装有2mL乙腈,需注意尽量减少样品中所携带气泡),振荡提取5min后继续以15000rpm,4℃条件下离心10min,结束后转移上清液于新的试管中。
1.6样品净化及测定
操作步骤示意图如图1所示,即净化装置预处理示意图,将萃取净化管上端密封塞及下端硅胶套摘下,将针筒推杆装入固相萃取柱中并推至底部。将固相萃取柱底端伸入样品提取液液面以下,同时将推杆缓慢向上拉伸(注意速度不可过快,一般将取样时间控制在1min左右)。取样结束后继续向上拉伸推杆,使部分空气进入净化管中,随后将0.22μm亲水性PTFE微孔滤膜安装在固相萃取柱底端同时将推杆向下压,在弃去前3-4滴液体后收集剩余滤液。准确吸取净化后的滤液500μL于进样瓶中,加入500μL纯水混合后完成制样,HPLC-MS/MS进样分析。
实施例2超高效液相色谱-串联质谱条件优化
芬太尼类药物极性中等,其在离子源中的离子化效率与流动性pH条件存在显著联系。当流动相中不添加甲酸或者加入氨水呈碱性后,所有芬太尼类药物几乎均不出峰,而在水相及有机相中加入0.1%甲酸后,各目标化合物响应值显著增加。而对比甲醇与乙腈作为流动相则效果差异不显著,考虑到采用乙腈作为样品提取剂后,最终选择0.1%甲酸-水溶液和0.1%甲酸-乙腈作为流动相组合,典型标准溶液(5.0ng/mL)MRM谱图如图2所示。
实施例3萃取净化管中混合净化剂条件优化
1、吸附剂的筛选
选取常用的QuEChERS净化剂,包括C18吸附剂,PSA(N-丙基乙二胺固相吸附剂),EMR(Bond Elut EMR-Lipid增强型脂质去除剂),碱性硅藻土,中性硅藻土,Florisil硅酸镁吸附剂,酸性氧化铝,中性氧化铝,碱性氧化铝,NH
表3净化剂回收率测试结果
从上表结果可知,Florisil硅酸镁吸附剂、酸性氧化铝及GCB填料会几乎吸附所有目标化合物,因此上述三种净化剂并不适用于净化芬太尼类药物。同时,PSA、碱性硅藻土、碱性氧化铝三种填料的回收率均在80%以上,表明其适用于芬太尼类药物的萃取,其中中性硅藻土萃取效果最佳;另一方面,C18吸附剂、EMR、NH
2、显著性因素筛查试验
显著性因素筛查试验即Plackett-Burman筛选试验,可针对当实验待优化因素超过4个时的情形,其基于非完全平衡块原理,能在实验次数较少的前提下选出对实验结果有显著影响的关键因素。若使用常规单因素控制变量法,所需试验次数多,且无法对因素的影响程度进行评估。在排除三种净化剂后,对剩余的净化剂如C18吸附剂、EMR、NH
表4Plackett-Burman试验设计矩阵
对所得结果进行分析,得到各目标化合物关键性因素:乙酰芬太尼(NH
3、响应曲面-中心组合设计
在确定对实验结果存在显著性影响的关键因素后,对其进行中心组合响应曲面设计,每个因素进行五个不同水平的实验来减少操作中偶然误差的影响,实验矩阵设计与结果如表5所示。
表5中心组合实验设计因素、水平及结果
利用软件结合实验结果进行模型拟合度分析,在本实验中,优选模型为二次多项式(Quadratic polynomial)模型。随后对所得数据进行二次多元回归拟合,整理得到对实验因素一次项、交互项和二次项进行评估的回归方程:
式中,Y为预测响应值,Xi和Xj代表独立变量,δ
表6二次多项式模型方差分析结果
通过结果可以发现,所有芬太尼药物的方程模型项P值均远远小于0.05表明其为极显著项;模型矢拟项均大于0.05,表明该实验所得数据受到误差影响的可能性低于0.01%;多项式模型方程的拟合程度和模型质量由确定系数(R
在确定净化剂种类及用量配比后,将净化剂充分混合均匀,填入空固相萃取柱中,两端以10μm规格筛板固定。固相萃取柱底端以硅胶塞密封,上端在净化管内充满N
4、方法学参数验证
在设计、确定净化管方案后,对整个预处理流程的方法学参数进行验证。首先,针对净化管在血浆样品中的净化效果进行了比较:(1)净化管使用时可显著吸附血浆中显黄色杂质,最终得到的处理液无肉眼可见明显色素成分;(2)质谱表现结果以总离子流图(见图4)展示,图4-A是使用萃取净化管后效果;图4-B是提取后未经净化直接进样;可以发现在萃取净化管净化后,总离子流图基线整体更加平整,保留时间1.5min、4min-5min之间的杂质峰干扰更低,后者也处于三种芬太尼类药物(异丁酰芬太尼、戊酰芬太尼和呋喃芬太尼)的保留时间附近。
随后对三种基质的基质效应进行考察,评价方法:取用预处理后的空白样品溶液,往其中加入适当浓度的标准溶液,配制成与标准曲线相同的基质匹配曲线(浓度分别为0.05ng/mL、0.10ng/mL、0.20ng/mL、0.50ng/mL、1.0ng/mL、2.0ng/mL、5.0ng/mL、10.0ng/mL、20.0ng/mL、50.0ng/mL,共10点),进样分析后计算基质匹配曲线与标准溶液曲线斜率,并取两者比值即为基质效应。结果总结于表7中,可以发现目标化合物在三种基质中基质(抑制或增强)效应不显著,基质效应均处于81.5%-108%之间,基本可忽略,但为了提高定量准确性仍建议使用基质匹配曲线外标法定量。
表7曲线线性及基质效应结果
注:上述所有曲线线性相关系数均大于0.999。
随后在三种基质中进行三浓度水平加标实验(n=6)以考察方法测定结果的准确性和重现性,浓度水平分别为1.0μg/kg、5.0μg/kg及10.0μg/kg(见图5)。
结果表明,在大部分情况下,三种基质中芬太尼类药物的回收率均在90%以上,方法准确度高;同时,RSD小于8.2%,证实方法重现性良好,结果令人满意。最后,在上述三种基质中对方法的灵敏度进行考察,以3倍信噪比定值检出限,方法为以接近检出限的浓度进行加标确认。最终除芬太尼检出限为0.2μg/kg、定量限为0.5μg/kg外,其余6种芬太尼类药物的检出限均为0.1μg/kg,定量限为0.3μg/kg,方法灵敏度极高,完全可应对人体体液样本中的痕量水平芬太尼药物的测定。
以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
- 一种测定生物样品中芬太尼类药物的方法及试剂盒
- 一种直接测定生物样品中未经分离的小核酸的方法及检测试剂盒