一种人血浆中利奈唑胺的LC-MS/MS检测方法
文献发布时间:2023-06-19 11:29:13
技术领域
本发明涉及人血浆中利奈唑胺浓度检测技术领域,尤其涉及是一种人血浆中利奈唑胺的LC-MS/MS检测方法。
背景技术
利奈唑胺(linezolid)是一种应用于临床的新型唑烷酮类抗菌药物,具有独特的药理机制,与其他药物无交叉耐药性,对大多数革兰阳性菌包括多重耐药的肠球菌属、葡萄球菌属和肺炎链球菌等引起的感染具有较好的治疗作用。由于感染患者在病理生理条件下存在影响药物体内分布和排泄的多种因素(如水肿、液体治疗、胸腔积水、烧伤等),可能影响药物在体内过程的改变。因此,在临床上建立一种简便、快捷、灵敏、可靠的利奈唑胺血药浓度的监测方法,可以更好地确保药物的有效治疗,优化利奈唑胺的临床使用,减少耐药菌株的产生及不良反应的发生。
目前,人血浆中测定利奈唑胺的方法主要有HPLC、LC-MS/MS及UPLC-MS/MS法等。然而,这些方法分别存在着灵敏度不够、样品处理烦琐、对仪器要求较高等限制。
因此,亟待开发一种灵敏性好、准确度高、测定范围广、样品处理简便的LC-MS/MS法,以检测人血浆中利奈唑胺的药物浓度,从而满足临床血药浓度监测及药动学研究的需要。
发明内容
针对现有技术存在的问题,本发明提出的一种人血浆中利奈唑胺的LC-MS/MS检测方法,首先,将待测人血浆样品通过蛋白沉淀法处理;然后,采用适当的有机溶剂和缓冲溶液作为混合流动相进行LC-MS/MS分析测定。
优选地,所述有机溶剂为甲醇,所述缓冲溶液为5mM乙酸铵水溶液。
该方法具体包括以下步骤:
S1,配制标准工作溶液,得到利奈唑胺工作溶液、利奈唑胺平行工作溶液、内标工作液、校正标样和质控样品。
进一步地,在利奈唑胺中加入甲醇,配制成浓度为2.000mg/mL的利奈唑胺储备液,将所述利奈唑胺储备液用体积比为1:1的甲醇-水溶液稀释为多份利奈唑胺工作溶液,其浓度范围为0.600~600.000μg/mL;按上述步骤重复一次,配置成多份利奈唑胺平行工作溶液,其浓度范围为0.600~450.000μg/mL;在内标中,加入甲醇配置成浓度为1.00mg/mL的内标储备液,再用体积比为1:1的甲醇-水溶液稀释成2.500μg/mL的内标工作液;所有储备液、工作液均在-20℃下保存,备用;取健康人空白血浆,分别加入所述利奈唑胺工作溶液,涡旋混匀,配置成多份相应浓度的校正标样,其浓度范围为30~30000ng/mL;取健康人空白血浆,分别加入所述利奈唑胺平行工作溶液,涡旋混匀,配置成多份相应浓度的质控样品,其浓度范围为30.000~22500.000ng/mL。所述内标采用利奈唑胺-d3。
更进一步地,所述利奈唑胺工作溶液浓度为:0.600μg/mL、1.200μg/mL、6.000μg/mL、30.000μg/mL、120.000μg/mL、240.000μg/mL、480.000μg/mL和600.000μg/mL;所述利奈唑胺平行工作溶液浓度为:0.600μg/mL、1.800μg/mL、18.000μg/mL、180.000μg/mL和450.000μg/mL;所述校正标样和所述质控样品为新鲜配制;所述校正标样的浓度为:30.000ng/mL、60.000ng/mL、300.000ng/mL、1500.000ng/mL、6000.000ng/mL、12000.000ng/mL、24000.000ng/mL、30000.000ng/mL;所述质控样品的浓度为:30.000ng/mL、90.000ng/mL、900.000ng/mL、9000.000ng/mL、22500.000ng/mL。
S2,取等体积的校正标样、质控样品、待测人血浆样品和空白人血浆,向校正标样、质控样品、待测人血浆样品和空白人血浆中分别加入所述内标工作液,向空白人血浆加入体积比为1:1的甲醇-水溶液,涡旋混匀、离心,取上清。通过在人血浆样品中加入甲醇沉淀剂,较传统固相萃取前处理方法,操作更简单,处理速度快,引进误差小,分离效果好,检测效率高。
进一步地,所述涡旋混匀的时间为10min;所述离心的条件为:离心时温度为4℃,离心转速为4200r/min,离心时间为10min。
S3,采用LC-MS/MS法测定步骤S2处理后的校正标样、质控样品的上清,以利奈唑胺和内标的色谱峰面积比为纵坐标,以人血浆中利奈唑胺浓度为横坐标制作标准曲线。通过校正标样及随行质控样品确定标准曲线,提高了检测的精密度和准确度。
进一步地,色谱条件:色谱柱:4.6×50mm,5μm;柱温:40℃;进样体积:2μL;流动相A:含有0.1%甲酸的5mM乙酸铵水溶液;流动相B:甲醇,洗脱,自动进样器温度为4℃,流动相的流速为0.8mL/min。
更进一步地,所述色谱柱采用Welch Ultimate XB-C18。
更进一步地,所述洗脱的程序为:
质谱条件:离子源:电喷雾电离源ESI;喷雾电压5500V;离子源温度:500℃;CUR:30.00psi;扫描模式:正离子多反应监测+MRM,利奈唑胺和利奈唑胺-d3离子反应分别为m/z338.2→195.1和m/z 342.3→298.2,碰撞能量CE分别为30和26V。通过采用多反应监测模式进行定性和定量,能够有效提高定性准确度和方法的灵敏度。
S4,将经步骤S2处理后的待测人血浆样品,按照步骤S3中所得的标准曲线方程计算,得到待测人血浆样品中利奈唑胺的浓度。
本发明的有益效果:1、本发明采用多反应监测模式进行定性和定量,能够有效提高定性准确度和方法的灵敏度,最低检测限可达到纳克级水平,分析时间短,所需样品量少;2、本发明在人血浆样品中加入甲醇沉淀剂,较传统固相萃取前处理方法,操作更简单,处理速度快,引进误差小,分离效果好,检测效率高;3、本发明利用校正标样及随行质控样品确定标准曲线,提高了检测的精密度和准确度;4、本发明的检测方法的专属性强、稳定性高、回收率高达95%以上,既能满足回收率的要求,又可以提高检测效率。
附图说明
图1为实施例1提供的人血浆中利奈唑胺的LC-MS/MS检测方法中利奈唑胺的产物离子扫描质谱图;
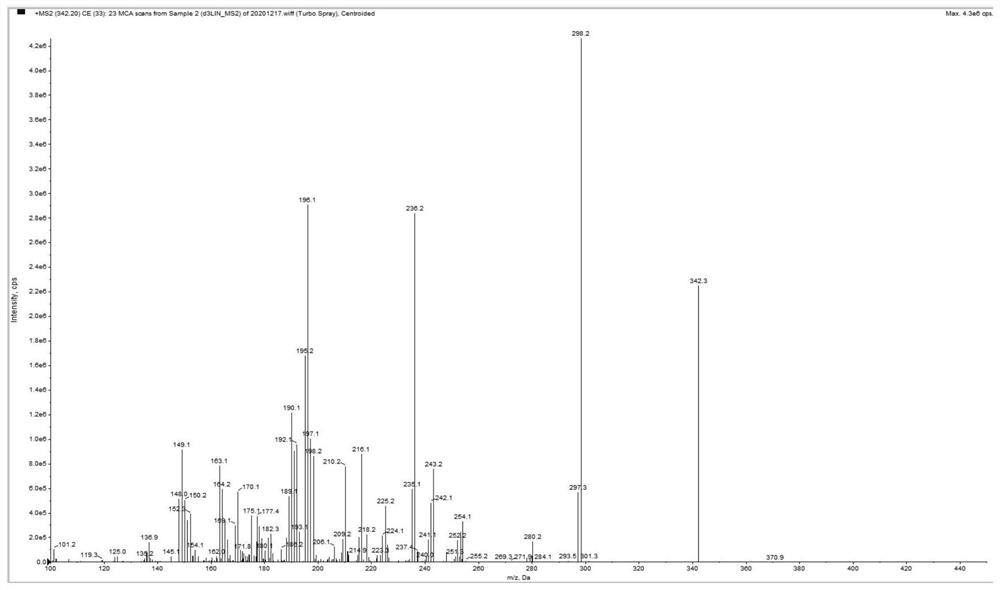
图2为实施例1提供的人血浆中利奈唑胺的LC-MS/MS检测方法中利奈唑胺-d3的产物离子扫描质谱图;
图3为实施例1中空白血浆样品中利奈唑胺(左)和利奈唑胺-d3(右)MRM色谱图;
图4为实施例1中LLOQ血浆样品中利奈唑胺(左)和利奈唑胺-d3(右)MRM色谱图。
具体实施方式
下面结合附图和具体实施方式对本发明作进一步详细的说明。本发明的实施例是为了示例和描述起见而给出的,而并不是无遗漏的或者将本发明限于所公开的形式。很多修改和变化对于本领域的普通技术人员而言是显而易见的。选择和描述实施例是为了更好说明本发明的原理和实际应用,并且使本领域的普通技术人员能够理解本发明从而设计适于特定用途的带有各种修改的各种实施例。
实施例1:
如图1至图4所示,一种人血浆中利奈唑胺的LC-MS/MS检测方法。
1、溶液及样品的配制
1.1标准系列样品:精密称取利奈唑胺照品适量,以甲醇溶解并定容,精密吸取各自储备液适量,以甲醇:水(1:1,v/v)逐级稀释得到标准系列工作溶液,以人空白血浆稀释该工作溶液,制成标准曲线样品利奈唑胺浓度范围为30.000~30000.000ng/mL,用于绘制标准曲线。
1.2质控样品:采用与标准系列样品相似方法配制利奈唑胺五个浓度水平质控样品,定量下限(LLOQ)浓度30.000ng/mL,低质控(LQC)浓度为90.000ng/mL,中1质控(M1QC)浓度为900.000ng/mL,中2质控(M2QC)浓度为9000.000ng/mL,高质控(HQC)浓度为22500.000ng/mL。
1.3内标溶液:精密称取适量的利奈唑胺-d3标准品,经质量校正系数校正后,将其溶于甲醇中,得到最终浓度为1.000mg/mL的储备液,精密吸取内标储备液适量,加甲醇:水(1:1,v/v)稀释,获得利奈唑胺-d3浓度分别为2.500μg/mL的内标工作溶液。
2、样品前处理
将室温下解冻的样品或新配制的样品,涡旋混匀。在96孔板的孔中加入50μL样品(标准曲线,质控样品,系统适用性样品或待测人血浆样品,对于双空白样品或者零点样品,加入50μL的空白基质样品);对于双空白样品或ULOQWithout IS样品,加入50μL50%甲醇溶液,对于其他样品加入50μL内标工作液(2.500μg/mL),涡旋混匀;每一个样品孔中加入400μL甲醇,封板,混匀10min;样品在4℃,4200r/min离心10min;取离心后上清液50μL至另一96孔收集板中,加入450μL 0.5%甲酸,封板,混匀10min;样品在4℃,4200r/min离心10min,待进样。
3、检测仪器及分析条件
3.1主要设备仪器如下表2
表2:主要设备仪器
3.2分析条件
3.2.1色谱条件:色谱柱采用Welch Ultimate XB-C18 4.6×50mm 5μm,柱温40℃,自动进样器设置为4℃,流动相A为含0.1%甲酸的5mM醋酸铵水溶液,流动相B为甲醇,洗针液:80%甲醇(含0.1%甲酸);柱塞洗液:10%甲醇,流速0.8mL/min,进样量2μL。
3.2.2质谱条件:电喷雾电离源ESI;喷雾电压5500V;离子源温度:500℃;CUR:30psi;扫描模式:正离子多反应监测+MRM,监测利奈唑胺和利奈唑胺-d3离子反应分别为m/z 338.2→195.1和m/z 342.3→298.2,碰撞能量CE分别为30和26V。
4、方法学验证
按照中国药典及美国FDA指导原则对本方法进行了方法学验证,内容包括稳定性、选择性、线性、准确度、精密度、残留效应、回收率、基质效应和稀释可靠性。
4.1选择性
取六个来源不同的空白血浆及各自配制的LLOQ样品处理后进样分析,获得空白血浆样品中利奈唑胺MRM色谱图图3和LLOQ样品利奈唑胺MRM色谱图图4,色谱共流出干扰物的峰面积均小于LLOQ待测物峰面积的20%,小于内标峰面积的5%。
4.2精密度和准确度
方法验证分析每一批测定五个浓度的质控样本各六个样本,连续测定三批,计算批内和批间精密度和准确度,LLOQ批内、批间精密度以相对标准差(RSD)计算小于20%方可接受,准确度以相对偏差计算(RE)在±20%之间方可接受,其余各浓度水平的QC样品各成分批内、批间精密度需小于15%方可接受,准确度在±15%之间方可接受,结果见表3。
表3:测定人血浆中利奈唑胺的精密度和准确度
4.3标准曲线
以待测物理论浓度为横坐标(x),待测物与内标物的峰面积比为纵坐标(y),用加权(W=1/X2)最小二乘法进行回归运算,求得的直线回归方程即为标准曲线,方法验证每一分析批对标准曲线样品双样本分析,获得利奈唑胺血浆样品标准曲线线性方程式y=0.00104x+0.00269(r=0.9981)表示利奈唑胺在30.000~30000.000ng/mL的浓度范围内线性关系良好。
4.4残留效应
残留效应验证为在高浓度样本检测后以空白样本进样,分析利奈唑胺和内标出峰时间处仪器的响应值。在定量上限样品后进样空白血浆样品,空白样品利奈唑胺保留时间处的色谱峰面积均小于当批次标准曲线定量下限峰面积的20%,内标保留时间处色谱峰面积均小于当批次标曲定量下限内标峰面积的5%。
4.5提取回收率
配制低、中2、高三个浓度质控血浆样品,每一浓度进行6样本分析,另取空白血浆,除不加内标外,进行相同处理,向获得的上清液中加一定浓度对照溶液,使待测物和内标最终浓度分别与低、中2、高质控样品处理后的理论浓度相同,每一浓度进行6样本分析,以每一浓度两种处理方法的峰面积计算回收率,利奈唑胺低、中2、高三个浓度的回收率分别为96.8%、98.2%和100.9%。内标的提取回收率为100.7%。
4.6基质效应
考察低、中2、高三个QC浓度水平下的基质效应。取6个不同来源空白血浆,除不加内标外,按血浆样品预处理方法操作,向获得的上清液中加一定浓度对照溶液,使得待测物和内标最终浓度分别与低、中2、高QC样品处理后的理论浓度相同,进行3样本分析,同时配制待测物和内标最终浓度分别与低、中2、高QC样品处理后的理论浓度相同的纯溶液样品,同法操作后进行6样本分析,分别计算两种处理方式下内标归一化基质因子,通过基质因子的精密度评估基质效应,不同来源血浆内标归一化基质因子的RSD小于15%方可接受。
利奈唑胺在低、中2、高浓度的内标归一化基质效应因子均值分别为0.960、0.997和0.990,不同来源血浆内标归一化基质因子的RSD均小于4.2%,以上结果表明,基质不干扰利奈唑胺的测定。
4.7稳定性
稳定性考核覆盖整个样本的检测过程,设置低浓度90.000ng/mL和高浓度22500.000ng/mL两个浓度,每一个浓度重复6个样本,考察全血冰浴、室温分别放置2小时稳定性、血浆样本室温放置20小时稳定性、-70℃放置20天稳定性、5次反复冻融稳定性、提取后样本4℃放置稳定性,相对偏差(%)在±15%之间方可接受,结果见表4。
表4:利奈唑胺血浆稳定性
4.8.稀释可靠性
制备利奈唑胺浓度为900.000μg/mL的纯溶液样本,制备利奈唑胺浓度为45000.000ng/mL的血浆样品,用空白血浆稀释两倍后,取50.0μL血浆进行预处理,处理完毕后进样分析,每个浓度制备6个样本,测得利奈唑胺稀释质控样品的精密度和准确度分别为94.6%和5.4%。
显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域及相关领域的普通技术人员在没有作出创造性劳动的前提下所获得的所有其他实施例,都应属于本发明保护的范围。显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域及相关领域的普通技术人员在没有作出创造性劳动的前提下所获得的所有其他实施例,都应属于本发明保护的范围。
- 一种人血浆中利奈唑胺的LC-MS/MS检测方法
- LC-MS/MS联用检测人血浆中帕唑帕尼药物浓度的方法