一种改性阴离子型植物油基水性聚氨酯乳液及其制备方法和应用
文献发布时间:2023-06-19 13:45:04
技术领域
本发明属于高分子材料技术领域,尤其涉及一种改性阴离子型植物油基水性聚氨酯乳液及其制备方法和应用。
背景技术
几十年来,聚氨酯(PU)一直被认为是用途最广泛的聚合物之一,并应用于各种工业领域,如油墨、粘合剂、泡沫、密封剂等。然而,在生产和应用过程中,传统溶剂型聚氨酯总是使用大量的挥发性有机化合物(VOC)和有害空气污染物(HAP),对环境和健康造成严重危害。因此,水性聚氨酯(WPU)被认为是传统溶剂型聚氨酯的替代品。随着原油储量有限的问题日益受到关注,利用生物基原料,包括纤维素、木质素和植物油作为石油基对应物的替代品,是新的有前途的领域。其中,植物油因其低成本、可再生性和易得性而成为最有前途的选择之一。植物油分子中的三个酯和0-7个碳碳双键,为化学改性提供了反应位点。蓖麻油是唯一一种含有羟基的天然植物油,可直接用作多元醇无需进一步改性即可制备WPU,而其他植物油则必须进行改性以引入羟基。然而,基于植物油的多元醇的柔性长链脂肪酸和相对较低的-OH值(主要为70-300mg KOH/g)导致其所得WPU薄膜受到一定的机械和热性能限制,而影响植物油基水性聚氨酯实际应用。
针对以上问题,发明人提出了通过分子链结构的设计,通过改变其分子结构来提高植物油基水性聚氨酯的物理化学性能,并用于实际应用,使其制备的材料能具有较好的机械性能、玻璃化转变温度,并具有良好的防紫外透过性和防腐蚀性能。
发明内容
本发明的首要目的在于提供一种改性阴离子型植物油基高交联密度水性聚氨酯乳液。
本发明的另一目的在于提供上述水性聚氨酯乳液的的制备方法,利用司盘这一具有高活性反应位点的生物基单体增加水性聚氨酯的交联密度,操作简单,相比同类型的材料,具有较好的机械性能、玻璃化转变温度、防紫外透过性和防腐蚀性能。
为实现上述目的,本发明提供如下技术方案:
一种改性阴离子型植物油基水性聚氨酯,包括如下按重量份计的组分:蓖麻油30~80份,司盘3~25份,异弗尔同二异氰酸酯4~50份,2,4-二羟甲基丁酸10~15份,三乙胺5~10份,水800~1100份,催化剂0.05~0.2份。
本发明中司盘(失水山梨醇脂肪酸酯)采用市售司盘系列产品均可,例如司盘-85、司盘-80、司盘-60、司盘-40、司盘-20等。
本发明同时提供所述改性阴离子型植物油基水性聚氨酯的制备方法,包括如下步骤:
S1、将司盘和异弗尔同二异氰酸酯预聚反应得到以-NCO基团封端的预聚体;
S2、将蓖麻油和2,4-二羟甲基丁酸加入到步骤S1制备的预聚体中,并添加催化剂反应至当反滴定测定体系中NCO基团降至10%以下时,加入有机溶剂将预聚体稀释成溶液继续反应2~4h;
S3、在步骤S2反应后的溶液中加入中和剂中和,然后加水乳化,最后旋蒸,获得所述水性聚氨酯乳液。
作为一种优选的技术方案,步骤S1中所述预聚反应的温度为60~90℃,反应时间为1-2h。
作为一种优选的技术方案,步骤S2中所述蓖麻油与步骤S1中所述司盘的羟基摩尔比例为9:1~4:6。
作为一种优选的技术方案,步骤S2中所述催化剂为二月桂酸二丁基锡,催化温度为60~90℃,以加速-NCO基团与-OH反应速率。
作为一种优选的技术方案,步骤S3中所述加水乳化的机械转速为300~500r/min,搅拌时间为2h。
本申请同时要求保护所述改性阴离子型植物油基水性聚氨酯乳液在制备防紫外及防腐蚀材料中的应用。
进一步地,将此水性聚氨酯材料进行应用时,所述水性聚氨酯乳液的固含量为15~20%。
本申请通过实验发现,司盘可用于提升水性聚氨酯防紫外和防腐蚀材料中的应用,更具体地,将司盘和植物油作为多元醇进行水性聚氨酯合成反应。
本发明中水性聚氨酯在特殊波长段原本没有抗紫外性,通过分子链结构的设计使其与其他材料复合,通过改变植物油基水性聚氨酯分子结构,明显提高了材料的抗紫外透过性和防腐蚀性能。
与现有技术相比,本发明的有益效果是:
(1)本发明以司盘(失水山梨醇脂肪酸酯)作为多元醇,利用这一具有高活性反应位点的生物基单体增加水性聚氨酯涂层的交联密度,改性得到的水性聚氨酯具有良好的抗紫外透过性和防腐蚀性能,可以用于制备具有防紫外和防腐蚀性能的材料中。
(2)本发明利用原有制备工艺进行反应,仅以司盘(失水山梨醇脂肪酸酯)替代部分蓖麻油进行改性,操作简便,反应程度高,而且相比同类型的材料,具有较好的机械性能、玻璃化转变温度、防紫外和防腐性能,进一步拓宽了其应用领域,应用前景好。
附图说明
图1为本发明制备技术路线。
图2为本发明实施例及对比例的水性聚氨酯膜机械性能图。
图3为本发明实施例及对比例的水性聚氨酯膜紫外透过图。
图4为本发明实施例及对比例的水性聚氨酯凝胶率测试图。
图5为本发明实施例及对比例的水性聚氨酯电化学腐蚀极化曲线图。
具体实施方式
下面将结合本发明实施例和对比例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
下述实施例中所使用的试验方法如无特殊说明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。
实施例1
一种失水山梨醇单油酸酯改性阴离子型植物油基高交联密度水性聚氨酯乳液,按以下方法制备得到:
将异佛尔酮二异氰酸酯IPDI(4.4186g)和计算量的SP(0.3341g)加入干燥的二颈烧瓶中,并加入适量的二月桂酸二丁基锡DBTDL(0.01g)作为催化剂,在78℃下(250rpm)搅拌反应2小时。加入蓖麻油多元醇(7.2g)和2,4-二羟基丁酸(1.19g)继续反应1小时,然后加入少量丁酮MEK(20ml)降低体系粘度,继续反应。当反滴定测定体系中NCO基团降至10%以下时,将体系温度降至室温,加入三乙胺TEA(0.7919g)中和30分钟。最后在双口瓶中加入去离子水(102.22mL),剧烈搅拌(300r/min~500r/min),使聚氨酯反乳化2小时。通过旋转蒸发仪去除MEK后,得到所述水性聚氨酯乳液。
实施例2
一种失水山梨醇单油酸酯改性阴离子型植物油基高交联密度水性聚氨酯乳液,按以下方法制备得到:
将IPDI(4.4186g)和计算量的SP(0.6682g)加入干燥的二颈烧瓶中,并加入适量的DBTDL(0.01g)作为催化剂,在78℃下(250rpm)搅拌反应2小时。加入蓖麻油多元醇(6.4g)和2,4-二羟基丁酸(1.19g)继续反应1小时,然后加入少量MEK(20ml)降低体系粘度,继续反应。当反滴定测定体系中NCO基团降至10%以下时,将体系温度降至室温,加入TEA(0.7919g)中和30分钟。最后在双口瓶中加入去离子水(98.81mL),剧烈搅拌(500r/min),使聚氨酯反乳化2小时。通过旋转蒸发仪去除MEK后,得到所述水性聚氨酯乳液。
实施例3
一种失水山梨醇单油酸酯改性阴离子型植物油基高交联密度水性聚氨酯乳液,按以下方法制备得到:
将IPDI(4.4186g)和计算量的SP(1.0024g)加入干燥的二颈烧瓶中,并加入适量的DBTDL(0.01g)作为催化剂,在78℃下(250rpm)搅拌反应2小时。加入蓖麻油多元醇(5.6g)和2,4-二羟基丁酸(1.19g)继续反应1小时,然后加入少量MEK(20ml)降低体系粘度,继续反应。当反滴定测定体系中NCO基团降至10%以下时,将体系温度降至室温,加入TEA(0.7919g)中和30分钟。最后在双口瓶中加入去离子水(95.39mL),剧烈搅拌(500r/min),使聚氨酯反乳化2小时。通过旋转蒸发仪去除MEK后,得到所述水性聚氨酯乳液。
实施例4
一种失水山梨醇单油酸酯改性阴离子型植物油基高交联密度水性聚氨酯乳液,按以下方法制备得到:
将IPDI(4.4186g)和计算量的SP(1.3365g)加入干燥的二颈烧瓶中,并加入适量的DBTDL(0.01g)作为催化剂,在78℃下(250rpm)搅拌反应2小时。加入蓖麻油多元醇(4.8g)和2,4-二羟基丁酸(1.19g)继续反应1小时,然后加入少量MEK(20ml)降低体系粘度,继续反应。当反滴定测定体系中NCO基团降至10%以下时,将体系温度降至室温,加入TEA(0.7919g)中和30分钟。最后在双口瓶中加入去离子水(91.97mL),剧烈搅拌(500r/min),使聚氨酯反乳化2小时。通过旋转蒸发仪去除MEK后,得到所述水性聚氨酯乳液。
实施例5
一种失水山梨醇单油酸酯改性阴离子型植物油基高交联密度水性聚氨酯乳液,按以下方法制备得到:
将IPDI(4.4186g)和计算量的SP(1.6706g)加入干燥的二颈烧瓶中,并加入适量的DBTDL(0.01g)作为催化剂,在78℃下(250rpm)搅拌反应2小时。加入蓖麻油多元醇(4g)和2,4-二羟基丁酸(1.19g)继续反应1小时,然后加入少量MEK(20ml)降低体系粘度,继续反应。当反滴定测定体系中NCO基团降至10%以下时,将体系温度降至室温,加入TEA(0.7919g)中和30分钟。最后在双口瓶中加入去离子水(88.56mL),剧烈搅拌(500r/min),使聚氨酯反乳化2小时。通过旋转蒸发仪去除MEK后,得到所述水性聚氨酯乳液。
实施例6
一种失水山梨醇单油酸酯改性阴离子型植物油基高交联密度水性聚氨酯乳液,按以下方法制备得到:
将IPDI(4.4186g)和计算量的SP(2.0047g)加入干燥的二颈烧瓶中,并加入适量的DBTDL(0.01g)作为催化剂,在78℃下(250rpm)搅拌反应2小时。加入蓖麻油多元醇(3.2g)和2,4-二羟基丁酸(1.19g)继续反应1小时,然后加入少量MEK(20ml)降低体系粘度,继续反应。当反滴定测定体系中NCO基团降至10%以下时,将体系温度降至室温,加入TEA(0.7919g)中和30分钟。最后在双口瓶中加入去离子水(85.14mL),剧烈搅拌(500r/min),使聚氨酯反乳化2小时。通过旋转蒸发仪去除MEK后,得到所述水性聚氨酯乳液。
对比例1(不添加失水山梨醇单油酸酯进行改性制备)
将IPDI(4.42g)加入干燥的二颈烧瓶中,并加入适量的DBTDL(0.01g)作为催化剂,在78℃下(250rpm)搅拌反应2小时。加入蓖麻油多元醇(8g)和2,4-二羟基丁酸(1.19g)继续反应1小时,然后加入少量MEK降低体系粘度,继续反应。当反滴定测定体系中NCO基团降至10%以下时,将体系温度降至室温,加入TEA(0.7919g)中和30分钟。最后在双口瓶中加入去离子水(105.65mL),剧烈搅拌(500r/min),使聚氨酯反乳化2小时。通过旋转蒸发仪去除MEK后,得到所述水性聚氨酯乳液。
对比例1与实施例X不同的之处在于,不添加失水山梨醇单油酸酯进行改性。
为了比较各实施例和对比例制备的水性聚氨酯的综合性能,通过以下实验例进行测试。
实验例1:力学性能测试
样品制备:将所得乳液室温干燥后的膜样品剪裁成30mm×10mm(长×宽)的矩形样品。在拉力机上以100mm/min的延伸速率测量薄膜的拉伸性能。在测试中对所有样品进行了三份以上的重复试验,并将数据记录为平均值±标准偏差。测试结果见表1。
表1植物油基水性聚氨酯力学性能测试结果
表1结果说明,实施例样品的综合机械性能优于对比例,随着实施例中SP摩尔比例增加,水性聚氨酯的力学性能显着提高。具体来说,不含SP的聚氨酯薄膜的拉伸强度较低,为10.76MPa。但随着聚氨酯中SP含量从0%增加到50%,WPU-SP50的拉伸强度增加到26.32MPa。SP含量对聚氨酯拉伸强度的增加主要是由于在聚合物链段中引入了SP,增加了聚合物链段的硬链段含量和聚合物网络的交联密度。同时,将刚性呋喃环与SP一起引入聚氨酯网络中,增加了网络段的刚性。高硬链段含量、交联密度和刚性结构可以促进高分子聚合物的微相分离程度,从而显着提高材料的拉伸强度。总的来说,将SP根据其特征结构引入水性聚氨酯网络中,调控了聚氨酯材料的强度。
实验例2紫外透射性能测试:
使用DU 800紫外/可见分光光度计(Beckman Coulter,美国)在200-800nm范围内测量内径为40mm×40mm,厚度为500μm的水性聚氨酯乳液涂层薄膜的吸光度。所有薄膜测试都使用基线校正进行空气测量。测量结果见图3。图3显示了不同SP摩尔比的水性聚氨酯薄膜的紫外透射光谱,所有厚度在0.5mm左右的水性聚氨酯薄膜,其紫外光谱(200-800nm)从可见光区到近紫外区的紫外透过率逐渐降低,并随着SP含量的增加而逐渐降低。聚氨酯薄膜紫外线透过率逐渐降低。然而,所有薄膜在550-800nm的紫外线透过率都在80%以上,表明所有聚氨酯薄膜都具有优异的透明度。详细地说,所有薄膜样品在UVC(200-300nm)区域的UV透过率为0%,这表明所有水性聚氨酯薄膜都可以抵抗UVC辐射。更值得一提的是,在UVB(280-320nm)区域,只有WPU薄膜具有紫外线透过性,而其他含有SP的聚氨酯薄膜则是不透过紫外线的,这说明多元醇中SP的引入增加了材料对紫外线辐射的抵抗力意义重大。同时还可以观察到,WPU薄膜在400nm光谱范围内的UV透射率为70%,而WPU-SP10的UV透射率为38%,而SP含量较高的聚氨酯薄膜的UV透过率更为降低。这说明在UVA(320-400nm)区域,含SP的聚氨酯薄膜的紫外吸收能力远高于WPU薄膜。并且WPU-SP50和WPU-SP60在近紫外区域(200-380nm)表现出完全的抗紫外线能力。以SP为多元醇制备的SP/蓖麻油基水性聚氨酯薄膜在UVA(320-400nm)、UVB(280-320nm)和UVC(200-280nm)区域具有较强的抗紫外线能力。总的来说,SP较高的羟值导致更大的交联网络和更高的交联密度,提高了材料的内部致密性。这种结构使材料对紫外线产生强烈的折射和反射,从而降低聚氨酯薄膜的透光率。并且随着SP含量的增加,聚氨酯薄膜的透光率逐渐降低。
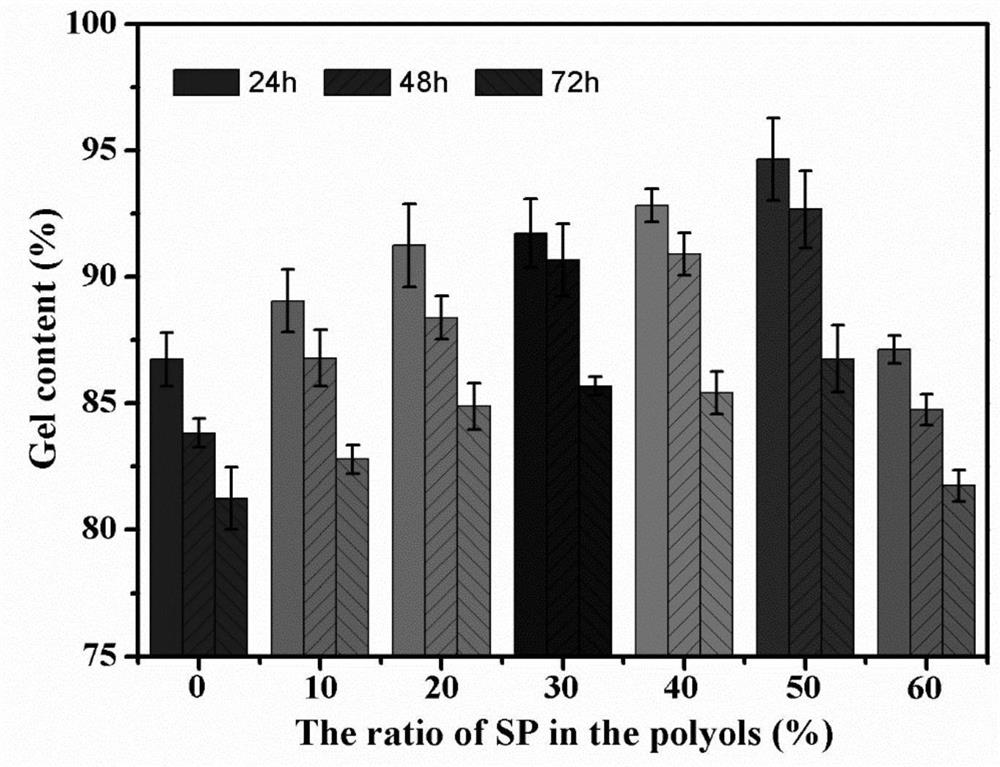
实验例3凝胶率测试:
凝胶含量用于计算聚合物中各种组分的反应程度。将干燥后的聚氨酯薄膜(m
根据以下公式计算样品凝胶含量:凝胶含量(%)=m
其中“m
由图4可知,在THF中浸泡24小时、48小时和72小时后,所有膜样品都显示出类似的凝胶化下降趋势。具体而言,WPU薄膜在THF中浸泡24h后的凝胶率为86.74%,72h后的凝胶率为81.24%。由于SP作为多元醇添加到水性聚氨酯中,薄膜的凝胶分数增加并随着SP含量的增加而增加。WPU-SP50薄膜的凝胶率在24h和72h分别达到94.65%和86.76%。这进一步说明,与以纯蓖麻油为多元醇制备的WPU相比,将分子量相对较低的SP代替部分蓖麻油引入水性聚氨酯结构中,提供更多的交联位点,使水性聚氨酯产生更多的交联。网络结构。这在聚氨酯薄膜的机械性能测试和热物理行为分析中也得到了证实。但是,当SP的摩尔比占多元醇的60%时,薄膜的凝胶率远低于其他样品,仅为80.12%。这可能与水性聚氨酯的合成工艺有关。在这项研究中,SP首先与IPDI反应形成小的刚性基团分子,最后用DMBA进行扩链反应形成聚合物。此时,DMBA主要与刚性小分子相连,导致分子扩链过程中硬链段集中聚集,反应基团的积累增加了空间位阻,阻碍了水性聚氨酯的链增长过程。最终,大量的小分子反应基团没有反应并溶解在THF中,导致WPU-SP60的凝胶率下降。
实验例4电化学腐蚀测试:
样品的塔菲尔分析数据由电化学工作站(上海晨华CHI 600E,中国)记录。将本发明中各实施例及对比例中制备的水性聚氨酯乳液以50±5μm的厚度涂覆在马口铁上以制备样品。3.5wt%NaCl水溶液为电解质,三电极体系[饱和Ag/AgCl电极为参比电极,铂(Pt)电极为对电极,分别以未涂层或涂层马口铁作为工作电极]用于样品和空白样品的1×1cm
表2植物油基水性聚氨酯力电化学腐蚀测试结果
图5显示,本发明中涂覆的水性聚氨酯样品实施例比对比例样品具有明显更高的Ecorr和更小的Icorr(见表2),这可以通过水性聚氨酯涂料的阻隔性来解释腐蚀性颗粒。还可以观察到,多元醇中SP的比例对水性聚氨酯涂料的防腐性能有重要影响。WPU的IE为89.60%,而WPU-SP的IE可以达到95.39%。同时,WPU-SP还具有较高的Ecorr和较小的Icorr(见表4)。这可以归因于在水性聚氨酯中引入SP以构建更致密的交联网络结构,这强烈阻碍了电解质中的腐蚀性颗粒与金属基材的接触。此外,涂层的表面接触角和凝胶率也是影响涂层防腐性能的重要因素。WPU-SP60比WPU-SP50更低的IE和更高的Icorr可以从凝胶含量来解释。
本发明的上述实施例仅仅是为了清楚地说明本发明技术方案的所作的举例,而并非是对本发明的具体实施方式的限定。凡在本发明权利要求书的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。