一种低介电高粘度LCP材料的制备方法
文献发布时间:2023-06-19 13:49:36
技术领域
本发明涉及高分子材料技术领域,特别是涉及一种低介电高粘度LCP材料的制备方法。
背景技术
LCP(Liquid Crystal Polymer,液晶聚合物)是一类在一定条件下能以液晶相存在的新型高分子材料,这类材料具有优异机械性能、尺寸稳定性、光学性能和电学性能,并且耐热性好,热膨胀系数和介电常数较低。近年来随着5G通讯技术的不断发展,对材料的综合性能提出了更加严格的要求,开发高性能介质材料,以减少互连延迟、能耗和串扰显得尤为重要,LCP材料因其具有较低介电常数受到了通讯类产品的关注和青睐。
目前,在通讯类产品中应用最为广泛LCP材料为聚氨酯类,传统的聚氨酯类LCP材料的合成方法,主要分为熔融缩聚和溶液缩聚两类。其中熔融缩聚方法通常是直接将单体按一定的比例混合,在惰性气体的保护下加热到熔融状态进行缩合反应;而溶液缩聚方法根据合成温度的不同,又分为低温酰化法和高温溶剂缩聚法,其中低温酰化法,是利用单体所带羧基与SO
采用传统的熔融缩聚方法合成液晶聚氨酯,需要较高的反应温度,在此温度下,不仅单体容易分解,聚合物本身也会发生降解,导致合成的材料粘度偏低,并不能满足使用需求。与熔融缩聚法相比,溶液缩聚法工艺条件相对简单,但由于单体的浓度较低,使得分子量难以提升,因而较难用该法合成性能良好的液晶聚芳酯。此外,不论是熔融缩聚法,还是溶液缩聚法,合成的LCP材料介电常数相对较高,不利于通讯信号传输,并不能满足使用需求。
发明内容
为解决上述问题,本发明提供一种低介电高粘度LCP材料的制备方法,其工艺流程简单,制备容易,制得的LCP材料介电常数低,粘度高,可以更好地满足应用需求。
本发明采用的技术方案是:
一种低介电高粘度LCP材料的制备方法,包括以下步骤:
S1:制备预聚物
将对乙酰氧基苯甲酸、2-乙酰氧基-6-萘甲酸、对苯二甲酸、双(4-乙酰氧基苯基)六氟丙烷和乙酸锌加入反应器中,先在氮气氛围下,于170-190℃恒温反应0.5-2h,去除反应体系中残留的乙酸成分,接着升高温度至225~235℃,回流1.5~2.5h,之后继续升高温度至285~295℃,逐渐降低反应体系压力至3~8kpa,并持续反应0.5~1.5h后停止反应,在氮气保护氛围下降至室温,得到LCP预聚物;
S2:制备LCP材料
将LCP预聚物研磨粉碎成均匀的颗粒,并转移到连续式固相反应器中,在真空及氮气气氛下,预热后,升高温度至170-190℃,反应6-8h,接着升高温度至210~230℃,之后在氮气氛围下降温至室温,即可得到LCP材料。
进一步地,在步骤S1中,由2,2-双(4-乙酰氧基苯基)丙烷替代2,2-双(4-乙酰氧基苯基)六氟丙烷。
进一步地,在步骤S1中,加入的对乙酰氧基苯甲酸、2-乙酰氧基-6-萘甲酸、对苯二甲酸和双(4-乙酰氧基苯基)六氟丙烷的摩尔比例为1:1:(0.2~0.3):(0.2~0.3)。
进一步地,在步骤S1中,乙酸锌的添加量为加入的乙酰氧基苯甲酸、2-乙酰氧基-6-萘甲酸、对苯二甲酸和双(4-乙酰氧基苯基)六氟丙烷总量的3%~8%。
进一步地,在步骤S1中,需要搅拌升温至170-190℃,进行恒温反应。
进一步地,在步骤S1中,将反应温度由225~235℃升至285~295℃时,控制升温速率控制为10-20℃/h。
进一步地,在步骤S2中,真空度为20-100pa,氮气流速为550-650Nm
进一步地,在步骤S2中,反应温度由170-190℃升高至220℃时,控制升温速率为5-10℃/h。
进一步地,在步骤S2中,将LCP预聚物研磨粉碎成粒径尺寸不同的颗粒。
进一步地,在步骤S2中,LCP预聚物研磨形成的颗粒的粒径范围为1-10mm。
本发明的有益效果如下:
本发明的制备方法,通过两步法制备LCP材料,首先通过单体的预聚,引入含氟元素的单体,单体在惰性气体氛围下逐步升温发生酯交换反应,脱除大部分的乙酸,制备低介电LCP预聚体;接着是预聚体的固相聚合,其用意在于能将预聚体在高温下,高分子链末端未反应的基团间继续碰撞缩合,从而达到提高分子量,增加材料粘度,并且固相聚合时的固相反应温度一般都是低于LCP材料的熔点,因此可以有效的避免单体分解、聚合物降解等多种缺陷,从而使得本发明制备的LCP材料介电常数低,粘度高,可以更好地满足应用需求,此外,本发明的制备方法,整体工艺简单,容易操作,满足大批量生产制造需求,具有良好的应用前景。
附图说明
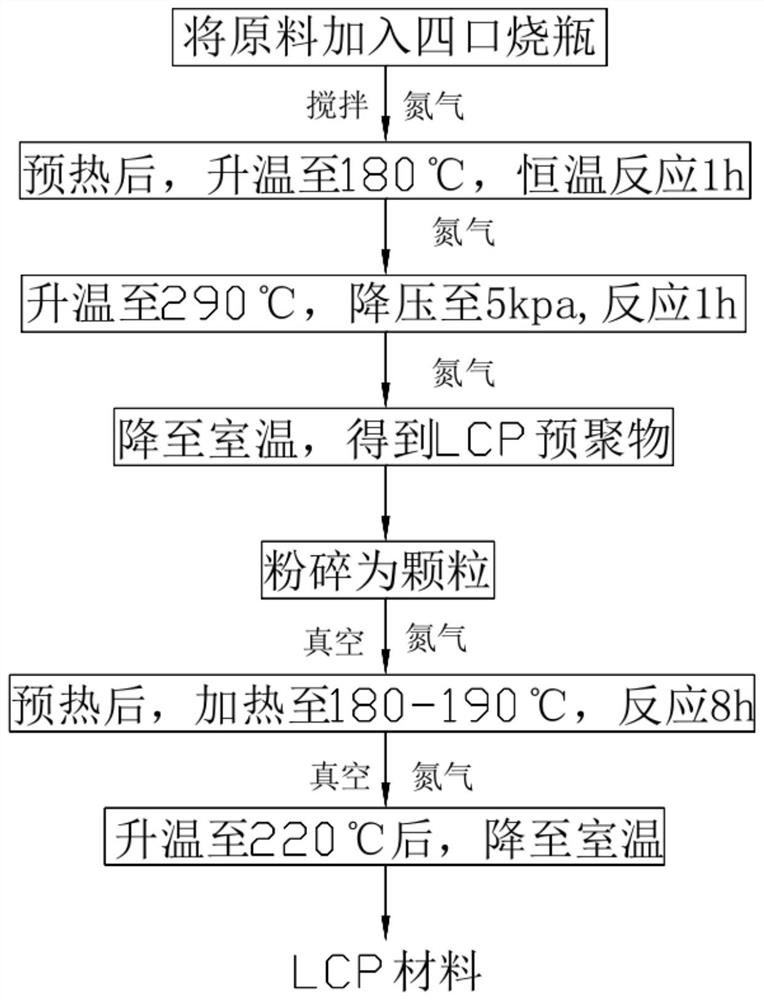
图1为本发明实施例1-6中的制备流程图;
具体实施方式
为了便于理解本发明,下面将参照实施例对本发明进行更全面的描述,以下给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。实施例中使用到的各类原料,除非另有说明,均为常见市售产品。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
本发明实施例中揭露的数值是近似值,而并非确定值。在误差或者实验条件允许的情况下,可以包括在误差范围内的所有值而不限于本发明实施例中公开的具体数值。
本发明实施例中揭露的数值范围用于表示在混合物中的组分的相对量以及其他方法实施例中列举的温度或者其他参数的范围。
本申请的低介电高粘度LCP材料的制备方法,包括以下步骤:
S1:制备预聚物
将对乙酰氧基苯甲酸、2-乙酰氧基-6-萘甲酸、对苯二甲酸、双(4-乙酰氧基苯基)六氟丙烷和乙酸锌加入反应器中,先在氮气氛围下,于170-190℃恒温反应0.5-2h,去除反应体系中残留的乙酸成分,接着升高温度至225~235℃,回流1.5~2.5h,之后继续升高温度至285~295℃,逐渐降低反应体系压力至3~8kpa,并持续反应0.5~1.5h后停止反应,在氮气保护氛围下降至室温,得到LCP预聚物;
S2:制备LCP材料
将LCP预聚物研磨粉碎成均匀的颗粒,并转移到连续式固相反应器中,在真空及氮气气氛下,预热后,升高温度至170-190℃,反应6-8h,接着升高温度至210~230℃,之后在氮气氛围下降温至室温,即可得到LCP材料。
进一步地,在步骤S1中,由2,2-双(4-乙酰氧基苯基)丙烷替代2,2-双(4-乙酰氧基苯基)六氟丙烷。
进一步地,在步骤S1中,加入的对乙酰氧基苯甲酸、2-乙酰氧基-6-萘甲酸、对苯二甲酸和双(4-乙酰氧基苯基)六氟丙烷的摩尔比例为1:1:(0.2~0.3):(0.2~0.3)。
进一步地,在步骤S1中,乙酸锌的添加量为加入的乙酰氧基苯甲酸、2-乙酰氧基-6-萘甲酸、对苯二甲酸和双(4-乙酰氧基苯基)六氟丙烷总量的3%~8%。
进一步地,在步骤S1中,需要搅拌升温至170-190℃,进行恒温反应。
进一步地,在步骤S1中,将反应温度由225~235℃升至285~295℃时,控制升温速率控制为10-20℃/h。
进一步地,在步骤S2中,真空度为20-100pa,氮气流速为550-650Nm
进一步地,在步骤S2中,反应温度由170-190℃升高至220℃时,控制升温速率为5-10℃/h。
进一步地,在步骤S2中,将LCP预聚物研磨粉碎成粒径尺寸不同的颗粒。
进一步地,在步骤S2中,LCP预聚物研磨形成的颗粒的粒径范围为1-10mm。
本申请的制备方法,通过两步法制备LCP材料,首先通过单体的预聚,引入含氟元素的单体,单体在惰性气体氛围下逐步升温发生酯交换反应,脱除大部分的乙酸,制备低介电LCP预聚体;接着是预聚体的固相聚合,其用意在于能将预聚体在高温下,高分子链末端未反应的基团间继续碰撞缩合,从而达到提高分子量,增加材料粘度,并且固相聚合时的固相反应温度一般都是低于LCP材料的熔点,因此可以有效的避免单体分解、聚合物降解等多种缺陷,从而使得本发明制备的LCP材料介电常数低,粘度高,可以更好地满足应用需求,此外,本发明的制备方法,整体工艺简单,容易操作,满足大批量生产制造需求,具有良好的应用前景。
下面为本申请的具体制备例:
实施例1:
S1:制备预聚物
取0.19mol的对乙酰氧基苯甲酸、0.19mol的2-乙酰氧基-6-萘甲酸、0.044mol的对苯二甲酸和0.044mol的2,2-双(4-乙酰氧基苯基)丙烷及乙酰氧基苯甲酸、2-乙酰氧基-6-萘甲酸、对苯二甲酸和双(4-乙酰氧基苯基)丙烷总质量4%的乙酸锌催化剂加入到带有搅拌器、温度计、氮气导入管和回流冷凝装置的四口烧瓶中,在氮气氛围下搅拌升温到180℃,恒温反应1h,去除反应体系中残留的乙酸成分;然后继续升高温度至230℃,并在该温度下回流反应2h;然后以15℃/h的升温速率继续升高温度至290℃,最后缓慢降低反应体系压力至5kpa并持续反应1h后停止反应,在氮气保护氛围下降低反应体系温度至室温得到LCP预聚物;
S2:制备LCP材料
将S1中的LCP预聚物通过手动研磨或机械球磨的方法粉碎成均匀的颗粒,颗粒直径范围控制在1-3mm。将研磨好LCP预聚物颗粒转移到连续式固相反应器中,在氮气保护和高真空度下对LCP预聚物进行升温预热,其中,真空度控制在80pa,氮气流速控制550Nm
实施例2:
S1:制备预聚物
取0.19mol的对乙酰氧基苯甲酸、0.19mol的2-乙酰氧基-6-萘甲酸、0.044mol的对苯二甲酸和0.044mol的2,2-双(4-乙酰氧基苯基)六氟丙烷及乙酰氧基苯甲酸、2-乙酰氧基-6-萘甲酸、对苯二甲酸和双(4-乙酰氧基苯基)六氟丙烷总质量4%的乙酸锌催化剂加入到带有搅拌器、温度计、氮气导入管和回流冷凝装置的四口烧瓶中,在氮气氛围下搅拌升温到180℃,恒温反应1h,去除反应体系中残留的乙酸成分;然后继续升高温度至230℃,并在该温度下回流反应2h;然后以15℃/h的升温速率继续升高温度至290℃,最后缓慢降低反应体系压力至5kpa并持续反应1h后停止反应,在氮气保护氛围下降低反应体系温度至室温得到LCP预聚物;
S2:制备LCP材料
将S1中的LCP预聚物通过手动研磨或机械球磨的方法粉碎成均匀的颗粒,颗粒直径范围控制在1-3mm。将研磨好LCP预聚物颗粒转移到连续式固相反应器中,在氮气保护和高真空度下对LCP预聚物进行升温预热,其中,真空度控制在80pa,氮气流速控制550Nm
实施例3:
S1:制备预聚物
取0.19mol的对乙酰氧基苯甲酸、0.19mol的2-乙酰氧基-6-萘甲酸、0.044mol的对苯二甲酸和0.044mol的2,2-双(4-乙酰氧基苯基)六氟丙烷及乙酰氧基苯甲酸、2-乙酰氧基-6-萘甲酸、对苯二甲酸和双(4-乙酰氧基苯基)六氟丙烷总质量6%的乙酸锌催化剂加入到带有搅拌器、温度计、氮气导入管和回流冷凝装置的四口烧瓶中,在氮气氛围下搅拌升温到180℃,恒温反应1h,去除反应体系中残留的乙酸成分;然后继续升高温度至230℃,并在该温度下回流反应2h;然后以15℃/h的升温速率继续升高温度至290℃,最后缓慢降低反应体系压力至5kpa并持续反应1h后停止反应,在氮气保护氛围下降低反应体系温度至室温得到LCP预聚物;
S2:制备LCP材料
将S1中的LCP预聚物通过手动研磨或机械球磨的方法粉碎成均匀的颗粒,颗粒直径范围控制在1-3mm。将研磨好LCP预聚物颗粒转移到连续式固相反应器中,在氮气保护和高真空度下对LCP预聚物进行升温预热,其中,真空度控制在80pa,氮气流速控制550Nm
实施例4:
S1:制备预聚物
取0.19mol的对乙酰氧基苯甲酸、0.19mol的2-乙酰氧基-6-萘甲酸、0.044mol的对苯二甲酸和0.044mol的2,2-双(4-乙酰氧基苯基)六氟丙烷及乙酰氧基苯甲酸、2-乙酰氧基-6-萘甲酸、对苯二甲酸和双(4-乙酰氧基苯基)六氟丙烷总质量6%的乙酸锌催化剂加入到带有搅拌器、温度计、氮气导入管和回流冷凝装置的四口烧瓶中,在氮气氛围下搅拌升温到180℃,恒温反应1h,去除反应体系中残留的乙酸成分;然后继续升高温度至230℃,并在该温度下回流反应2h;然后以15℃/h的升温速率继续升高温度至290℃,最后缓慢降低反应体系压力至5kpa并持续反应1h后停止反应,在氮气保护氛围下降低反应体系温度至室温得到LCP预聚物;
S2:制备LCP材料
将S1中的LCP预聚物通过手动研磨或机械球磨的方法粉碎成均匀的颗粒,颗粒直径范围控制在7-10mm。将研磨好LCP预聚物颗粒转移到连续式固相反应器中,在氮气保护和高真空度下对LCP预聚物进行升温预热,其中,真空度控制在80pa,氮气流速控制550Nm
实施例5:
S1:制备预聚物
取0.19mol的对乙酰氧基苯甲酸、0.19mol的2-乙酰氧基-6-萘甲酸、0.044mol的对苯二甲酸和0.044mol的2,2-双(4-乙酰氧基苯基)六氟丙烷及乙酰氧基苯甲酸、2-乙酰氧基-6-萘甲酸、对苯二甲酸和双(4-乙酰氧基苯基)六氟丙烷总质量6%的乙酸锌催化剂加入到带有搅拌器、温度计、氮气导入管和回流冷凝装置的四口烧瓶中,在氮气氛围下搅拌升温到180℃,恒温反应1h,去除反应体系中残留的乙酸成分;然后继续升高温度至230℃,并在该温度下回流反应2h;然后以15℃/h的升温速率继续升高温度至290℃,最后缓慢降低反应体系压力至5kpa并持续反应1h后停止反应,在氮气保护氛围下降低反应体系温度至室温得到LCP预聚物;
S2:制备LCP材料
将S1中的LCP预聚物通过手动研磨或机械球磨的方法粉碎成均匀的颗粒,颗粒直径范围控制在7-10mm。将研磨好LCP预聚物颗粒转移到连续式固相反应器中,在氮气保护和高真空度下对LCP预聚物进行升温预热,其中,真空度控制在20pa,氮气流速控制550Nm
实施例6:
S1:制备预聚物
取0.19mol的对乙酰氧基苯甲酸、0.19mol的2-乙酰氧基-6-萘甲酸、0.044mol的对苯二甲酸和0.044mol的2,2-双(4-乙酰氧基苯基)六氟丙烷及乙酰氧基苯甲酸、2-乙酰氧基-6-萘甲酸、对苯二甲酸和双(4-乙酰氧基苯基)六氟丙烷总质量6%的乙酸锌催化剂加入到带有搅拌器、温度计、氮气导入管和回流冷凝装置的四口烧瓶中,在氮气氛围下搅拌升温到180℃,恒温反应1h,去除反应体系中残留的乙酸成分;然后继续升高温度至230℃,并在该温度下回流反应2h;然后以15℃/h的升温速率继续升高温度至290℃,最后缓慢降低反应体系压力至5kpa并持续反应1h后停止反应,在氮气保护氛围下降低反应体系温度至室温得到LCP预聚物;
S2:制备LCP材料
将S1中的LCP预聚物通过手动研磨或机械球磨的方法粉碎成均匀的颗粒,颗粒直径范围控制在7-10mm。将研磨好LCP预聚物颗粒转移到连续式固相反应器中,在氮气保护和高真空度下对LCP预聚物进行升温预热,其中,真空度控制在20pa,氮气流速控制650Nm
将上述实施例制备得到的LCP材料进行介电常数和特性粘度测试,其中介电常数按GB/T 5597-1999标准测试,测试频率为1GHz;特性粘度按GB/T14190-2008标准测试。
性能测试结果如下表所示:
参见上表可知,使用本发明的制备方法制得的LCP材料,其介电常数均远远低于同类材料,而特性粘度远远高于同类材料,此外,通过对上述实施例进行对比,可以看出,在制备LCP预聚物时,通过引入含氟单体,用2-双(4-乙酰氧基苯基)六氟丙烷替代2,2-双(4-乙酰氧基苯基)丙烷,以及协同催化剂乙酸锌共同改善介电性能,从3.68降低到2.73;在固相反应聚合时,通过控制预聚体的直径大小、真空度、氮气流速以及固相反应温度和升温速率来进行LCP材料的粘度改善,特性粘度从0.62增加到0.96dL/g,在实际应用中,通过同时调整乙酸锌催化剂的添加量和预聚体的固相聚合步骤的工艺条件,制备出介电常数低和特性粘度高的LCP材料,以更好地满足使用需求。本申请的制备方法,整体工艺简单,容易操作,满足大批量生产制造需求,具有良好的应用前景。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 一种低介电高粘度LCP材料的制备方法
- 一种5G低介电强度LCP复合材料及其制备方法