一种硝酸硫康唑起始原料咪唑乙醇有关物质的检测方法
文献发布时间:2024-04-18 20:00:50
技术领域
本发明涉及药物分析技术领域,特别是涉及一种硝酸硫康唑起始原料咪唑乙醇有关物质的检测方法。
背景技术
硝酸硫康唑是一种咪唑衍生物,具有广谱抗真菌作用,可抑制包括红色毛癣菌、须癣毛癣菌、絮状表皮癣菌和犬小孢子菌等常见致病皮肤真菌的生长,也可以抑制造成花斑癣的微生物、糠秕马拉色菌以及某些革兰阳性菌。其结构式如下:
立体结构:硝酸硫康唑有一个手性中心,结构式中用*标注,产品为硝酸硫康唑消旋体。
作为合成硝酸硫康唑的起始原料咪唑乙醇,其结构式如下所示:
咪唑乙醇中可能存在并需要进行研究的有关物质包括:杂质ii-1、ii-2、ii-3、ii-4、ii-5、ii-6、ii-7、ii-8、ii-9和Ⅱ-3,共10个杂质。具体信息如下表所示:
咪唑乙醇为合成硝酸硫康唑关键步骤的起始原料,为了保证药物的安全性,有必要对其有关物质进行研究。其中ii-4、ii-5为主成分(咪唑乙醇)的二氯代苯环位置异构体,杂质ii-6、ii-7及ii-8互为单甲基取代咪唑环位置异构体,上述位置异构体杂质因极性差异较小,难以采用十八烷基硅烷键合硅胶色谱柱进行分离。咪唑(杂质ii-2)为制备咪唑乙醇最后一步反应用到的关键物料,是咪唑乙醇中存在的主要工艺杂质,应对其进行重点控制。
通过相关文献及专利调研发现:咪唑(杂质ii-2)及其结构类似物的检测方法包括紫外分光光度法、气相色谱法、流动相中加入离子对试剂的高效液相色谱法、柱前衍生化法及LC-MS/GC-MS法等。刘娜等发表的《克霉唑阴道片中微量咪唑测定方法》,以己烷磺酸钠作为离子对试剂,甲醇-磷酸二氢钾/磷酸缓冲液为流动相进行梯度洗脱;程利侠等采用柱前衍生-高效液相色谱法测定烟用添加剂中微量咪唑的含量,以丹磺酰氯为衍生试剂,柱前衍生,经乙酸乙酯萃取、浓缩、乙腈定容后,经C18色谱柱分离,用乙腈-水为流动相梯度洗脱,荧光检测器测定;专利文献《一种测定碱性反应液中咪唑含量的高效液相色谱分析方法与流程》,以十二烷磺酸钠作为离子对试剂,乙腈-磷酸二氢钾/磷酸缓冲液为流动相进行梯度洗脱。咪唑的结构简单,其结构和极性与咪唑乙醇及其他杂质差异较大,可以考虑采用上述相关报道中的检测方法单独控制咪唑含量,但难以同步测定咪唑(杂质ii-2)、异构体杂质(杂质ii-4、ii-5、ii-6、ii-7、ii-8)和其他极性较小的杂质,且一旦使用相关报道中的离子对试剂(己烷磺酸钠、十二烷磺酸钠等)后,离子对将永久残留于色谱柱中,造成色谱柱重现性和使用寿命下降,使得色谱柱不可继续用于分离检测其他无需加入离子对试剂到流动相中的化合物,导致检验成本大幅增加。柱前衍生化法、LC-MS/GC-MS法亦存在操作过程相对复杂、系统适用性及专属性差等缺陷。现有文献中均未有较为满意的方法公开。
发明内容
为了解决现有检测方法的上述问题,本发明提供一种硝酸硫康唑起始原料咪唑乙醇有关物质的检测方法。本发明方法能够有效分离并同步测定杂质ii-1、ii-2、ii-3、ii-4、ii-5、ii-6、ii-7、ii-8、ii-9和Ⅱ-3,共10个杂质;方法简单,不影响色谱柱的重现性和使用寿命,成本低;同时具备专属性、系统适用性、灵敏度、线性、精密度、准确度及耐用性良好等优势。
本发明的技术方案如下:
一种硝酸硫康唑起始原料咪唑乙醇有关物质的检测方法,采用高效液相色谱法,其色谱条件包括:全氟苯基硅烷键合硅胶色谱柱,以活性炭为填料的鬼峰捕集柱;以六氟磷酸钾水溶液为流动相A,乙腈为流动相B。
根据本发明优选的,流动相A六氟磷酸钾水溶液的浓度为20~30mmol/L。优选的,流动相A六氟磷酸钾水溶液的浓度为25mmol/L;六氟磷酸钾水溶液的配制方法为:将六氟磷酸钾溶于水中,用磷酸调节pH值至3.7,即得。
咪唑乙醇及其有关物质多为含咪唑结构的路易斯碱性化合物,当流动相中缓冲盐选用六氟磷酸钾,并调整pH至小于待测物的pKa-2,使含咪唑环的杂质转换成离子态时,六氟磷酸根在反向色谱中可发挥一定的弱离子对效能,从而增强极性较大的咪唑(杂质ii-2)在反向色谱中的保留,且六氟磷酸钾不会永久残留于色谱柱中,较己烷磺酸钠等离子对试剂有显著优势。本发明在流动相A缓冲盐种类的筛选试验中发现,当选用六氟磷酸钾溶液浓度为20~30mmol/L,并选用全氟苯基硅烷键合硅胶色谱柱时,可以在一个分析方法下,同步检测咪唑、苯环位置异构体、咪唑环位置异构体和其他杂质。
根据本发明优选的,流动相采用如下梯度程序洗脱:
在0~2分钟内,流动相A和流动相B的体积比保持80:20不变;在2~5分钟内,流动相A和流动相B的体积比由80:20线性渐变至65:35;在5~15分钟内,流动相A和流动相B的体积比保持65:35不变;在15~22分钟内,流动相A和流动相B的体积比由65:35线性渐变至20:80;在22~30分钟内,流动相A和流动相B的体积比保持20:80不变;在30~30.1分钟内,流动相A和流动相B的体积比由20:80线性渐变至80:20;在30.1~40分钟内,流动相A和流动相B的体积比保持80:20不变。
根据本发明优选的,色谱柱选择:ACE Excel 3 C18-PFP,4.6mm×250mm,3μm;捕集柱:Welch Ghost-Buster Column,4.6mm×50mm。
根据本发明优选的,色谱条件包括:采用紫外检测器,检测波长为220nm;柱温选自40~50℃,优选为45℃;流动相流速选自0.8~1.2m/min,优选为1.0ml/min;进样体积:10μl。
根据本发明优选的,采用乙腈的水溶液作为稀释剂配制供试品溶液、对照溶液,其中,乙腈和水的体积比为1:1;精密量取对照溶液、供试品溶液,注入液相色谱仪,记录色谱图。
根据本发明优选的,按照加校正因子的自身对照品法计算咪唑乙醇中各杂质的含量。
根据本发明,一种优选的技术方案,所述检测方法包括:
(1)色谱条件为:
仪器:高效液相色谱仪-紫外检测器;色谱柱:ACE Excel 3 C18-PFP,4.6mm×250mm,3μm;捕集柱:Welch Ghost-Buster Column,4.6mm×50mm;流动相A:25mmol/L、pH为3.7的六氟磷酸钾水溶液;流动相B:乙腈;流动相流速:1.0ml/min;检测波长:220nm;柱温:45℃;进样体积:10μl;
梯度洗脱程序:
(2)溶液配制:
稀释剂:乙腈的水溶液,其中,乙腈和水的体积比为1:1。
供试品溶液:精密称定咪唑乙醇,用稀释剂超声溶解并稀释制成每1ml溶剂中含咪唑乙醇0.5mg的溶液,摇匀。
对照溶液:精密量取供试品溶液,用稀释剂稀释制成每1ml溶剂中含咪唑乙醇0.5μg的溶液,摇匀。
(3)测定法:
精密量取稀释剂、对照溶液及供试品溶液,注入液相色谱仪,记录色谱图,按照加校正因子的自身对照品法计算咪唑乙醇中各杂质的含量。
本发明的技术特点及有益效果如下:
1、本发明通过对多种色谱柱型号的系统筛选,最终确定了选用ACE Excel 3 C18-PFP,4.6mm×250mm,3μm色谱柱,对咪唑乙醇中的位置异构体杂质分离效能最佳。本方法检测波长为220nm,因接近末端吸收,存在基线波动情况,本方法通过使用鬼峰捕集柱基本消除了基线波动对检测的干扰。
2、本发明未选用文献报道的强离子对试剂(己烷磺酸钠、十二烷磺酸钠等),而是选用了具备弱离子对效能的六氟磷酸钾,并摸索出了最佳的缓冲盐浓度、pH范围,从而实现了采用一种检测方法同步准确测定咪唑乙醇中的咪唑和其他杂质含量的目的,且本方法因未使用离子对试剂,不影响色谱柱的重现性和使用寿命,降低了检验成本,提高了方法的重现性和适用性。本发明使用的具备弱离子对效能的缓冲盐六氟磷酸钾酸性流动相体系和ACE Excel 3 C18-PFP色谱柱尚未在测定咪唑乙醇中的杂质咪唑和异构体杂质含量的相关文献或专利报道中出现,为同步测定化学药物、起始原料及中间体中的咪唑和位置异构体类杂质提供了新思路。
3、本发明方法通过对色谱柱、流动相、洗脱程序等色谱条件的筛选,实现了采用一种分析方法对咪唑乙醇中可能存在的多个苯环/咪唑环位置异构体和在反向色谱中几乎无保留的杂质咪唑的同步测定。本发明方法作为一个整体,各步骤、条件共同作用使得本发明能够有效分离并同步测定杂质ii-1、ii-2、ii-3、ii-4、ii-5、ii-6、ii-7、ii-8、ii-9和Ⅱ-3,共10个杂质;方法简单,不影响色谱柱的重现性和使用寿命,成本低;同时具备专属性、系统适用性、灵敏度、线性、精密度、准确度及耐用性良好等优势。
附图说明
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
图1为对比例1中混合溶液色谱图;
图2为对比例2中不同pH值流动相A的供试品溶液色谱图;
图3为对比例3中不同缓冲盐浓度流动相A的杂质ii溶液色谱图;
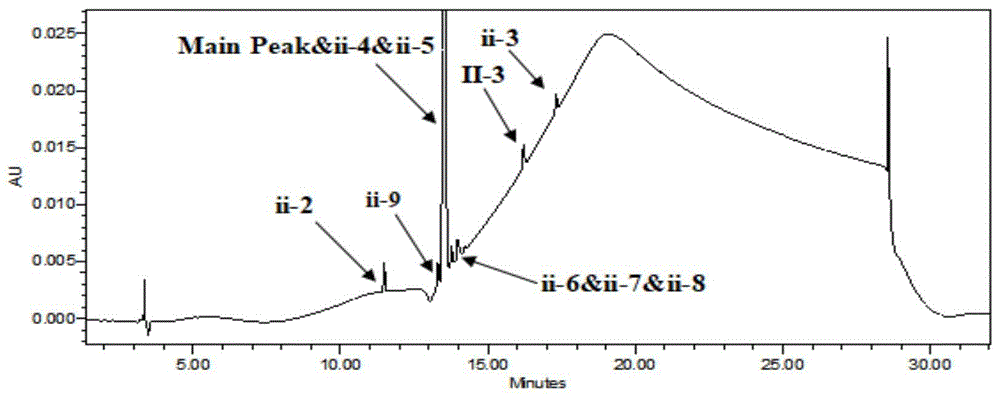
图4为实施例1中代表性批次供试品溶液色谱图,至下而上依次为230201批、230202批、230203批;
图5为实施例2专属性试验中混合溶液色谱图;
图6为实施例2耐用性试验中不同流速的混合溶液色谱图,至下而上依次为流速0.9ml/min、1.0ml/min、1.1ml/min;
图7为实施例2耐用性试验中不同柱温的混合溶液色谱图,由上至下依次为柱温40℃、45℃、50℃;
图8为实施例2耐用性试验中不同pH流动相的混合溶液色谱图,由上至下依次为3.5、3.7、3.9;
图9为实施例2耐用性试验中同品牌同类型不同序列号的混合溶液色谱图,由下至上依次为色谱柱1、色谱柱2。
具体实施方式
应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。
本实施例检测了采用池州中瑞化工有限公司生产的230201批、230202批、230203批咪唑乙醇中有关物质的含量,方法学验证试验中采用池州中瑞化工有限公司生产的Y210907301批样品进行。其他试剂均为市售产品。
对比例1:
1、色谱条件:
色谱柱:YMC Triart C18,4.6mm×150mm,3μm;
检测波长:220nm,紫外检测器;流速:1.0ml/min;柱温:30℃;进样量:20μl;
流动相A:磷酸盐溶液(取磷酸二氢钾6.8g,己烷磺酸钠6g,加水溶解并稀释至1000ml,用磷酸调节pH值至3.5)-乙腈(体积比90:10),流动相B:乙腈;
洗脱梯度:
2、溶液配制:
混合溶液:分别取咪唑乙醇、杂质ii-1、ii-2、ii-3、ii-4、ii-5、ii-6、ii-7、ii-8、ii-9、Ⅱ-3对照品适量,精密称定并置于同一量瓶中,用适量乙腈-水(体积比1:1)超声溶解并稀释制成每1ml溶剂中含咪唑乙醇约0.5mg、各杂质约0.5μg的溶液,摇匀。
精密量取混合溶液注入液相色谱仪,记录色谱图;结果见图1。
结论:当选用相关文献报道的C18色谱柱,并在流动相A中加入己烷磺酸钠作为离子对试剂时,能够测定极性较大的杂质咪唑(ii-2),但因无法进一步提高有机相比例(乙腈比例大于70%时,流动相A中的己烷磺酸钠析出),极性较小的ii-1无法在该色谱条件下洗脱,且该色谱条件下,异构体杂质(ii-4、ii-5、ii-6、ii-7、ii-8)之间及其与主峰的分离度较差。综上,相关文献报道的咪唑含量检测方法无法用于咪唑乙醇中有关物质的测定,且使用过离子对试剂后,会导致色谱柱改性,重现性和使用寿命下降。
对比例2:
为增强杂质咪唑在反向色谱柱中的保留且避免使用对比例1中的强离子对试剂,考虑在流动相A中加入具备弱阴离子对效能且不会使色谱柱改性的六氟磷酸钾,流动相A分别选择pH为2.8、3.2和4.5。
1、色谱条件:
色谱柱:ACE Excel 3 C18-PFP,4.6mm×250mm,3μm;
检测波长:220nm,紫外检测器;流速:1.0ml/min;柱温:30℃;进样量:20μl
流动相A:10mM六氟磷酸钾水溶液(用磷酸调节pH分别为2.8、3.2和4.5)
流动相B:乙腈
洗脱梯度:
2、溶液配制
供试品溶液:取咪唑乙醇适量,精密称定,用适量乙腈-水(体积比1:1)超声溶解并稀释制成每1ml溶剂中含咪唑乙醇约0.5mg的溶液,摇匀。
精密量取供试品溶液注入液相色谱仪,记录色谱图;结果见表1和图2。
表1供试品溶液中主峰的拖尾因子-对比例2
结论:当流动相A选用pH为2.8、3.2的六氟磷酸钾溶液时,主峰拖尾因子为3.04、1.88(均大于1.5),峰形拖尾;当提高六氟磷酸钾溶液的pH为4.5时,主峰拖尾因子为0.54(小于0.8),峰形前延,需进一步在3.2~4.5之间选择最佳流动相A的pH,以改善主成分及各杂质的峰形。
对比例3:
1、色谱条件:
色谱柱:ACE Excel 3 C18-PFP,4.6mm×250mm,3μm;
检测波长:220nm,紫外检测器;流速:1.0ml/min;柱温:30℃;进样量:20μl
流动相A:5mM六氟磷酸钾水溶液(用磷酸调节pH3.7);15mM六氟磷酸钾水溶液(用磷酸调节pH3.7)
流动相B:乙腈
洗脱梯度:同对比例2
2、溶液配制:
杂质ii-2溶液:取杂质ii-2对照品适量,精密称定,用适量乙腈-水(体积比1:1)超声溶解并稀释制成每1ml溶剂中杂质ii-2约5μg的溶液,摇匀。
精密量取杂质ii-2溶液注入液相色谱仪,记录色谱图;结果见图3。
结论:当流动相A中六氟磷酸钾的浓度为5mmol/L时,杂质咪唑(ii-2)在色谱柱中几乎无保留,无法满足定量要求;当流动相A中六氟磷酸钾的浓度提高至15mmol/L时,杂质咪唑的保留时间延后,但仍与稀释剂峰重合,需进一步提高六氟磷酸钾的浓度。
实施例1:硝酸硫康唑起始原料咪唑乙醇有关物质的检测方法
1.1色谱条件为:
仪器:高效液相色谱仪-紫外检测器;
色谱柱:ACE Excel 3 C18-PFP,4.6mm×250mm,3μm;捕集柱:Welch Ghost-BusterColumn,50×4.6mm;流动相A:25mM六氟磷酸钾水溶液(pH3.7);流动相B:乙腈;流速:1.0ml/min;检测波长:220nm;柱温:45℃;进样体积:10μl。
梯度程序:
1.2溶液配制:
流动相A:称取六氟磷酸钾4.60g,加水1000ml使溶解,用磷酸调节pH值至3.7,摇匀。
稀释剂:乙腈-水(体积比1:1)。
供试品溶液:取本品咪唑乙醇适量,精密称定,用适量稀释剂超声溶解并稀释制成每1ml溶剂中含咪唑乙醇约0.5mg的溶液,摇匀。
对照溶液:精密量取供试品溶液适量,用稀释剂稀释制成每1ml溶剂中含咪唑乙醇约0.5μg的溶液,摇匀。
灵敏度溶液:精密量取供试品溶液适量,用稀释剂稀释制成每1ml溶剂中含咪唑乙醇约0.25μg的溶液,摇匀。
1.3测定法:
精密量取稀释剂、灵敏度溶液、对照溶液及供试品溶液,注入液相色谱仪,记录色谱图,按照加校正因子的自身对照品法计算咪唑乙醇中各杂质的含量,结果见图4和表2。
表2多批次咪唑乙醇中有关物质的检测结果
实施例2:方法学验证
2.1系统适用性
稀释剂:乙腈-水(体积比1:1)。
供试品溶液:取本品咪唑乙醇适量,精密称定,用适量稀释剂超声溶解并稀释制成每1ml溶剂中含咪唑乙醇约0.5mg的溶液,摇匀。
对照溶液:精密量取供试品溶液适量,用溶剂稀释制成每1ml溶剂中含咪唑乙醇约0.5μg的溶液,摇匀。
灵敏度溶液:精密量取供试品溶液适量,用溶剂稀释制成每1ml溶剂中含咪唑乙醇约0.25μg的溶液,摇匀。
精密量取上述溶液,注入液相色谱仪,色谱条件同实施例1,记录色谱图,结果见表3。
表3系统适用性试验结果
.结论:灵敏度溶液色谱图中,主峰S/N>10;供试品溶液色谱图中,主峰与相邻峰的最小分离度>1.5;对照溶液色谱图中,主峰拖尾因子在0.8~1.5之间。符合可接受标准。
2.2专属性
杂质贮备液:分别取杂质ii-1、ii-2、ii-3、ii-4、ii-5、ii-6、ii-7、ii-8、ii-9、Ⅱ-3对照品约10mg,精密称定,置不同的50ml量瓶中,加稀释剂超声使溶解并稀释至刻度,摇匀。
杂质定位溶液:分别精密量取各杂质贮备液3ml,置不同的50ml量瓶中,用稀释剂稀释至刻度,摇匀,分别精密量取2ml,置不同的50ml量瓶中,用稀释剂稀释至刻度,摇匀。
混合杂质贮备液:分别精密量取各杂质贮备液3ml,置同一50ml量瓶中,用稀释剂稀释至刻度,摇匀。
混合溶液:取本品咪唑乙醇约25mg,精密称定,置50ml量瓶中,加稀释剂超声使溶解,精密量取混合杂质贮备液2ml,置上述50ml量瓶中,用稀释剂稀释至刻度,摇匀。
供试品溶液:取本品咪唑乙醇适量,精密称定,用适量稀释剂超声溶解并稀释制成每1ml溶剂中含咪唑乙醇约0.5mg的溶液,摇匀。
精密量取上述各溶液,注入液相色谱仪,色谱条件同实施例1,记录色谱图,结果见图5和表4所示。
表4专属性试验结果
结论:稀释剂不干扰本品有关物质检测;混合溶液色谱图中主峰及各已知杂质峰与相邻峰的最小分离度>1.5,供试品溶液主峰峰纯度符合要求。符合可接受标准。
2.3定量限及检测限
咪唑乙醇贮备液:取咪唑乙醇对照品约10mg,精密称定,置50ml量瓶中,加稀释剂超声使溶解并稀释至刻度,摇匀。
定量限溶液:分别精密量取2.2项下各杂质贮备液3ml,咪唑乙醇贮备液3ml,置同一50ml量瓶中,用稀释剂稀释至刻度,摇匀,精密量取1ml,置50ml量瓶中,用稀释剂稀释至刻度,摇匀。
检测限溶液:精密量取定量限溶液3ml,置10ml量瓶中,用稀释剂稀释至刻度,摇匀。
精密量取上述溶液,注入液相色谱仪,色谱条件同实施例1,记录色谱图,结果见表5、表6。
表5定量限试验结果
/>
/>
表6检测限试验结果
结论:定量限溶液连续进样6针,咪唑乙醇、ii-1、ii-2、ii-3、ii-4、ii-5、ii-6、ii-7、ii-8、ii-9、Ⅱ-3峰的S/N均大于10,峰面积RSD均小于10%,定量限浓度约为0.25μg/ml,相当于供试品浓度的0.05%;表明含量≥0.05%的各杂质均能被准确定量;检测限溶液中的咪唑乙醇、ii-1、ii-2、ii-3、ii-4、ii-5、ii-6、ii-7、ii-8、ii-9、Ⅱ-3峰的S/N均大于3,检测限浓度约为0.07μg/ml,相当于供试品浓度的0.02%,表明含量≥0.02%的各杂质均能被检出,本方法灵敏度良好。
2.4线性与范围
线性贮备液:分别精密量取2.2项下各杂质贮备液3ml,2.3项下咪唑乙醇贮备液3ml,置同一50ml量瓶中,用稀释剂稀释至刻度,摇匀。
线性溶液L1~L4:分别精密量取线性贮备液2ml、3ml、4ml、6ml,置不同的100ml量瓶中,用稀释剂稀释至刻度,摇匀。
线性溶液L5、L6:分别精密量取线性贮备液2ml、4ml,置不同的25ml量瓶中,用稀释剂稀释至刻度,摇匀。
精密量取上述各线性溶液,注入液相色谱仪,色谱条件同实施例1,记录色谱图,结果见表7。
表7线性与范围试验结果
/>
/>
/>
结论:咪唑乙醇及各已知杂质在限度浓度的LOQ~400%浓度范围内,相关系数r均大于0.99。符合可接受标准,表明浓度与峰面积线性关系良好。
2.5准确度
50%、100%、150%、250%回收率溶液:分别取本品咪唑乙醇约25mg,精密称定,置4个不同的50ml量瓶中,加稀释剂超声使溶解,精密量取2.2项下的混合杂质贮备液1、2、3、5ml,置上述4个不同的50ml量瓶中,用稀释剂稀释至刻度,摇匀。每个浓度水平重复制备3份。
对照溶液:精密量取100%回收率溶液1ml,置100ml量瓶中,用稀释剂稀释至刻度,摇匀,精密量取2ml,置20ml量瓶中,用稀释剂稀释至刻度,摇匀。
精密量取上述溶液,注入液相色谱仪,色谱条件同实施例1,记录色谱图,结果见表8。
表8准确度试验结果
/>
/>
结论:各已知杂质回收率的单值及均值均在85~115%之间,RSD均小于10%。符合可接受标准,表明本方法准确度良好。
2.6精密度
重复性溶液:同2.5项下100%回收率溶液。
对照溶液:精密量取重复性溶液1ml,置100ml量瓶中,用稀释剂稀释至刻度,摇匀,精密量取2ml,置20ml量瓶中,用稀释剂稀释至刻度,摇匀。
精密量取上述溶液,注入液相色谱仪,色谱条件同实施例1,记录色谱图,结果见表9。
表9重复性试验结果
/>
结论:按加校正因子的主成分自身对照法计算,6份重复性溶液中,各已知杂质含量及总杂含量RSD均小于10%。符合可接受标准,表明本方法精密度良好。
2.7耐用性
2.7.1溶液稳定性
混合溶液:取本品咪唑乙醇约25mg,精密称定,置50ml量瓶中,加稀释剂超声使溶解,精密量取2.2项下混合杂质贮备液2ml,置上述50ml量瓶中,用稀释剂稀释至刻度,摇匀。
对照溶液:精密量取混合溶液1ml,置100ml量瓶中,用稀释剂稀释至刻度,摇匀,作为对照贮备液,精密量取对照贮备液2ml,置20ml量瓶中,用稀释剂稀释至刻度,摇匀。
分别精密量取对照溶液、混合溶液于不同时间点注入液相色谱仪,色谱条件同实施例1,记录色谱图,结果见表10。
表10对照溶液及混合溶液稳定性试验结果
结论:对照溶液及混合溶液在室温条件下放置25h,各时间点主峰峰面积与0h峰面积的比值均在0.9~1.1之间。混合溶液在室温条件下放置25h,各时间点各杂质含量与0h含量差值的绝对值均小于0.05%,且均未出现≥0.05%的新增杂质。符合可接受标准,表明对照溶液和混合溶液在室温条件下放置25h稳定性良好。
2.7.2色谱条件耐用性
混合溶液:同2.7.1项下。
对照溶液:精密量取混合溶液1ml,置100ml量瓶中,用稀释剂稀释至刻度,摇匀,作为对照贮备液,精密量取对照贮备液2ml,置20ml量瓶中,用稀释剂稀释至刻度,摇匀。
灵敏度溶液:精密量取对照贮备液1ml,置20ml量瓶中,用稀释剂稀释至刻度,摇匀。
在实施例1色谱条件基础上变更色谱条件,并保持单一变量,其它色谱条件通实施例1:
A:流速(1.0ml/min±0.1ml/min);
B:柱温(45℃±5℃);
C:流动相缓冲盐pH值(3.7±0.2);
D:色谱柱:同品牌同类型不同批号色谱柱;
依次在正常条件(即实施例1的色谱条件)、变更上述色谱条件下,精密量取上述溶液进样,记录色谱图,分别如图6-9所示。
表11色谱条件耐用性试验结果
结论:微调各色谱条件,灵敏度溶液色谱图中,主峰S/N均大于10;混合溶液色谱图中,主峰及各已知杂质峰与相邻峰的最小分离度均大于1.5。混合溶液中各杂质含量与正常条件含量差值的绝对值均小于0.05%;总杂含量与正常条件下总杂含量差值的绝对值均小于0.1%;符合可接受标准,系统适用性良好。
- 一种基于改进下垂控制的直流微电网暂态电压改善方法
- 一种用于直流微电网的基于SOC的改进下垂控制方法