工程大肠杆菌合成牡荆素和荭草苷的应用
文献发布时间:2023-06-19 19:28:50
技术领域
本发明涉及工程大肠杆菌合成牡荆素和荭草苷的应用,属于生物工程技术领域。
背景技术
类黄酮是广泛分布于植物中的酚类化合物,基本骨架是一个15碳的苯丙烷核(C6-C3-C6),它有A和B两个芳环,由一个杂环吡喃环(C)连接。由于糖链参与细胞分化、发育、免疫、衰老、信息传递等几乎所有生命过程,糖基化可以改变类黄酮的生物活性,增加水溶性,减少毒副作用,提高特异性。在自然界中类黄酮一般以糖苷的形式存在,类黄酮经过糖基化修饰后,通常表现出比其骨架更好的特性,甚至可能具有不同的特性。牡荆素和荭草苷是许多中草药的有效活性成分,牡荆素已知在许多癌症异种移植模型和细胞系中表现出广泛的抗氧化,抗炎,镇痛和抗肿瘤活性。荭草苷可以通过改善抗氧化防御机制来缓解小鼠由于电磁辐射引起的学习和记忆障碍,并且还具有抗心肌缺血、抗细胞凋亡、抗辐射、抗肿瘤和抗衰老等功能。
蔗糖合成酶能利用反应的副产物二磷酸尿苷(Uridine diphosphate,UDP)和蔗糖合成尿苷二磷酸葡萄糖(UDP-glucose,UDPG)。因此,将糖基转移酶和蔗糖合成酶耦合原位再生UDPG可有效降低反应成本。植物来源的蔗糖合成酶的偏好受体为UDP,因此,利用蔗糖合成酶和类黄酮糖基转移酶偶联反应的产物多为类黄酮葡萄糖苷。糖基转移酶和蔗糖合酶偶联反应虽然能提高UDP的利用次数,在一定程度上降低了生产成本,但反应体系中仍然需要添加UDP,并且前期需要对酶进行纯化,耗费大量精力,该生产方式并不是十分经济。微生物合成法具有反应条件温和、易于控制等优点,用于合成类黄酮糖苷具有较高潜力。
由于糖基供体UDPG合成的前体G-6-P绝大部分流入糖酵解途径,导致胞内UDPG浓度较低,并且UDPG在胞内参与多种代谢反应,进一步降低了UDPG在胞内的积累。目前针对胞内UDPG浓度过低的问题,主要是通代谢工程的手段强化UDPG前体G-6-P的供应,敲除糖酵解途径中的关键基因6-磷酸葡萄糖异构酶(pgi)和磷酸戊糖途径中的关键基因6-磷酸葡萄糖1-脱氢酶(zwf),从而使G-6-P在流向UDPG合成。Eiteman等使用敲除pgi基因的工程菌并表达UGT73B3,用于合成槲皮素-3-葡萄糖苷,该方法也分别在槲皮素-3-鼠李糖苷,槲皮素-3-木糖苷,柚皮素-7-木糖苷,大黄素葡萄糖苷等。除了抑制消耗途径,UDPG合成通路基因的过表达也能提高类黄酮糖苷的生物合成。
在之前的工作中构建了糖基转移酶和蔗糖合成酶偶联的方法,并用于牡荆素和荭草苷的合成(《Efficient Production of Orientin and Vitexin from Luteolin andApigenin Using Coupled Catalysis of Glycosyltransferase and SucroseSynthase》)。酶偶联法虽然能提高UDP的利用次数,在一定程度上降低了生产成本,但仍然需要添加UDP,并不十分经济。大肠杆菌拥有内源UDPG合成路径,如果能强化该途径,构建一株能充足供应UDPG的工程菌,对糖基化产物合成具有积极意义。
发明内容
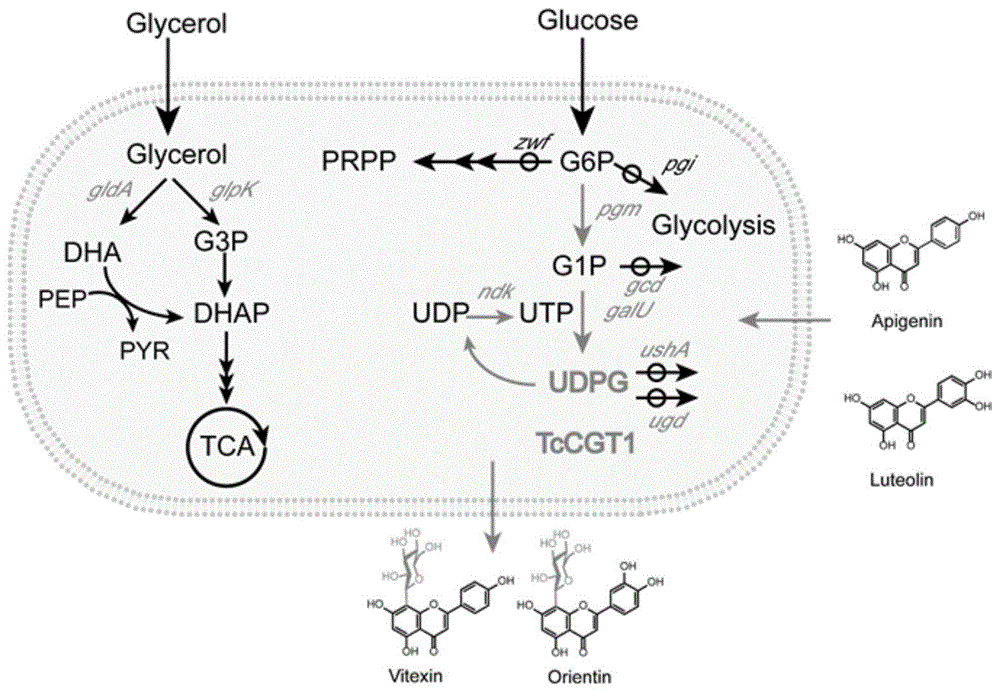
为了解决糖基供体供应不足的问题,本发明构建一株能充足供应UDPG的菌株,使用CRISPR-Cas9系统敲除糖酵解途径关键基因6-磷酸葡萄糖异构酶基因(pgi)和6-磷酸葡萄糖1-脱氢酶基因(zwf),在此基础上敲除UDPG合成路径的旁路基因UDP-葡萄糖6-脱氢酶(UGD),葡萄糖脱氢酶(GCD)和海藻糖-6-磷酸合酶(OtsA)。为了进一步提高内源UDPG供体,UDPG关键基因磷酸葡萄糖变位酶基因(PGM)和UTP-葡萄糖-1-磷酸尿苷酸转移酶(galU)使用pETDuet质粒过表达,并使用pACYCDuet质粒游离表达UDP尿苷激酶(ndk),构建一株能充足供应UDPG菌株,在此菌株表达金莲花(Trollius chinensis Bunge)的8位碳苷糖基转移酶TcCGT1,用于合成牡荆素和荭草苷(图1)。
本发明的第一个目的是提供一株高产尿苷二磷酸葡萄糖(UDP-glucose,UDPG)的工程菌,所述工程菌以大肠杆菌为宿主,敲除或抑制基因组上糖酵解途径关键基因以及UDPG合成路径的旁路基因,游离表达磷酸葡萄糖变位酶、UTP-葡萄糖-1-磷酸尿苷酸转移酶和UDP尿苷激酶。
在一种实施方式中,所述糖酵解途径关键基因包括6-磷酸葡萄糖异构酶编码基因pgi和/或6-磷酸葡萄糖1-脱氢酶编码基因zwf。
在一种实施方式中,所述UDPG合成路径的旁路基因包括UDP-葡萄糖6-脱氢酶编码基因ugd,葡萄糖脱氢酶编码基因gcd和/或海藻糖-6-磷酸合酶编码基因otsA。
在一种实施方式中,利用质粒pRSFDuet-1、pCDFDuet-1或pACYCDuet-1游离表达了磷酸葡萄糖变位酶编码基因、UTP-葡萄糖-1-磷酸尿苷酸转移酶编码基因和/或UDP尿苷激酶编码基因。
在一种实施方式中,利用质粒pCDFDuet-1游离表达了磷酸葡萄糖变位酶编码基因pgm、UTP-葡萄糖-1-磷酸尿苷酸转移酶编码基因galU,利用pACYCDue-1游离表达了UDP尿苷激酶编码基因。
在一种实施方式中,所述pgi的GenBank号为X15196.1,所述zwf的GenBank号为M55005.1,所述ugd的GenBank号为WP_000704871,所述gcd的GenBank号为X51323.1,所述otsA的GenBank号为X69160.1。
在一种实施方式中,所述磷酸葡萄糖变位酶编码基因pgm的GenBank号为U08369.1,所述UTP-葡萄糖-1-磷酸尿苷酸转移酶编码基因galU的GenBank号为M98830.1。
在一种实施方式中,所述UDP尿苷激酶编码基因的GenBank号为X71492.1。
在一种实施方式中,所述工程菌以大肠杆菌BL21(DE3)为出发菌株。
本发明的第二个目的是提供一种制备高产糖基产物的重组菌株,所述重组菌株是在上述工程菌的基础上,游离表达金莲花来源的8位碳苷糖基转移酶TcCGT1。
在一种实施方式中,利用质粒pCDFDuet-1表达TcCGT1。
在一种实施方式中,所述质粒pCDFDuet-1上携带上述基因pgm和galU。
在一种实施方式中,所述TcCGT1编码基因的GenBank号为MK644229.1。
本发明的第三个目的是提供一种提高糖基产物产量的方法,所述方法为以大肠杆菌为宿主,敲除或抑制基因组上糖酵解途径关键基因以及UDPG合成路径的旁路基因,游离表达磷酸葡萄糖变位酶、UTP-葡萄糖-1-磷酸尿苷酸转移酶、金莲花来源的8位碳苷糖基转移酶TcCGT1和UDP尿苷激酶。
在一种实施方式中,所述糖酵解途径关键基因包括6-磷酸葡萄糖异构酶编码基因pgi和/或6-磷酸葡萄糖1-脱氢酶编码基因zwf。
在一种实施方式中,所述UDPG合成路径的旁路基因包括UDP-葡萄糖6-脱氢酶编码基因ugd,葡萄糖脱氢酶编码基因gcd和/或海藻糖-6-磷酸合酶编码基因otsA。
在一种实施方式中,利用质粒pRSFDuet-1、pCDFDuet-1或pACYCDuet-1游离表达了磷酸葡萄糖变位酶编码基因、UTP-葡萄糖-1-磷酸尿苷酸转移酶编码基因、金莲花来源的8位碳苷糖基转移酶TcCGT1和/或UDP尿苷激酶编码基因。
在一种实施方式中,利用质粒pCDFDuet-1游离表达磷酸葡萄糖变位酶编码基因pgm、UTP-葡萄糖-1-磷酸尿苷酸转移酶编码基因galU和TcCGT1编码基因,利用pACYCDue-1游离表达UDP尿苷激酶编码基因。
在一种实施方式中,所述pgi的GenBank号为X15196.1,所述zwf的GenBank号为M55005.1,所述ugd的GenBank号为WP_000704871,所述gcd的GenBank号为X51323.1,所述otsA的GenBank号为X69160.1。
在一种实施方式中,所述磷酸葡萄糖变位酶编码基因pgm的GenBank号为U08369.1,所述UTP-葡萄糖-1-磷酸尿苷酸转移酶编码基因galU的GenBank号为X59940.1,所述TcCGT1编码基因的GenBank号为MK644229.1。
在一种实施方式中,所述UDP尿苷激酶编码基因的GenBank号为X71492.1。
在一种实施方式中,所述工程菌以大肠杆菌BL21(DE3)为出发菌株。
本发明的第四个目的是提供上述工程菌在制备糖基产物中的应用,所述应用为以黄酮或其他类黄酮为底物,在上述工程菌的基础上,过表达金莲花来源的8位碳苷糖基转移酶TcCGT1得到重组菌株,再以重组菌为发酵菌株进行糖基产物的发酵制备。
在一种实施方式中,所述黄酮或其他类黄酮包括木犀草素、芹菜素。
本发明的第五个目的是提供上述重组菌株在制备糖基产物中的应用,所述应用为以黄酮或其他类黄酮为底物,以上述重组菌株为发酵菌株进行糖基产物的发酵制备。
在一种实施方式中,所述黄酮或其他类黄酮包括木犀草素、芹菜素。
本发明的有益效果:
本发明构建了一株能充足供应糖基供体UDPG的菌株,通过使用CRISPR-Cas9系统敲除糖酵解途径关键基因pgi和zwf,以及敲除了UDPG合成路径的旁路基因ugd,gcd和otsA;使用pETDuet-1质粒过量表达大肠杆菌内源的UDPG关键基因磷酸葡萄糖变位酶基因(pgm)和UTP-葡萄糖-1-磷酸尿苷酸转移酶(galU),并使用pACYCDuet质粒游离表达UDP尿苷激酶(ndk),构建一株能充足供应UDPG菌株,并在此菌株的基础上表达金莲花来源的8位碳苷糖基转移酶TcCGT1,以黄酮或其他类黄酮为底物用于合成糖基产物,在本发明中以木犀草素、芹菜素为例进行糖基产物牡荆素和荭草苷的发酵生产,最终,在5L发酵罐中牡荆素和荭草苷的产量分别达到17.25g/L和36.52g/L。
附图说明
图1:改造大肠杆菌牡荆素和荭草苷示意图。
图2:敲除糖酵解途径关键基因促进糖基产物合成。
图3:UDPG旁路基因敲除。
图4:UDPG合成关键基因过表达。
图5:通过优化基因表达强度提高糖基产物合成。
图6:强化UTP循环促进糖基化产物合成。
图7:5L-发酵罐扩大培养。
具体实施方式
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
LB培养基:5.0g/L酵母粉、10.0g/L胰蛋白胨、10.0g/L NaCl。
TBG培养基(发酵培养基),由12g/L蛋白胨,24g/L酵母粉,12.54g/L K
牡荆素和红草苷检测方法;牡荆素和荭草苷采用Thermo Fisher C18色谱柱(4.6mm×250mm,5μm)进行检测,柱温箱温度设置为40℃,进样量为10μL,流动相为55%的甲醇,PDA检测器检测波长为290和350nm。
摇瓶发酵培养方法:在培养时,将平板上活化的单菌落接种到3mL LB培养基中生长10h,将种子液按2%(v/v)比例接种到发酵培养基中,在37℃下培养。当OD
发酵罐发酵培养方法:重组菌株的补料分批发酵在含有2.5L发酵培养基的迪必尔5L发酵罐中进行。在培养时,将平板上活化的单菌落接种到25mL LB培养基中生长10h,将种子液按10%(v/v)接种至发酵培养基中,发酵过程在溶氧和转速关联模式下进行控制,初始温度、搅拌转速和通气量分别设定为37℃、200rpm和2.5vvm。当溶氧开始反弹时,开始补料,将500mL含有100g/L酵母提取物和300g/L甘油的补充培养基以10ml/L的流速流入发酵液中。当OD
底物芹菜素或木犀草素以100g/L的浓度储存在DMSO中,并且在整个发酵过程中控制底物浓度不超过5g/L。通过自动添加50%(w/v)NH
菌株信息如表1所示:
表1本发明中涉及的菌株、基因
基因敲除方法
使用CRISPR/Cas9系统对大肠杆菌BL21(DE 3)进行基因编辑,参考文献Li Q,SunB,Chen J,Zhang Y,Jiang Y,Yang S.A modified pCas/pTargetF system for CRISPR-Cas9-assisted genome editing in Escherichia coli.Acta Biochim Biophys Sin(Shanghai).2021Apr15;53(5):620-627.。在目的基因(pgi、zwf、ugd、gcd、otsA、ushA)上下游选500bp左右片段作为同源臂,利用表2中目的基因相对应的引物,使用融合PCR将上下游同源臂连接作为供体基因。将携带pEcCas质粒的菌株接种至含有卡那霉素的LB培养基中,在37℃培养到OD
本文所有基因敲除用的pTarget质粒均通过PCR介导的点突变技术构建,模板pTarget质粒来源于实验室保存(记载于Jiang,Y.,Chen,B.,Duan,C.,Sun,B.,Yang,J.,Yang,S.,2015.Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system.Applied and Environmental Microbiology.81(7):2506-2514.)。使用http://www.rgenome.net/网站在线预测目的基因(pgi、zwf、ugd、gcd、otsA、ushA)的靶特异性序列20nt,用带有20nt同源臂的引物(表2)对pTarget质粒进行线性化,随后,按照操作说明使用DpnI消除模板质粒,将纯化后的PCR产物转化至大肠杆菌JM109,挑阳性菌落进行测序,对测序正确的菌落甘油保藏。将200ng的pTarget质粒和800ng的供体DNA与100μL感受态细胞混合,冰上静置20min后进行电转化。转化后混合物立即悬浮在1mL新鲜LB培养基中。细胞在37℃孵育1h后,涂于含有卡那霉素(50μg/mL)和氯霉素(50μg/mL)的LB固体培养基平板上,将平板在37℃下培养过夜培养,随机挑选单个菌落,并通过菌落PCR和DNA测序进行验证。
表2构建质粒使用引物
质粒固化方法
将目的基因敲除成功的菌株接种在含有10mM的鼠李糖和50μg/mL卡那霉素的2mLLB培养基中,在220rpm下过夜培养,然后在含有卡那霉素(50μg/mL)的固体LB培养基上划线,在37℃培养过夜后长出的单菌落,通过平板影印的方式,分别在只含有卡那霉素的LB平板上,以及含有卡那霉素和链霉素的LB平板上进行筛选。在卡那霉素单抗平板上生长,在卡那霉素和链霉素双抗性平板上无法生长的菌落表明pTarget质粒被消除。将消除pTarget的菌落接种在含有5g/L葡萄糖的液体LB培养基中,并在220rpm,37℃下摇动过夜。将培养液在含有5g/L葡萄糖和10g/L蔗糖的LB板上划线,并在37℃下培养过夜,然后随机挑选单个菌落在卡那霉素的LB平板以及无抗LB平板进行平板影印,在无抗LB平板上正常生长,在卡那霉素的LB平板上无法生长的菌落,即为消除pEcCas质粒的菌株
质粒构建
所有质粒均使用无缝克隆的方式构建。引物P1-F/R用于扩增pETDuet-1质粒,引物P2-F/R以pET28a-TcCGT1模板扩增TcCGT1(质粒pET28a-TcCGT1公开于文献Liu S,Lyu Y,YuS,Cheng J,Zhou J.Efficient Production of Orientin and Vitexin from Luteolinand Apigenin Using Coupled Catalysis of Glycosyltransferase and SucroseSynthase.J Agric Food Chem.2021Jun16;69(23):6578-6587.),使用无缝克隆酶(上海生工)对PCR产物进行连接,构建pETDuet-TcCGT1。使用引物P3-F/R用于扩增pETDuet-TcCGT1质粒获得线性化载体,引物P4-F/R和P5-F/R用于从大肠杆菌BL21基因组中扩增PGM和galU,引物P6-F/R用于从酿酒酵母C800基因组中扩增UGP1,将线性化载体pETDuet-TcCGT1与基因片段连接分别获得质粒pETDuet-TcCGT1-PGM-galU和pETDuet-TcCGT1-PGM-UGP1。引物P7-F/R和P8-F/R用于从酿酒酵母C800基因组中扩增PGM2和UGP1,P9-F/R用于从大肠杆菌BL21基因组中扩增galU,将线性化载体pETDuet-TcCGT1和基因片段连接分别获得质粒pETDuet-TcCGT1-PGM2-galU和pETDuet-TcCGT1-PGM2-UGP1。P10-F/R从pETDuet-TcCGT1-PGM2-galU扩增TcCGT1-PGM2-galU,P11-F/R用于扩增pRSFDuet-1,pCDFDuet-1,pACYCDuet-1质粒获得线性化载体,将线性化载体和基因片段连接分别获得质粒pRSFDuet-TcCGT1-PGM2-galU,pCDFDuet-TcCGT1-PGM2-galU,pACYCDuet-TcCGT1-PGM2-galU。P12-F/R用于从大肠杆菌BL21基因组中扩增ndk基因,P13-F/R用于扩增pACYCDuet-1质粒获得线性化载体,将线性化载体和目的基因片段连接获得质粒pACYCDuet-ndk。
表3构建质粒使用引物
/>
实施例1:敲除糖酵解途径关键基因促进糖基产物合成
在大肠杆菌中删除磷酸葡萄糖异构酶基因(pgi),从6磷酸葡萄糖(G-6-P)到6-磷酸果糖(F-6-P)的途径被阻断,G-6-P被导向1磷酸葡萄糖(G-1-P)用于合成UDPG。为了进一步提高胞内UDPG的积累,以E.coli BL21(DE3)为出发菌株,使用CRISPR/Cas9系统敲除方法,对EMP途径和PPP途径的关键基因6-磷酸葡萄糖异构酶基因(pgi)和6-磷酸葡萄糖1-脱氢酶基因(zwf)进行了单基因敲除和组合敲除,并转入表达金莲花来源的8位碳苷糖基转移酶的质粒pETDuet-TcCGT1,构建得到EG01,EG02,EG03菌株(表1),以原始菌株EG00作为对照。
本发明中以芹菜素作为底物验证相关基因敲除对糖基供体UDPG和糖基化产物合成的影响。从摇瓶发酵的结果可以看出,在72h芹菜素糖基化产物牡荆素积累最高(图2)。EG00、EG01、EG02和EG03牡荆素的产量在72h分别为0.17g·L
实施例2:UDPG旁路基因敲除
在实施例1构建的EG03的基础上,使用CRISPR/Cas9系统敲除方法,对相关旁路基因进行单敲除和组合敲除,构建菌株EG04~EG10(表1)。以芹菜素作为底物,摇瓶发酵结果如图3所示,与双敲除菌株EG03相比最终OD
实施例3:UDPG合成关键基因过表达
为强化UDPG的合成,选择来自大肠杆菌和酿酒酵母的磷酸葡萄糖变位酶基因(pgm和pgm2)和UTP-葡萄糖-1-磷酸尿苷酸转移酶(galU和UGP1)进行组合表达,构建了重组质粒pETDuet-TcCGT1-PGM-galU、pETDuet-TcCGT1-PGM-UGP1、pETDuet-TcCGT1-PGM2-galU和pETDuet-TcCGT1-PGM2-UGP1,分别电转至不含有质粒pETDuet-TcCGT1的重组菌株EG10,构建EG11~EG14四株菌。
以芹菜素作为底物,EG11、EG12、EG13和EG14四株菌在摇瓶发酵72h分别积累了3.04、2.79、2.25、1.91g/L的牡荆素(图4),EG11与EG10相比牡荆素产量提高了37.6%。大肠杆菌内源的磷酸葡萄糖变位酶因效果要明显高于酿酒酵母来源的磷酸葡萄糖变位酶,选择过表达内源磷酸葡萄糖变位酶和UTP-葡萄糖-1-磷酸尿苷酸转移酶显著提高UDPG在胞内的积累,进一步提升糖基化产物产量。
实施例4:通过优化基因表达强度提高糖基产物合成
选择不同的质粒载体pRSFDuet-1、pCDFDuet-1和pACYCDuet-1表达磷酸葡萄糖变位酶基因和UTP-葡萄糖-1-磷酸尿苷酸转移酶基因,构建重组质粒pRSFDuet-TcCGT1-PGM2-galU,pCDFDuet-TcCGT1-PGM2-galU,pACYCDuet-TcCGT1-PGM2-galU,将重组质粒分别电转至不含有质粒pETDuet-TcCGT1的重组菌株EG10,得到菌株EG15~EG17。
以芹菜素作为底物,摇瓶发酵结果显示,菌株EG15、EG16和EG17的牡荆素产量分别为3.24、3.35和2.62g/L(图5)。与EG11相比,使用pCDFDuet-1作为表达载体的重组菌EG16的牡荆素产量提高了10.1%。
实施例5:通过优化基因表达强度提高糖基产物合成
UDPG在结构上由葡萄糖分子和尿苷二磷酸分子组成,大肠杆菌内源的UDPG水解酶将UDPG降解为尿苷一磷酸和葡萄糖-1-磷酸,敲除该基因可以提高胞内UDPG的供应。在实施例4构建的菌株EG16基础上敲除基因ushA得到菌株EG18。摇瓶发酵结果显示,与EG16相比菌株EG18的牡荆素产量没有提升。
UTP在大肠杆菌中是合成UDPG的直接底物,UDPG在糖基转移酶的作用下合成糖基产物和UDP,UDP由尿苷激酶(ndK)催化合成UTP,使用pACYCDuet-1过表达该基因,构建得到重组质粒pACYCDuet-ndK分别导入实施例3和实施例4构建的重组菌EG11、EG15和EG16中,产生重组菌EG19、EG20和EG21。
以芹菜素作为底物,摇瓶发酵结果显示,重组菌EG19、EG20和EG21的牡荆素产量分别为3.15、3.20和3.53g/L(图6)。
实施例6:5L-发酵罐扩大培养
为了获得更高的牡荆素浓度,使用补充有10g/L葡萄糖的TBG培养基在5L生物反应器中进行工程菌株EG21的补料分批发酵。
以500g/L甘油和100g/L酵母粉为补料培养基。当溶氧开始反弹时,开始补料,生长12h后OD
实施例7:工程菌株制备糖基化产物的扩展应用
由于TcCGT1底物混杂性,可以催化36种黄酮和其他类黄酮的8-C-糖基化,还可以催化各种酚的O-糖基化。为了进一步扩展该工程细胞的应用,本发明以木犀草素作为底物催化合成荭草苷为例,进一步说明本发明提供的工程细胞在糖基化产物生产中的应用。按照实施例6条件进行荭草苷的发酵生产。生长12h后OD
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。