一种基于单原子催化剂诱导非自由基路径的水处理方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及催化材料及水处理领域,特别涉及一种基于单原子催化剂诱导非自由基路径的水处理方法及单原子催化剂用于有机物降解的用途。
背景技术
随着人口增长与社会发展,人类对水资源的需求快速扩大。水资源短缺与水污染严重是当今社会面临的重大难题之一。非均相催化氧化是一种有前景的水深度处理技术,它主要利用臭氧、过氧化、过硫酸盐等氧化剂在催化剂表面分解产生的活性氧物种降解水中有机污染物。催化剂的设计和构建为该技术的核心之一。
为避免金属污染物、提高催化活性、最大化原子利用率及探索反应机理,最有前景的方法之一是将过渡金属基催化位点分散到原子水平。单原子催化剂(SACs)即活性金属以单个原子的形式负载于载体表面,并主要是通过与异原子键合方式联接在载体表面的具有优异催化性能的催化剂。单原子催化剂因其高原子利用效率、可调的配位结构使其具有高活性、高稳定性和高选择性。专利文献1(CN113620407A)公开了一种用单原子催化剂催化臭氧氧化处理污水的方法,具体是将过渡金属以单原子形式负载于改性多孔活性纳米碳上,制备单原子催化剂,再将单原子催化剂涂覆在碳化硅蜂窝陶瓷板上,涂覆后的碳化硅蜂窝陶瓷板应用于臭氧氧化污水处理设备上,进行污水处理,该申请认为其制备的单原子催化剂的电子不饱和效应可高效催化臭氧分解成更高级氧化剂——羟基自由基,且羟基自由基氧化选择性比臭氧小,能降解更多污染物,大大提高污水中的COD和氨氮降解率,提高污水中的细菌杀灭率和水藻去除率,进而提高污水处理效果。然而考虑到实际复杂废水很多情况下具有高的盐含量,氯离子等具有低的氧化还原电位的阴离子容易使得羟基自由基发生淬灭的问题,从而导致催化剂的降解活性下降。如何设计高效稳定的适于水处理用的单原子催化剂具有实际研究意义。
发明内容
发明要解决的问题
为了解决上述技术问题,本发明的目的在于提供一种基于单原子催化剂诱导非自由基路径的水处理方法,该方法高效稳定且使用的单原子催化剂对水中的杂质尤其是阴离子表现出强的抗干扰性能。
进一步地,本发明的目的还在于提供一种单原子催化剂用于有机物降解的用途。
用于解决问题的方案
通过发明人长期的研究,发现通过如下技术方案的实施能够解决上述技术问题:
[1]一种基于单原子催化剂诱导非自由基路径的水处理方法,其中,所述方法利用单原子催化剂分解氧化剂产生非自由基活性氧物种以降解水中有机物,所述单原子催化剂为过渡金属基单原子负载的杂原子掺杂碳催化剂。
[2]根据[1]所述的水处理方法,其中,所述过渡金属选自锰、铁、钴、镍、铜、铈、锌中的一种或多种,进一步地,与所述过渡金属配位的杂原子数为1~6。
[3]根据[1]或[2]所述的水处理方法,其中,所述非自由基活性氧物种选自表面吸附态原子氧和单线态氧中的一种或两种。
[4]根据[1]~[3]任一技术方案所述的水处理方法,其中,所述氧化剂选自臭氧和/或过硫酸盐,进一步优选氧化剂选自臭氧。
[5]根据[4]所述的水处理方法,其中,所述氧化剂选自臭氧,所述单原子催化剂投加量为0.02~5g/L,所述臭氧的浓度为2~20mg/L,所述臭氧的流量为0.1~2L/min。
[6]根据[1]~[5]任一技术方案所述的水处理方法,其中,所述单原子催化剂的制备方法包括将除锌盐外的其他过渡金属盐、锌盐、2-甲基咪唑溶于溶剂中,在常温常压下反应,干燥获得前驱体,在惰性气体保护下对所述前驱体进行热解制得。
[7]根据[6]所述的水处理方法,其中,所述除锌盐外的其他过渡金属盐与锌盐的摩尔比为(0.002~0.6):1,所述锌盐与2-甲基咪唑的摩尔比为1:(2~10),所述溶剂相对于锌盐的加入量为(5-50)mL/mmol。
[8]根据[1]~[7]任一技术方案所述的水处理方法,其中,所述水包括饮用水或有机废水,进一步地有机废水包括煤化工废水、石油化工废水、制药废水、垃圾渗滤液、印染废水、造纸废水中的一种或多种。
[9]一种单原子催化剂用于有机物降解的用途,其中,所述单原子催化剂为过渡金属基单原子负载的氮掺杂碳催化剂,利用所述单原子催化剂分解臭氧产生非自由基活性氧物种以降解有机物。
发明的效果
本发明的基于单原子催化剂诱导非自由基路径的水处理方法,利用过渡金属基单原子负载的杂原子掺杂碳催化剂分解氧化剂尤其是臭氧,可以定向诱导产生非自由基活性氧物种,其抗干扰性能强,能够在常温常压下有效地降解和去除水中难降解有机物,去除效率高,在复杂实际废水的深度处理方面具有潜在优势。同时,本发明制备单原子催化剂的方法简单且在常温常压下在温和反应体系下就可以获得所需单原子催化剂,相比较水热法等方式来说具有更好的安全性。
需要说明的是,上述的记载并不是公开了本发明的全部实施方式和本发明的全部优点。
附图说明
图1:实施例1涉及的单原子铁催化剂前驱体扫描电镜和透射电镜图
图2:实施例1制得的单原子铁催化剂扫描电镜和透射电镜图
图3:实施例1制得的单原子铁催化剂透射电镜暗场像及元素分布图
图4:实施例1制得的单原子铁催化剂高角环形暗场扫描透射电镜图
图5:实施例1制得的单原子铁催化剂X射线吸收精细结构谱图
图6:实施例1制得的单原子铁催化剂Fe-N4位点结构图(黄色:Fe;蓝色:N;青色:C)
图7:实施例2涉及的单原子钴催化剂前驱体扫描电镜图
图8:实施例2制得的单原子钴催化剂扫描电镜图
图9:实施例1、比较例1和比较例2制备的催化剂及臭氧对草酸的降解效果对比图
图10:实施例2、比较例3和比较例4制备的催化剂及臭氧对草酸的降解效果对比图
图11:实施例1、比较例1和比较例2制备的催化剂及臭氧对对羟基苯甲酸的降解效果对比图
图12:实施例2、比较例3和比较例4制备的催化剂及臭氧对对羟基苯甲酸的降解效果对比图
图13:实施例1制得的单原子铁催化剂催化臭氧深度处理制药废水图
图14:实施例2制得的单原子钴催化剂催化臭氧深度处理制药废水图
图15:实施例1制得的单原子铁催化臭氧深度处理垃圾渗滤液图
图16:不同体系的电子顺磁共振波谱(以DMPO为自旋捕获剂,以水为溶剂)
图17:不同体系的电子顺磁共振波谱(以DMPO为自旋捕获剂,以DMSO为溶剂)
图18:不同体系的电子顺磁共振波谱(以TEMP为自旋捕获剂,以水为溶剂)
图19:不同捕获剂对草酸降解的影响图
图20:不同捕获剂对对羟基苯甲酸降解的影响图
图21:单原子铁催化臭氧诱导非自由基路径图(黄色:Fe;蓝色:N;青色:C;红色:O)
图22:单原子钴催化臭氧诱导非自由基路径图(粉色:Co;蓝色:N;青色:C;红色:O)
具体实施方式
以下对本发明的实施方式进行说明,但本发明不限定于此。本发明不限于以下说明的各构成,在发明请求保护的范围内可以进行各种变更,而适当组合不同实施方式、实施例中各自公开的技术手段而得到的实施方式、实施例也包含在本发明的技术范围中。另外,本说明书中记载的文献全部作为参考文献在本说明书中进行援引。
除非另有定义,本说明书中所用的技术和科学术语具有与本发明所属技术领域中的普通技术人员所通常理解的相同含义。
在描述本说明书的上下文中(尤其在所附权利要求的上下文中),术语“一个”、“一种”和“该(所述,the)”和类似的语言将被解释为覆盖单数和复数,除非本文另外指示或与上下文明显矛盾。
本说明书中,使用“数值A~数值B”或“数值A-数值B”表示的数值范围是指包含端点数值A、B的范围。
本说明书中,使用“可以”表示的含义包括了进行某种处理以及不进行某种处理两方面的含义。本说明书中,“任选的”或“任选地”是指接下来描述的事件或情况可发生或可不发生,并且该描述包括该事件发生的情况和该事件不发生的情况。
本说明书中,所提及的“一些具体/优选的实施方式”、“另一些具体/优选的实施方式”、“一些具体/优选的技术方案”、“另一些具体/优选的技术方案”等是指所描述的与该实施方式有关的特定要素(例如,特征、结构、性质和/或特性)包括在此处所述的至少一种实施方式中,并且可存在于其它实施方式中或者可不存在于其它实施方式中。另外,应理解,所述要素可以任何合适的方式组合在各种实施方式中。
本发明的说明书和权利要求书中的术语“包括”以及它们任何变形,意图在于覆盖不排他的包含。例如包含一系列步骤或单元的过程、方法或系统、产品或设备没有限定于已列出的步骤或单元,而是可选地还包括没有列出的步骤或单元,或可选地还包括对于这些过程、方法、产品或设备固有的其它步骤或单元。
<水处理方法>
本发明提供了一种基于单原子催化剂诱导非自由基路径的水处理方法,其利用单原子催化剂分解氧化剂产生非自由基活性氧物种以降解水中有机物。
单原子催化剂
本发明的单原子催化剂为过渡金属基单原子负载的杂原子掺杂碳催化剂。本发明以碳材料为基底,利用杂原子锚定过渡金属,使其以单个原子分散于基底表面,制备过渡金属基单原子负载的杂原子掺杂碳催化剂。在本发明的一些具体实施方式中,过渡金属选自锰、铁、钴、镍、铜、铈、锌中的一种或多种,进一步地,优选地过渡金属选自铁、钴、锌中一种或多种。在本发明的一些具体实施方式中,杂原子选自氧、硫、氮、磷、硼中的一种或多种。进一步地,杂原子选自氮、磷、硫中的一种或多种,更进一步地,杂原子选自氮。在本发明的一些具体实施方式中,与过渡金属配位的杂原子数为1~6,进一步地,与过渡金属配位的杂原子数优选为4,更进一步地,单原子催化剂主要活性位点为过渡金属-N4基团,即指过渡金属单原子通过4个氮原子配位卡入多孔碳基底上形成的发挥催化活性的位点。在本发明的一些具体实施方式中,单原子催化剂呈正十二面体形状,分散性良好,催化剂的平均粒径为50~1000nm,进一步优选地平均粒径为150~500nm,更优选地粒径为300~400nm,平均粒径采用imageJ软件通对SEM图中的纳米颗粒大小统计分析得出。在本发明的一些具体实施方式中,单原子催化剂中过渡金属的质量百分比为0.1~10wt%,优选为1.0~5.0wt%,更进一步优选为2%~3.0wt%。在本发明的一些具体实施方式中,单原子催化剂中杂原子的掺杂量为2~20wt%,优选为5~15wt%,更优选为10~15wt%。
在本发明的一些实施方式中,单原子催化剂的制备可以在常温常压下进行。更具体地,单原子催化剂的制备方法包括将除锌盐外的其他过渡金属盐、锌盐、2-甲基咪唑溶于溶剂中,在常温常压下反应,干燥获得前驱体,在惰性气体保护下对所述前驱体进行热解制得。其中除锌盐外的其他过渡金属盐的种类不受特别的限制,比如可选自硝酸盐、卤素盐、醋酸盐、乙酰丙酮盐或其水合物中的一种或多种。以锌盐为锌源,锌盐的种类不受特别的限制,比如可选自硝酸锌、氯化锌、醋酸锌或其水合物中的一种或多种。2-甲基咪唑作为有机连接剂,溶剂优选甲醇、水或N,N-二甲基甲酰胺(DMF)。在本发明的一些具体实施方式中,除锌盐外的其他过渡金属盐与锌盐的摩尔比为(0.002~0.6):1,进一步地优选为(0.02~0.5):1,如果锌盐用量过大会导致其分散过渡金属的作用不足,催化剂中过渡金属颗粒有团聚的倾向;锌盐与2-甲基咪唑的摩尔比为1:(2~10),进一步地优选为1:(5~9);溶剂相对于锌盐的加入量为(5~50)mL/mmol,进一步地优选为(10~30)mL/mmol,当溶剂用量过低时,多孔晶体难以合成,当溶剂用量过大,合成的催化剂有团聚的倾向。本发明的单原子催化剂的制备无需额外进行加温和加压,可在常温(室温)常压下将除锌盐外的其他过渡金属盐、锌盐、2-甲基咪唑溶于溶剂中搅拌反应即可,具有良好的安全性且能耗低,操作更为便捷。本发明的一些实施方式中,反应时间为15~35h,进一步地为20~30h,多孔晶体经历成核生长过程。反应之后离心干燥获得粉末状的前驱体,本发明的一些实施方式中,离心干燥的优选采用高速离心机进行离心分离,转速为8000~12000rpm,离心8~15分钟,在50~70℃烘箱干燥10~15h。最后在惰性气体保护下对前驱体进行热解制得本发明的单原子催化剂,惰性气体优选为氮气或者氩气,热解的温度优选为800~1200℃,进一步地为900~1100℃,热解时间是2~5h,进一步地为3~4h,热解优选在管式炉中进行。本发明的单原子催化剂的制备方法操作工艺简单、安全性好、成本低廉,重复性较好,制备得到的催化剂稳定性好。
氧化剂和非自由基活性氧物种
本发明的氧化剂是指能在催化剂表面分解产生的活性氧物种降解水中有机污染物的物质。在本发明的一些具体实施方式中,本发明的氧化剂包括臭氧和/或过硫酸盐。进一步优选地,本发明的氧化剂选自臭氧。臭氧(O
水处理方法
本发明的基于单原子催化剂诱导非自由基路径的水处理方法,是将单原子催化剂填充到反应器中,导入氧化剂和待处理的水进行水中有机物的降解。在本发明的一些具体实施方式中,相对于含过渡金属纳米颗粒的杂原子掺杂碳催化剂以及杂原子掺杂碳催化剂,本发明的过渡金属基单原子负载的杂原子掺杂碳催化剂具有优越的臭氧催化氧化特性,特别适合在臭氧催化氧化反应体系的应用。具体地,将单原子催化剂填充到反应器中,通入臭氧和待处理的水进行水中有机物的降解。为了兼顾降解效果和成本的考虑,单原子催化剂优选投加量为0.02~5g/L,进一步优选为0.04~2g/L,更进一步为0.05~0.5g/L,臭氧的浓度为2~20mg/L,进一步为3~20mg/L,臭氧的流量为0.1~2L/min,进一步为0.2~1L/min。本发明的待处理的水包括生活或工业的有机废水或者饮用水,更进一步地,有机废水包括煤化工废水、石油化工废水、制药废水、垃圾渗滤液、印染废水、造纸废水中的一种或多种。本发明能够在常温常压下有效地降解和去除待处理的水中难降解有机物,即无需对反应器进行额外的加温加压就能达到高的有机物去除效率,在复杂实际废水的深度处理方面具有潜在优势。进一步地,由于本发明是非自由基路径,本发明对于高盐废水(总含盐质量分数至少1%的废水,以NaCl计)具有优异的深度处理效果,在本发明的一些优选实施方式中,本发明处理效果较纯臭氧体系提升25%以上,深度处理出水COD值稳定低于50mg/L。
<用途>
本发明进一步提供了一种单原子催化剂用于有机物降解的用途。本发明的单原子催化剂为过渡金属基单原子负载的氮掺杂碳催化剂,该单原子催化剂能分解臭氧产生非自由基活性氧物种以降解有机物。进一步地,非自由基活性氧物种选自表面吸附态原子氧和/或单线态氧,有机物主要是指难降解有机物,一般为含有多环芳烃类化合物、杂环类化合物、氯化芳香族化合物、有机氰化物、药物、农药等。本发明的催化剂相对于杂原子掺杂碳基底和过渡金属纳米颗粒催化剂而言,催化臭氧去除有机物的去除速率大大提升。在本发明的一些优选实施方式中,针对实际废水深度处理,单原子催化臭氧体系去除速率,较杂原子掺杂碳基底催化臭氧体系提升50%以上,较过渡金属纳米颗粒掺杂碳催化臭氧提升30%以上。以草酸为例,去除速率较杂原子掺杂碳基底催化臭氧体系提升220%以上,较过渡金属纳米颗粒掺杂碳催化臭氧提升140%以上。以对羟基苯甲酸为例,去除速率较杂原子催化臭氧提升40%以上,较过渡金属纳米颗粒催化臭氧提升25%以上。
实施例
以下将通过具体的实施例对本发明做出进一步的说明:
实施例1:
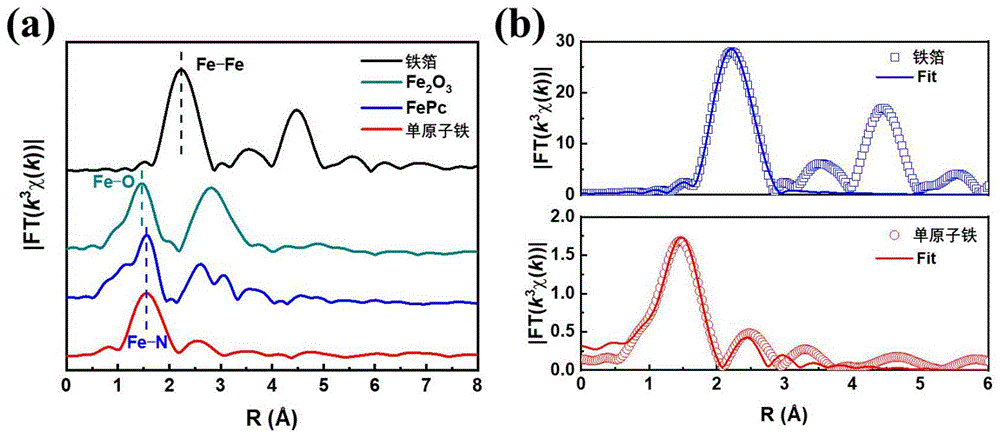
将5mmol乙酰丙酮铁、10mmol硝酸锌、80mmol 2-甲基咪唑溶于200mL甲醇中,在常温常压下搅拌1小时,反应24小时,采用高速离心机进行离心分离,转速为10000rpm,离心10分钟。干燥条件60℃烘箱干燥12小时,制备单原子铁催化剂前驱体,其扫描电镜(上)和透射电镜图(下)见图1,将获得的前驱体在氩气保护下1000℃无氧热解3小时制备得到单原子铁催化剂,其扫描电镜(上)和透射电镜(下)图见图2,其透射电镜暗场像及元素分布图见图3,高角环形暗场扫描透射电镜图见图4,X射线吸收精细结构谱图见图5。
如图1和图2所示,单原子铁催化剂前驱体呈现规则分散统一的正十二面体形状,晶型良好。热解后获得单原子铁催化剂仍保留正十二面体形状,分散性良好,且催化剂表面变得粗糙。图3显示碳、氮、铁等元素均匀分布在单原子铁催化剂表面。图4可以看出铁在催化剂上呈现单原子分散状态。通过X射线吸收精细结构谱(图5a)分析,单原子铁催化剂仅有观察到Fe-N的峰,并没有Fe-Fe的峰,再次证明铁原子为单原子分散状态。进一步通过拟合分析(图5b),单原子铁催化剂中,与铁配位的氮原子数为4,因此其主要活性位点为Fe-N4基团,其结构图如图6所示。
实施例2:
将0.25mmol硝酸钴、10mmol硝酸锌、80mmol 2-甲基咪唑溶于200mL甲醇中,在常温常压下搅拌1小时,反应24小时,采用高速离心机进行离心分离,转速为10000rpm,离心10分钟。干燥条件60℃烘箱干燥12小时,制备单原子钴催化剂前驱体,其扫描电镜图见图7,将获得的前驱体在氩气保护下1000℃无氧热解3小时制备单原子钴催化剂,其扫描电镜图见图8。
比较例1:
与实施例1相比,不添加乙酰丙酮铁,其余步骤相同,制备得到氮掺杂碳基底1。
比较例2:
与实施例1相比,乙酰丙酮铁的添加量变为10mmol,其余步骤相同,制备得到含铁纳米颗粒的氮掺杂碳催化剂。
比较例3:
与实施例2相比,不添加硝酸钴,其余步骤相同,制备得到氮掺杂碳基底2。
比较例4:
与实施例2相比,硝酸钴的添加量变为5mmol时,制备得到含钴纳米颗粒的氮掺杂碳催化剂。
性能测试
(仪器和实验方法)
化学需氧量(COD):利用水质多参数测量仪(联华5B-3BV8)进行检测,测量方法为重铬酸钾快速消解分光光度法。
草酸:利用高效液相色谱仪(Agilent1200),配合色谱柱(Waters Atlantis T3)测定。流动相为20.0mM磷酸二氢钠溶液,并用磷酸将pH调至2.5左右,流速为1.0mL/min。柱温箱温度为30℃,采用DAD检测器,波长为210nm。
对羟基苯甲酸:利用高效液相色谱仪(Agilent1200),配合色谱柱(C18反相色谱柱)。流动相A为纯甲醇,流动相B为‰1磷酸水溶液,流速为1.0mL/min。柱温箱温度为30℃,采用DAD检测器,波长为280nm。
去除效率(去除率)=处理前后目标物的浓度变化值(C-C
深度处理效率=处理前后COD的差值/处理前原始COD值×100%。
臭氧催化反应采用序批式或半序批式运行方式进行粉末流化床实验,投加一定量单原子催化剂于柱形反应器的污水中,同时臭氧发生器(Longevity EXT120)以纯净氧气为发生源,生成臭氧经臭氧浓度检测器(同林3S-J5000)通过曝气头布气通入到反应溶液中,启动催化反应,定时取样过滤,测量污水中的有机物。
草酸模拟废水进行催化剂的性能评价:草酸模拟废水浓度100mg/L,不调节pH,初始pH约为3.3。单次处理的目标废水体积为250mL,臭氧浓度为10mg/L,气体流量为0.2L/min,反应60min。
对羟基苯甲酸模拟废水进行催化剂的性能评价:对羟基苯甲酸模拟废水浓度50mg/L,不调节pH,初始pH约为4.3。单次处理的目标废水体积为250mL,臭氧浓度为3mg/L,气体流量为0.2L/min,反应60min。
(草酸的降解效果)
选取草酸作为典型的模式有机污染物,对比研究实施例1、比较例1和比较例2制备的催化剂催化臭氧降解草酸的特性,实验结果见图9。由图9可知,在0.2L/min气体流量,10mg/L臭氧浓度条件下,反应60分钟后,100mg/L草酸的去除效率仅为5%。在比较例1制得的氮掺杂碳基底1催化臭氧的体系中,草酸的去除效率为53%。当投加0.05g/L比较例2制得的铁纳米颗粒的氮掺杂碳催化剂时,草酸去除效率为64%。而当投加0.05g/L实施例1制得的单原子铁催化剂,在单原子铁催化臭氧体系中,100mg/L草酸的去除效率达到91%以上,表明单原子铁催化臭氧体系可快速高效去除草酸。
对比研究实施例2、比较例3和比较例4制备的催化剂催化臭氧降解草酸的特性,实验结果见图10。由图10可知,在0.2L/min气体流量,10mg/L臭氧浓度条件下,反应60分钟后,100mg/L草酸的去除效率仅为5%。在比较例3制得的氮掺杂碳基底2催化臭氧的体系中,草酸的去除效率为24%。当投加0.05g/L比较例4制得的钴纳米颗粒的氮掺杂碳催化剂时,草酸去除效率为86%。而当投加0.05g/L实施例2制得的单原子钴催化剂,在单原子钴催化臭氧体系中,100mg/L草酸的去除效率为100%,去除效率显著提升,表明单原子钴催化臭氧体系可快速高效去除草酸。
(对羟基苯甲酸的降解效果)
选取对羟基苯甲酸作为典型的酚类有机污染物,对比研究了实施例1、比较例1和比较例2制备的催化剂催化臭氧降解对羟基苯甲酸的特性,实验结果见图11。由图11可知,在0.2L/min气体流量,3mg/L臭氧浓度条件下,反应60分钟后,50mg/L对羟基苯甲酸的去除效率为90%。在比较例1制得的氮掺杂碳基底1催化臭氧的体系中,对羟基苯甲酸的去除效率为97%。当投加0.2g/L比较例2制得的铁纳米颗粒的氮掺杂碳催化剂时,对羟基苯甲酸去除效率为98%。而当投加0.2g/L单原子铁催化剂,在单原子铁催化臭氧体系中,50mg/L对羟基苯甲酸几乎被完全去除,且较氮掺杂碳基底1催化臭氧和铁纳米颗粒催化臭氧体系,其去除速率大大提升。
对比研究实施例2、比较例3和比较例4制备的催化剂催化臭氧降解对羟基苯甲酸的特性,实验结果见图12。由图12可知,在0.2L/min气体流量,3mg/L臭氧浓度条件下,反应60分钟后,50mg/L对羟基苯甲酸的去除效率为88%。而当投加0.2g/L单原子钴催化剂,在单原子钴催化臭氧体系中,50mg/L对羟基苯甲酸的去除效率几乎达到100%,且较氮掺杂碳基底2催化臭氧和钴纳米颗粒催化臭氧体系,其去除速率大大提升。
(对制药废水的深度处理性能)
以COD浓度约在92mg/L、盐浓度约为1.14g/L的某制药废水处理厂生化处理后的制药废水为目标废水。如图13所示,在0.5L/min气体流量,10mg/L臭氧浓度条件下,反应60分钟后,制药废水的深度处理效率为40%。而当投加0.2g/L实施例1制得的单原子铁催化剂,在单原子铁催化臭氧体系中,制药废水的深度处理效率达到54%以上,出水COD为42mg/L,稳定低于50mg/L,表明单原子铁催化臭氧体系对制药废水也具有良好的深度处理性能,反映了单原子铁催化臭氧体系的普适性。
针对同一制药废水,当投加0.2g/L实施例2制得的单原子钴催化剂,在单原子钴催化臭氧体系中,如图14所示,制药废水的深度处理效率达到58%以上,出水COD为39mg/L,稳定低于50mg/L,表明单原子钴催化臭氧对实际难降解有机废水具有更好深度处理特性。
(对垃圾渗滤液的深度处理性能)
以COD浓度约在110mg/L、盐浓度约为10g/L的某垃圾处理处理厂生化处理后的垃圾渗滤液为目标废水。对比研究了实施例1、比较例1和比较例2制备的催化剂催化臭氧深度处理垃圾渗滤液的特性,如图15所示,在0.5L/min气体流量,10mg/L臭氧浓度条件下,反应60分钟后,垃圾渗滤液的深度处理效率为38%。而当投加0.2g/L单原子铁催化剂,在单原子铁催化臭氧体系中,垃圾渗滤液的深度处理效率达到62%以上,出水COD为41mg/L,稳定低于50mg/L。单原子铁催化臭氧体系对垃圾渗滤液的深度处理性能也明显优于氮掺杂碳基底1催化臭氧体系和铁纳米颗粒催化臭氧体系。
(机理研究)
以5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)为自旋捕获剂,以水为溶剂,采用电子顺磁共振波谱检测不同反应体系中产生的自由基种类及丰度,结果见图16。由图16可知,在单独臭氧体系、比较例1制得的氮掺杂碳基底1/臭氧和实施例1制得的单原子铁催化剂/臭氧体系中均没观察到羟基自由基和超氧阴离子自由基的特征峰,表明自由基不是主要的活性物种。
以5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)为自旋捕获剂,以二甲基亚砜(DMSO)为溶剂,采用电子顺磁共振波谱检测不同反应体系中产生的自由基种类及丰度,结果见图17。图17可知,在单独臭氧体系并未观察到自由基的特征峰,而在比较例1制得的氮掺杂碳基底1/臭氧和实施例1制得的单原子铁催化剂/臭氧体系中均观察到超氧阴离子自由基的特征峰,且单原子铁催化臭氧体系中超氧阴离子自由基的丰度明显更高。由于在水溶剂中并没有观察到自由基的特征峰,而在DMSO溶剂中产生的超氧阴离子自由基,说明超氧阴离子自由基在水中快速转变成了其他活性氧物种,如单线态氧。
以2,2,6,6-四甲基哌啶(TEMP)为自旋捕获剂,以水为溶剂,采用电子顺磁共振波谱检测不同反应体系中产生单线态氧的丰度,结果见图18。图18可知,在单独臭氧体系、比较例1制得的氮掺杂碳基底1/臭氧和单原子铁催化剂/臭氧体系中均观察到单线态氧的特征峰,但单原子铁催化臭氧体系中单线态氧的丰度明显更高,说明单线态氧是反应体系中的主要活性氧物种,且超氧阴离子自由基是关键中间体。
基于淬灭原理,考察了单原子铁催化臭氧降解草酸的主要活性氧物种。叔丁醇可以捕获水溶液中的羟基自由基,甲醇可以捕获水溶液和催化剂表面的羟基自由基,二甲基亚砜可以捕获催化剂表面的吸附态原子氧。从图19可知,加入叔丁醇和甲醇后,草酸的去除过程几乎不受影响,说明羟基自由基不是降解草酸的主要活性氧物种,而加入二甲基亚砜后,草酸降解明显受到抑制,说明表面吸附态原子氧是去除草酸的主要活性氧物种。草酸的去除主要发生在催化剂表面,依赖非自由基表面吸附态原子氧。
基于淬灭原理,考察了单原子铁催化臭氧降解对羟基苯甲酸的主要活性氧物种。叔丁醇可以捕获水溶液中的羟基自由基,甲醇可以捕获水溶液和催化剂表面的羟基自由基,二甲基亚砜可以捕获催化剂表面的吸附态原子氧,而糠醇可以捕获水体中的单线态氧。从图20可知,加入叔丁醇和甲醇后,对羟基苯甲酸的去除过程并没收到抑制,说明羟基自由基不是降解对羟基苯甲酸的主要活性氧物种,加入二甲基亚砜后,对羟基苯甲酸的降解也几乎不受影响,表明表面吸附态原子氧也不是主要活性氧物种,而加入糠醇后,对羟基苯甲酸的降解明显受到抑制,说明单线态氧是去除对羟基苯甲酸的主要活性氧物种。对羟基苯甲酸的去除主要发生在水体溶液中,依赖非自由基单线态氧。
机理分析表明,单原子催化臭氧去除污染物的过程为非自由基氧化过程,主要依赖臭氧在单原子催化剂表面分解产生的表面吸附态原子氧和单线态氧,而不是羟基自由基。以实施例1制得的单原子铁催化剂催化臭氧为例,其分子动力学分析见图21,图21显示臭氧在单原子铁催化剂表面的Fe-N4活性位点分解,臭氧的一个端原子氧与Fe-N4基团的铁原子结合,之后臭氧中近表面的氧氧键被拉长,并发生断裂,产生表面吸附态原子氧和超氧阴离子自由基,超氧阴离子自由基进一步转化为单线态氧,因此在单原子铁催化臭氧体系中,有机物的去除主要包括两条非自由基路径,一条是污染物在催化剂表面去除,依靠表面吸附态原子氧的氧化过程,另一条是污染物在溶液中降解,依靠单线态氧的氧化。实施例2制得的单原子钴催化剂催化臭氧过程也符合非自由基氧化路径,其分子动力学分析见图22,臭氧受到单原子钴催化剂的吸引,向催化剂表面靠近,进一步,臭氧的一个端原子氧与Co-N4基团的钴原子结合,之后臭氧中近表面的氧氧键被拉长,并发生断裂,产生表面吸附态原子氧和单线态氧,因此在单原子钴催化臭氧体系也遵循非自由基路径,主要依赖臭氧在单原子催化剂表面分解产生的表面吸附态原子氧和单线态氧,而不是羟基自由基。
产业上的可利用性
本发明公开的技术方案可以在工业上应用。