一种槐角的处理方法、指纹图谱及其应用
文献发布时间:2024-04-18 19:58:26
技术领域
本发明涉及一种槐角的处理方法、指纹图谱及其应用。
背景技术
槐角为豆科植物槐Sophora japonica L.的干燥成熟果实,性寒、味苦,归肝、大肠经。具有清热泻火、凉血止血的功效,用于肠热便血、痣肿出血,肝热头痛、眩晕目赤等症。槐角主要成分有黄酮、异黄酮、生物碱、三萜皂苷、氨基酸及硬脂酸类等,其中以异黄酮及其苷类含量最高。槐角具有防癌抗癌及雌性激素样作用,对癌症、心血管疾病、骨质疏松和女性更年期综合征有很好的防治作用。槐角生品清热凉血力较强,用于凉血止血。槐角炭寒性大减,长于收敛止血。槐角经炮制其外型、功效都发生了变化,推测可能与炒炭过程中活性物质的改变相关。槐角炭在经典名方凉血地黄汤中为君药使用,而在验方选集、杨氏家藏方等中都有关于槐角炭的应用记载。目前对于槐角炭的炮制研究还不够系统深入,槐角炭存性标准尚未统一,仅凭主观经验判别,各地的炮制品质量相差很大,致使临床作用也相差很大。目前对槐角炒炭前后的成分鉴别研究较少,尚无对槐角炭止血物质基础的分析。
2021年中草药第52卷第21期出版了李晶峰等人的不同产地槐角HPLC指纹图谱建立及化学模式识别研究,方法是采用Agilent Zorbax SB C18色谱柱在Agilent 1260高效液相色谱仪上,检测波长为260nm,体积流量为0.6mL/min,甲醇-乙腈-0.07%磷酸水(12∶20∶68)为流动相进行等度洗脱,共得到9个共有峰,仅指认了芦丁、染料木苷、槐角苷3个色谱峰,所给出的化学信息稍显单薄。现有技术所提供的方法对于槐角药材成分信息不够全面,难以客观、准确、全面的评价槐角药材和槐角炭的质量。因此建立槐角药材和槐角炭指纹图谱方法对它们的质量控制具有重要意义。
发明内容
本发明所要解决的技术问题是为了克服现有技术中针对槐角生品或槐角炭的检测方法较少,且能够检测到的组分较少,以及缺少有效高效的评价槐角生品或槐角炭质量的方法的缺陷,提供了一种槐角的处理方法、指纹图谱及其应用,本发明所提供的指纹图谱对槐角生品或槐角炭的多个有效成分进行分离和定性鉴别可以快速、准确地评价槐角生品或槐角炭的质量,能够较为全面表征槐角和槐角炭中的化学信息,用于指导工业生产,更好的保证产品质量,为槐角生品或槐角炭的药效物质基础研究及质量控制提供科学有效的方法。
本发明主要通过以下技术方案解决上述技术问题。
本发明提供了一种处理方法,其包括以下步骤:
采用液相色谱法,将槐角待测物进行梯度洗脱,即可;
所述液相色谱法中,色谱条件如下:
色谱柱为十八烷基硅烷键合硅胶色谱柱;
流动相为流动相A-流动相B或流动相A-流动相B,所述流动相A为甲酸溶液,所述流动相B为甲醇,所述流动相C为乙腈;
当流动相为流动相A-流动相B时,以所述流动相的总体积为100%计,所述梯度洗脱的程序按下表的顺序从上到下依次进行:
其中,x1为83%-92%(85%、90%),x2为68%-82%(70%、80%),x3为48%-67%(50%、60%、65%),x4为38%-48%(40%、55%),x5为18%-45%(20%、40%),x6为83-92%(85%、90%),x1>x2>x3>x4>x5;
当流动相为流动相A-流动相C时,以所述流动相的总体积为100%计,所述梯度洗脱的程序按下表的顺序从上到下依次进行:
其中,y1为90%-95%(93%),y2为88%-92%(90%),y3为83%-87%(85%),y4为78%-82%(80%),y5为73%-77%(75%),y6为68%-72%(70%),y7为53%-58%(55%),y8为38%-43%(40%),y9为90%-95%(93%);
所述洗脱时长为该洗脱阶段的洗脱终点时间与洗脱起点时间的差值。
其中,所述洗脱时长为该洗脱阶段的洗脱终点时间与洗脱起点时间的差值。
所述色谱柱可为Waters ACQUITY CSH C18色谱柱(规格可为150mm×2.1mm,1.7μm)、1-ACQUITY CSH C18色谱柱(规格可为150mm×2.1mm,1.7μm)或2-Hypersi1 Gold(规格可为100×2.1mm,1.9μm)色谱柱;优选为Waters ACQUITY CSH C18色谱柱(规格可为150mm×2.1mm,1.7μm)。
所述色谱条件中,x1可为85%或90%。
所述色谱条件中,x2可为70%或80%。
所述色谱条件中,x3可为50%、60%或65%。
所述色谱条件中,x4可为40%或55%。
所述色谱条件中,x5可为20%或40%。
所述色谱条件中,x6可为85%或90%。
所述色谱条件中,y1可为93%。
所述色谱条件中,y2可为90%。
所述色谱条件中,y3可为85%。
所述色谱条件中,y4可为80%。
所述色谱条件中,y5可为75%。
所述色谱条件中,y6可为70%。
所述色谱条件中,y7可为55%。
所述色谱条件中,y8可为40%。
所述色谱条件中,y9可为93%。
所述色谱条件中,所述梯度洗脱的程序可为如下任一条件:
条件A:
条件B:
条件C:
条件D:
条件E:
条件F:
条件G:
条件H:
条件I:
。
优选的,所述色谱柱为Waters ACQUITY CSH C18色谱柱,所述流动相为流动相A-流动相B,所述梯度洗脱的程序如下表所示:
。
所述液相色谱法可为高效液相色谱法(HPLC)或超高效液相色谱法(UPLC)。
所述色谱条件中,所述流动相A中甲酸的浓度可为本领域常规,例如为0.05-0.5%甲酸溶液,又例如0.05%甲酸溶液。
所述色谱条件中,可使用本领域常规的检测器,例如检测器为UV-DAD检测器。
所述色谱条件中,可使用本领域常规的检测波长,例如检测波长为210-300nm,又例如检测波长为230、254或280nm,优选波长为278或280nm。
所述色谱条件中,流速可为0.12-0.18mL/min;例如0.12mL/min、0.15mL/min和0.18mL/min;优选为0.12-0.15mL/min。
所述色谱条件中,柱温可为30-40℃;例如30℃、35℃或40℃;优选为35℃。
所述色谱条件中,进样量可为本领域常规,例如进样量为1-5μL,又例如进样量为1μL。
所述色谱条件中,所述槐角待测物的进样浓度可为本领域常规,例如为0.004-0.1g/mL,又例如0.025g/mL。
所述槐角待测物在洗脱前可进行预处理,以符合进样标准。所述的预处理可为本领域常规的预处理,较佳地,所述预处理包括下述步骤:将所述槐角待测物溶解于溶剂中;较佳地,所述溶剂为甲醇。
所述预处理步骤中,还可包括以下步骤:将槐角加水(200mL)浸泡(30min)、煎煮(煮沸后煎煮30min加入160mL水继续煎煮20min)、过滤、滤液浓缩后冷冻干燥,得槐角待测物的冻干粉末,以冻干粉末的形式溶解于所述溶剂中。
所述槐角待测物可为各类槐角制品或其他含槐角成分的组合物,例如槐角的生品和/或槐角的炮制品;所述槐角的炮制品优选为槐角炭。
所述槐角炭可采用本领域常规方法制备,例如,所述槐角炭通过以下方法制备:将槐角生品进行炒制。较佳地,所述炒制的温度为200-300℃,例如200℃。较佳地,所述炒制的时间为5-50min;例如5、9、12、15、20、30、50min;更佳地;所述炒制的时间为15-20min。
所述处理方法用于检测或分离所述槐角待测物中的槐角成分。
较佳地,所述槐角成分包括以下组分:没食子酸甲酯、二氢山柰酚-3-O-葡萄糖苷、山柰酚-3-O-(2″-O-β-D-葡萄糖基)-β-D-芸香糖苷、山柰酚-3-O-槐糖、二氢芹菜素-7-O-葡萄糖苷、芹菜素-7-O-芸香糖苷、山柰酚-3-O-芸香糖苷、异鼠李素-3-O-β-D-芸香糖苷和柃木花甙中的一种或多种。更佳地,所述槐角成分还进一步包括以下组分:没食子酸、对羟基苯甲醇和染料木素-4′-O-(6″-乙酰基)-葡萄糖苷中的一种或多种。进一步地,所述槐角成分还进一步包括以下组分:(3,4,5-三羟基-3-呋喃基)-4-甲氧基苯甲酸、对双没食子酸、染料木素-7,4′-di-O-β-D-葡萄糖苷、染料木素-7-O-葡萄糖-4′-O-新橙皮糖苷、山柰酚-3-O-(3′,4′-二-O-葡萄糖基)-鼠李糖-7-O-鼠李糖苷、山柰酚-3-O-槐糖7-O-鼠李糖苷、染料木素-7-O-丙二酰葡萄糖-4′-O-葡萄糖苷、槲皮素-3-O-(3′-O-葡萄糖基)-芸香糖苷、染料木苷、芹菜素-7-O-(3″-乙酰基)-芸香糖苷、山柰酚-7-甲氧基-4′-O-葡萄糖基-3-O-(2-O-apio-呋喃酰基)葡萄糖、异鼠李素-3-O-槐糖、柚皮苷、芦丁、槐角苷、槐属双苷、染料木素-4′-O-(6″-丙二酰基)葡萄糖苷、山茶黄酮苷A、染料木素和山柰酚中的一种或多种。
较佳地,所述槐角成分包括没食子酸甲酯、二氢山柰酚-3-O-葡萄糖苷、山柰酚-3-O-(2″-O-β-D-葡萄糖基)-β-D-芸香糖苷、山柰酚-3-O-槐糖、二氢芹菜素-7-O-葡萄糖苷、芹菜素-7-O-芸香糖苷、山柰酚-3-O-芸香糖苷、异鼠李素-3-O-β-D-芸香糖苷、柃木花甙、没食子酸、对羟基苯甲醇、染料木素-4′-O-(6″-乙酰基)-葡萄糖苷、(3,4,5-三羟基-3-呋喃基)-4-甲氧基苯甲酸、对双没食子酸、染料木素-7,4′-di-O-β-D-葡萄糖苷、染料木素-7-O-葡萄糖-4′-O-新橙皮糖苷、山柰酚-3-O-(3′,4′-二-O-葡萄糖基)-鼠李糖-7-O-鼠李糖苷、山柰酚-3-O-槐糖7-O-鼠李糖苷、染料木素-7-O-丙二酰葡萄糖-4′-O-葡萄糖苷、槲皮素-3-O-(3′-O-葡萄糖基)-芸香糖苷、染料木苷、芹菜素-7-O-(3″-乙酰基)-芸香糖苷、山柰酚-7-甲氧基-4′-O-葡萄糖基-3-O-(2-O-apio-呋喃酰基)葡萄糖、异鼠李素-3-O-槐糖、柚皮苷、芦丁、槐角苷、槐属双苷、染料木素-4′-O-(6″-丙二酰基)葡萄糖苷、山茶黄酮苷A、染料木素和山柰酚。
本发明还提供了记载了利用上述所述处理方法获得的液相色谱图的产品。
所述产品可为计算机存储介质;或者,为绘制或印刷的说明书。
较佳地,所述液相色谱图中,在如下一个或多个保留时间处有特征峰:19.83±0.01min、28.24±0.06min、33.60±0.04min、34.99±0.05min、36.28±0.05min、40.90±0.04min、41.63±0.07min、42.09±0.05min和43.58±0.03min。
较佳地,所述液相色谱图中,还进一步在如下一个或多个保留时间处有特征峰:5.60±0.05min、6.56±0.01min和45.31±0.03min。
较佳地,所述液相色谱图中,还进一步在如下一个或多个保留时间处有特征峰:11.31±0.02min、16.89±0.01min、22.97±0.04min、25.92±0.01min、27.44±0.02min、27.91±0.02min、30.14±0.05min、30.53±0.03min、32.78±0.02min、33.15±0.03min、34.06±0.03min、34.70±0.03min、35.96±0.06min、37.45±0.05min、38.27±0.05min、39.89±0.06min、43.35±0.02min、46.28±0.02min、46.74±0.04min和49.21±0.04min。
较佳地,所述液相色谱图中,在如下保留时间处有特征峰:5.60±0.05min、6.56±0.01min、11.31±0.02min、16.89±0.01min、19.83±0.01min、22.97±0.04min、25.92±0.01min、27.44±0.02min、27.91±0.02min、28.24±0.06min、30.14±0.05min、30.53±0.03min、32.78±0.02min、33.15±0.03min、33.60±0.04min、34.06±0.03min、34.70±0.03min、34.99±0.05min、35.96±0.06min、36.28±0.05min、37.45±0.05min、38.27±0.05min、39.89±0.06min、40.90±0.04min、41.63±0.07min、42.09±0.05min、43.35±0.02min、43.58±0.03min、45.31±0.03min、46.28±0.02min、46.74±0.04min和49.21±0.04min。
本发明还提供了一种记载了槐角生品的指纹图谱的产品,其在如下一个或多个保留时间处有特征峰:19.83±0.01min、28.24±0.06min、33.60±0.04min、34.99±0.05min、36.28±0.05min、37.45±0.05min、40.90±0.04min、41.63±0.07min、42.09±0.05min和43.58±0.03min。
所述产品可为计算机存储介质;或者,为绘制或印刷的说明书。
较佳地,所述槐角生品的指纹图谱中,还进一步在如下一个或多个保留时间处有特征峰:22.97±0.04min、25.92±0.01min、27.44±0.02min、27.91±0.02min、30.14±0.05min、30.53±0.03min、32.78±0.02min、33.15±0.03min、34.06±0.03min、35.96±0.06min、38.27±0.05min、39.89±0.06min、44.34±0.04min、44.60±0.03min和46.74±0.04min。
较佳地,所述槐角生品的指纹图谱在如下保留时间处有特征峰:19.83±0.01min、22.97±0.04min、25.92±0.01min、27.44±0.02min、27.91±0.02min、28.24±0.06min、30.14±0.05min、30.53±0.03min、32.78±0.02min、33.15±0.03min、33.60±0.04min、34.06±0.03min、34.99±0.05min、35.96±0.06min、36.28±0.05min、37.45±0.05min、38.27±0.05min、39.89±0.06min、40.90±0.04min、41.63±0.07min、42.09±0.05min、43.58±0.03min、44.34±0.04min、44.60±0.03min和46.74±0.04min。
本发明还提供了一种记录了槐角炮制品的指纹图谱的产品,其在如下一个或多个保留时间处有特征峰:5.60±0.05min、6.56±0.01min、33.60±0.04min、34.99±0.05min、37.45±0.05min、40.90±0.04min、41.63±0.07min和45.31±0.03min。
所述产品可为计算机存储介质;或者,为绘制或印刷的说明书。
较佳地,所述槐角炮制品的指纹图谱中,还进一步在如下一个或多个保留时间处有特征峰:5.31±0.01min、9.14±0.03min、11.31±0.02min、16.89±0.01min、22.97±0.04min、25.92±0.01min、27.44±0.02min、27.91±0.02min、30.53±0.03min、32.78±0.02min、34.70±0.03min、35.96±0.06min、38.27±0.05min、39.89±0.06min、43.35±0.02min、46.28±0.02min、46.74±0.04min和49.21±0.04min。
较佳地,所述槐角炮制品的指纹图谱在如下保留时间处有特征峰:5.31±0.01min、5.60±0.05min、6.56±0.01min、9.14±0.03min、11.31±0.02min、16.89±0.01min、22.97±0.04min、25.92±0.01min、27.44±0.02min、27.91±0.02min、30.53±0.03min、32.78±0.02min、33.60±0.04min、34.70±0.03min、34.99±0.05min、35.96±0.06min、37.45±0.05min、38.27±0.05min、39.89±0.06min、40.90±0.04min、41.63±0.07min、43.35±0.02min、45.31±0.03min、46.28±0.02min、46.74±0.04min和49.21±0.04min。
较佳地,所述槐角炮制品的指纹图谱中,所述槐角炮制品指槐角生品炒制20min得到的槐角炭。
本发明还提供了上述槐角生品的指纹图谱作为标准指纹图谱在槐角生品检测与质量控制中的应用。
本发明还提供了上述槐角炮制品的指纹图谱作为标准指纹图谱在槐角炮制品检测与质量控制中的应用。
所述的应用中,还可包括以下步骤:
a、取待检测的槐角生品或槐角炮制品,采用如本发明任一项所述的处理方法得到对应的液相色谱图;
b、将步骤a得到的液相色谱图与如本发明任一项所述的指纹图谱进行相似度评价。
较佳地,以上步骤b中,若相似度≥0.90,则合格。
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明所用试剂和原料均市售可得。
本发明的积极进步效果在于:
(1)本发明所述的槐角和槐角炭的指纹图谱,各自包括25个共有峰,并确定了其中峰面积较大的12个共有峰的化学结构,本发明所提供的槐角和槐角炭指纹图谱均能够在统一波长下的一张图谱中体现,更为全面、准确、可靠地对槐角药材和槐角炭质量进行评价。
(2)本发明所述的槐角和槐角炭的指纹图谱,能更全面地反映药品质量,对指导工业生产控制药品质量更加有意义,更好保证产品的质量。
(3)本发明所述的槐角和槐角炭的指纹图谱的建立方法,采用超高效液相色谱法,通过合理选择色谱条件,实现槐角和槐角炭指纹图谱的构建,具有简便、稳定、精密度高、重现性好的优点。为进一步研究槐角和槐角炭化学成分和质量标准提供依据。
(4)本发明所提供的指纹图谱对槐角生品或槐角炭的多个有效成分例如没食子酸甲酯、二氢山柰酚-3-O-葡萄糖苷、山柰酚-3-O-(2”-O-β-D-葡萄糖基)-β-D-芸香糖苷等进行分离和定性鉴别,能充分反映产品疗效,对控制工艺稳定性、保障产品质量、稳定产品疗效具有重要意义。
附图说明
图1为本发明的10批槐角生品UPLC指纹图谱。
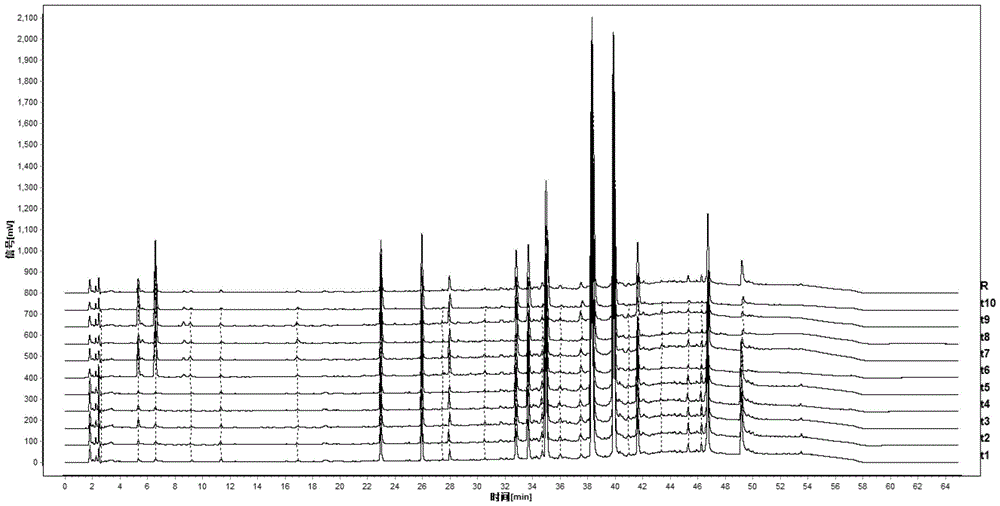
图2为本发明的10批槐角炒制20min炭品UPLC指纹图谱。
图3为对照品比对图,其中:1、没食子酸;2、山柰酚-3-O-β-D-槐二糖-7-O-α-L-鼠李糖苷;3、染料木苷;4、山柰酚-3-O-(2″-O-β-D-葡萄糖基)-β-D-芸香糖苷;5、山柰酚-3-O-β-D-槐糖;6、柚皮苷;7、芦丁;8、槐角苷;9、槐属双苷;10、山柰酚-3-O-芸香糖苷;11、异鼠李素-3-O-β-D-芸香糖苷;12、染料木素;13、山柰酚;
图4为本发明的槐角炭不同炮制时间UPLC指纹图谱。
图5为炮制前后槐角水煎液对小鼠出血时间的影响图。
图6为炮制前后槐角水煎液对小鼠凝血时间的影响图。
图7为各组小鼠凝血四项测定结果图。
图8为10批槐角生品和炒制20min炭品的PCA主成分分析图.
图9为主成分的ward法聚类分析验证图。
图10为色谱条件1得到的UPLC指纹图谱
图11为色谱条件2得到的UPLC指纹图谱
图12为色谱条件3得到的UPLC指纹图谱
图13为色谱条件4得到的UPLC指纹图谱
图14为色谱条件5得到的UPLC指纹图谱
图15为色谱条件6得到的UPLC指纹图谱
图16为色谱条件7得到的UPLC指纹图谱
图17为色谱条件8得到的UPLC指纹图谱
图18为不同检测波长得到的UPLC指纹图谱。
图19为不同色谱柱得到的UPLC指纹图谱。
图20为不同柱温得到的UPLC指纹图谱。
图21为不同流速得到的UPLC指纹图谱。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
1.试验仪器和材料
1.1仪器
美国Thremo scientific Vanquish高效液相色谱仪;PDA全波长紫外检测器(美国赛默飞世尔科技有限公司);SPSS 18.0统计分析软件;梅特勒MS105DU电子天平;超声波清洗器(KQ-250DE型),Millipore纯水机,URIT-600A半自动凝血分析仪。
1.2材料
对照品没食子酸(批号:110831-201605,90.8%)、芦丁(批号:100080-202012,91.6%)、山柰酚(批号:110861-202013,93.2%)、染料木素(批号:111704-201703,98.8%)、山柰酚-3-O-芸香糖苷(批号:112007-202103,94.0%)、柚皮苷(批号:110722-202116,93.5%)、染料木苷(批号:111709-201702,99.9%)、槐角苷(批号:111695-201703,99.6%)均购自中国食品药品检定研究院;槐属双苷(批号:ST82950105,≥98%)采购自上海诗丹德标准技术服务有限公司;山柰酚-3-O-β-D-槐糖(批号:21070121,≥98%)和山柰酚-3-O-(2″-O-β-D-葡萄糖基)-β-D-芸香糖苷(批号:21081031,≥98%)均购于上海同田生物技术有限公司;异鼠李素-3-O-β-D-芸香糖苷和山柰酚-3-O-β-D-槐二糖-7-O-α-L-鼠李糖苷为实验室自制,纯度>98%。
HPLC用甲醇和甲酸为色谱纯,水为超纯水,其余试剂为分析纯。
1.3实验动物
ICR小鼠由上海实验动物中心提供,给药前后,各组小鼠均分笼饲养,喂饲全价营养颗粒饲料,自由饮水,雌雄各半,体重19~21g。
2.制备槐角炭样品
分别从全国主要药材产区及道地产区购买处方药材共10批,药材的产地信息见表1。
表1药材产地信息
除非另有说明,实施例中槐角炭的制备方法如下:称取净制槐角生品约400g在锅温200℃左右投入锅中炒制20min,取出放凉备用,打粉过3号筛,即得槐角炭基准物质。
按表1所列药材批号取药材为原料制备槐角炭,并依次编号为t1-t10;t9为方法学验证用批次。
下面结合具体的实施例对本发明作进一步的解释和说明。
实施例1
1.对照品溶液的制备
取柚皮苷、槐角苷、槐属双苷、芦丁、山柰酚、染料木素、染料木苷、异鼠李素-3-O-β-D-芸香糖苷、山柰酚-3-O-β-D-芸香糖苷、山柰酚-3-O-β-D-槐糖、山柰酚-3-O-β-D-槐二糖-7-O-α-L-鼠李糖苷、山柰酚-3-O-(2”-O-β-D-葡萄糖基)-β-D-芸香糖苷对照品适量,加甲醇溶解,制成每1mL含各对照品10μg的溶液,作为对照品溶液。
2.待测样品溶液的制备精密量取槐角(s1-s10)或槐角炭粉末(t1-t10)约1g,精密称定,置50mL具塞锥形瓶中,精密加入70%甲醇30mL,密塞,称定重量,超声处理(功率250W,频率40KHz)60分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
在建立提取方法时,前期实验比较了水提法以及醇提法,在200-400nm全波长扫描下,甲醇提取比水和乙醇提取得到的峰数更多,且成本较低,因此选择甲醇提取,并在比较了不同浓度甲醇提取效果后,确定提取浓度为70%甲醇。
3.超高效液相法测定槐角的指纹图谱和槐角炭的指纹图谱
将按步骤1和2方法制备得到的10批槐角样品(s1-s10)、10批槐角炭样品(t1-t10)和对照品溶液分别注入超高效液相色谱仪,色谱条件为:Waters ACQUITY CSH C18色谱柱(150mm×2.1mm,1.7μm);流速0.15ml/min;柱温:35℃;采用紫外检测器检测,检测波长为280nm;理论塔板数按槐角苷峰计算不低于5000。流动相:0.05%甲酸水为A相、甲醇为B相,按下表2进行梯度洗脱。
表2
所得的槐角和槐角炭指纹图谱共35个色谱峰。
其中,槐角炭指纹图谱见图2。槐角炭指纹图谱包括25个共有峰,各峰的保留时间分别为:
1号峰5.31min,2号峰6.56min,3号峰9.14min,4号峰11.31min,5号峰16.89min,7号峰22.97min,8号峰25.92min,9号峰27.44min,10号峰27.91min,13号峰30.53min,14号峰32.78min,16号峰33.60min,18号峰34.70min,19号峰34.99min,20号峰35.96min,22号峰37.45min,23号峰38.27min,24号峰39.89min,25号峰40.90min,26号峰41.63min,28号峰43.35min,32号峰45.31min,33号峰46.28min,34号峰46.74min,35号峰49.21min。23号峰为对照峰。
槐角生品指纹图谱见图1。槐角指纹图谱包括25个共有峰,各峰的保留时间分别为:
6号峰19.83min,7号峰22.97min,8号峰25.92min,9号峰27.44min,10号峰27.91min,11号峰28.24min,12号峰30.14min,13号峰30.53min,14号峰32.78min,15号峰33.15min,16号峰33.60min,17号峰34.06min,19号峰34.99min,20号峰35.96min,21号峰36.28min,22号峰37.45min,23号峰38.27min,24号峰39.89min,25号峰40.90min,26号峰41.63min,27号峰42.09min,29号峰43.58min,30号峰44.34min,31号峰44.60min,34号峰46.74min。23号峰为对照峰。
各峰相对保留时间与对照峰相对保留时间比值是分别为:
1号峰相对保留时间RRT为0.14,2号峰相对保留时间RRT为0.17,3号峰相对保留时间RRT为0.24,4号峰相对保留时间RRT为0.30,5号峰相对保留时间RRT为0.44,6号峰相对保留时间RRT为0.52,7号峰相对保留时间RRT为0.60,8号峰相对保留时间RRT为0.68,9号峰相对保留时间RRT为0.72,10号峰相对保留时间RRT为0.73,11号峰相对保留时间RRT为0.74,12号峰相对保留时间RRT为0.79,13号峰相对保留时间RRT为0.80,14号峰相对保留时间RRT为0.86,15号峰相对保留时间RRT为0.87,16号峰相对保留时间RRT为0.88,17号峰相对保留时间RRT为0.89,18号峰相对保留时间RRT为0.91,19号峰相对保留时间RRT为0.92,20号峰相对保留时间RRT为0.94,21号峰相对保留时间RRT为0.95,22号峰相对保留时间RRT为0.98,23号峰相对保留时间RRT为1.00,24号峰相对保留时间RRT为1.05,25号峰相对保留时间RRT为1.07,26号峰相对保留时间RRT为1.09,27号峰相对保留时间RRT为1.10,28号峰相对保留时间RRT为1.13,29号峰相对保留时间RRT为1.14,30号峰相对保留时间RRT为1.16,31号峰相对保留时间RRT为1.17,32号峰相对保留时间RRT为1.18,33号峰相对保留时间RRT为1.21,34号峰相对保留时间RRT为1.23,35号峰相对保留时间RRT为1.28。其中23号峰为对照物的色谱峰。
其中:共有峰中10号峰为山柰酚-3-O-β-D-槐二糖-7-O-α-L-鼠李糖苷,14号峰染料木苷,16号峰山柰酚-3-O-(2”-O-β-D-葡萄糖基)-β-D-芸香糖苷,19号峰山柰酚-3-O-槐糖,20号峰为柚皮苷,22号峰是芦丁,23号峰为槐角苷,24号峰为槐属双苷,26号峰为山柰酚-3-O-芸香糖苷,27号峰为异鼠李素-3-O-β-D-芸香糖苷,34号峰为染料木素,35号峰为山柰酚。各对照品比对图见图3。
相似度结果
将积分信号导入中国药典委的“中药色谱指纹图谱相似度评价系统2012A版”软件,以S1为参照谱图,按中位数法计算得相似度结果,并生成标准对照指纹图谱(RFP),见图1和图2,相似度结果见表3。相似度小于0.90为不合格产品,对10批样品进行质量评价。
表3 10批槐角生品和炭品指纹图谱相似度
结果表明,10批样品均为合格产品。
生成对照指纹图谱的方法为中位数法,时间窗为0.1min,生成的对照指纹图谱见图3,经分析,10批样品共有25个共有峰。
4方法学考察
4.1精密度考察:
将编号t9样品按指纹图谱测定方法制备供试品溶液,连续进样6次,结果见表4和5。
表4精密度试验结果(保留时间,min)
/>
表5精密度试验结果(峰面积)
/>
结果显示,连续进样6针共有峰峰面积RSD值均小于4.0%,保留时间RSD值均小于1.0%,表明该方法的精密度良好。
4.2重复性考察:
取同一批号样品(t9),按照指纹图谱测定项下方法分别平行制备供试品溶液6份,依法测定,结果见表6和表7。
表6重复性试验结果(保留时间)
/>
表7重复性试验结果(峰面积)
结果显示测得6份样品中共有峰的峰面积的RSD值均小于5.0%,保留时间RSD值均小于1.0%,表明该方法的重复性良好。
4.3稳定性考察:
将t9样品按指纹图谱测定方法制备供试品溶液,在室温下放置0小时、2小时、4小时、8小时、12小时、18小时、24小时、48小时、72小时,分别依法测定,考察共有峰相对峰面积及相对保留时间RSD,结果见表8和9。
表8稳定性试验结果(保留时间)
表9稳定性试验结果(峰面积)
/>
结果显示48小时内各时间点分别测定供试品溶液中共有峰峰面积RSD值和保留时间RSD值均小于5.0%,表明供试品溶液在48小时内稳定。
5.指纹图谱的应用
5.1制备槐角炭样品
称取净制槐角生品(s1-s10)约400g,分为8份,其余7份在锅温200℃左右投入锅中,分别炒制5、9、12、15、20、30、50min,取出放凉备用,不同炒制程度样品各制备10批。
5.2.建立槐角炭不同炮制时间的UPLC指纹图谱
5.2.1槐角炭供试品溶液的制备
将槐角炭打粉过3号筛,各称取约1g,精密加入70%甲醇30mL,称重,超声(250w,70KHz)60min,放冷,再称,用70%甲醇补重,摇匀,过0.22μm微孔滤膜,即得供试品溶液1。
称取不同的槐角炭粗粉各10g,分别加200mL水浸泡30min,在煮沸后煎煮30min加入160mL水继续煎煮20min,过滤(单层滤布300目),滤液浓缩至100mL,冷冻干燥,即得槐角炮制品冻干粉末,用纯水现用现配,即得浓度为0.025g/mL的供试品溶液2。
根据前期样品前处理方法考察结果,供试品溶液1和2色谱峰基本一致。
云南白药供试品(批号为ZJA2014)的制备:云南白药临床给药剂量为2g/70kg,本实验云南白药给药剂量为临床等效剂量的2倍,即为0.52g/kg。精密称取云南白药2.6g于100mL容量瓶中,以生理盐水定容,摇匀即得。
5.2.2指纹图谱的建立:采用超高效液相色谱法对以上所得样品进行指纹图谱测定;色谱条件:采用Waters ACQUITY CSH C18色谱柱(150mm×2.1mm,1.7μm);流动相为:A为0.05%甲酸溶液,B为甲醇;采用梯度洗脱:0~9min,体积百分浓度10%→20%流动相B;9~27min,体积百分浓度20%→40%流动相B;27~30min,体积百分浓度40%流动相B;30~39min,体积百分浓度40%→60%流动相B;39~42min,体积百分浓度60%流动相B;42~48min,体积百分浓度60%→80%流动相B;48~50min,体积百分浓度80%流动相B;50~50.1min,体积百分浓度80%→10%流动相B;50.1~65min,体积百分浓度10%流动相B;流速:0.15mL/min;检测器:UV-DAD检测器;检测波长:280nm;柱温:35℃;进样量:1μl。
5.2.3通过《中药色谱指纹图谱相似度评价系统》软件分析得到特征峰数据:保留时间分别为:1号峰5.31min,2号峰5.60min,3号峰6.56min,4号峰9.14min,5号峰11.31min,6号峰16.89min,7号峰19.83min,8号峰22.97min,9号峰25.92min,10号峰27.44min,11号峰27.91min,12号峰28.24min,13号峰30.14min,14号峰30.53min,15号峰32.78min,16号峰33.15min,17号峰33.60min,18号峰34.06min,19号峰34.70min,20号峰34.99min,21号峰35.96min,22号峰36.28min,23号峰37.45min,24号峰38.27min,25号峰39.89min,26号峰40.90min,27号峰41.63min,28号峰42.09min,29号峰43.35min,30号峰43.58min,31号峰44.44min,32号峰44.60min,33号峰45.31min,34号峰46.28min,35号峰46.74min,36号峰49.21min。不同炮制时间特征峰色谱图见图4,22分钟以后黄酮苷类成分峰面积逐渐减少,苷元逐渐增加,因此根据前20分钟酚酸类成分呈现的先增加再减少的特点,发现15-20分钟之间达到最大,因此暂时认为15-20分钟为最佳炮制时间。
5.3对不同炮制时间样品进行药效学实验
5.3.1取体重19~21g ICR小鼠100只,雌雄各半,正常饲养3天后,随机分为10组,每组10只,分别编号称重。
5.3.2分组为:生理盐水组、云南白药组、槐角生品组(炒制0min)、炒制5min槐角样品组、炒制9min槐角样品组、炒制12min槐角样品组、炒制15min槐角样品组、炒制20min槐角样品组,炒制30min槐角样品组,炒制50min槐角样品组。
5.3.3出血时间(BT):以上各组每日灌胃给药(供试品溶液2)一次,给药体积:0.1mL/10g。正常对照组给予等体积0.9%生理盐水,连续灌胃6d。末次灌胃给药后1h,将小鼠置于固定器中,平放于手术台上,用剪刀于距离小鼠尾尖1mm处剪断,待血液开始自行溢出时计时,每30s用滤纸吸附尾尖血滴一次,直至再无血液流出(即滤纸吸附时无血迹出现)。从断尾后血液流出开始到出血时间停止,此段时间即为出血时间。计算出血时间,并进行组间比较。若出血时间超过10min,将不计入数据统计。
5.3.4凝血时间(CT):按照灌胃剂量为每只小鼠0.2mL/10g,连续6天。末次给药后1h,将小鼠固定于固鼠皿中,将小鼠剪尾采血,滴于载玻片,(弃去第1滴血,滴2滴,1滴复检)血滴直径约5mm,立即用秒表计时,每隔30s用清洁的7号注射针头自血滴边缘向里轻轻挑动1次,并观察有无血丝挑起。从采血开始至挑起血丝止,所历时间即为凝血时间。数据见下表10,药效图谱见图5、6。
表10炮制前后槐角水煎液对小鼠出血时间的影响
/>
表10中,与NS比较
从以上结果可以看出,炒制20min的槐角炭可以明显缩短止血、凝血时间,验证了止血作用,表明槐角炭炮制时间20min较为合适。
5.3.5凝血四项
抗凝血液的制备:自制采血管(小鼠给药1h后,摘取小鼠眼球,使其血液滴入到具有38g/L枸橼酸钠抗凝剂的EP管中(血液:抗凝剂=9:1))。
抗凝血浆的制备:将抗凝血液以3000r/min离心10min,所得上层血浆即为贫血小板血浆(PPP),所取得血浆样品需要在4h内完成检测。
5.3.5.1活化部分凝血活酶时间(APTT)的测定
APTT试剂平衡至室温,0.025mol/L的CaCl2溶液37℃孵育5min;取待测血浆50μL,加入平衡至室温的APTT试剂50μL,混匀,37℃水浴孵育3min;加入37℃预温的0.025mol/L的CaCl
5.3.5.2凝血酶原时间(PT)的测定
使用前预热PT试剂至37℃;取待测血浆50μL,混匀,37℃孵育3min,加入37℃预温的PT试剂100μL,混匀,立即启动秒表,记录凝固时间,即为PT值(s)。每个样品重复实验2次,取其平均值。
5.3.5.3血浆凝血酶时间(TT)的测定
将TT试剂平衡至室温;取待测血浆100μL,混匀,37℃孵育3min;加入平衡至室温的TT试剂50μL,混匀,立即启动秒表,记录凝固时间,即为TT值(s)。每个样品重复实验2次,取其平均值。
5.3.5.4纤维蛋白原(FIB)的测定
将待测血浆用咪唑缓冲溶液10倍稀释,取待测稀释血浆100μL,37℃孵育3min,加入平衡至室温的FIB测定试剂50μL,混匀,立即启动秒表,记录凝固时间。每个样品重复实验2次,取其平均值。
药效数据见表11,图谱见图7,由PT、FIB实验结果可知,槐角炭和生理盐水组、槐角生品组相比有显著性差异,可缩短PT时间和增加FIB含量,表明槐角炭止血作用可能与外源性凝血途径和纤维蛋白途径有关。
表11炮制前后槐角水煎液对小鼠血浆APTT、PT、TT、FIB的影响
表11中,与NS比较
6.谱效相关性分析
6.1方法
6.1.1双变量相关分析
以量化峰面积为自变量,药效指标为因变量,采用SPSS 18.0统计软件的双变量相关分析方法进行数据处理,得到指纹图谱中各相关峰与药效的皮尔森相关系数。
6.1.2多元线性回归分析
以量化峰面积为自变量,药效指标为因变量,用SPSS 18.0对数据进行线性回归分析,采用逐步引入法进行模型拟合。
6.1.3PCA分析及ward聚类分析
通过SPSS 18.0PCA分析对槐角生品、炭品(经炒制)共35个共有峰进行PCA分析,从而寻找出相关峰对槐角生品、炭品间差异有较大影响且较大累计贡献率的指标,使谱效关系评价更准确,并且通过ward聚类分析验证PCA分析结果。
6.2结果
62.1双变量相关分析结果
如下表12,有21个色谱峰与止血药效指标具有相关性,相关性较好的色谱峰为2、3、7、12、17、20、22、23、27、30和31,其中色谱峰2与BT呈现强相关性,色谱峰7、12、31与CT呈现强相关性,色谱峰3与PT值呈现强相关性、而色谱峰20与FIB呈现强相关性。结合对照品和相对保留时间比对,紫外可见图谱和质谱数据比对指认色谱峰2为没食子酸、峰3为对羟基苯甲醇、峰7为没食子酸甲酯、峰12为二氢山柰酚-3-O-葡萄糖苷、峰17为山柰酚-3-O-(2″-O-β-D-葡萄糖基)-β-D-芸香糖苷、峰20为山柰酚-3-O-槐糖、峰22为二氢芹菜素-7-O-葡萄糖苷、峰23为芦丁、峰27为山柰酚-3-O-芸香糖苷,峰30为柃木花甙,结构式如下:
表12相关峰峰面积
表13双变量相关性分析结果
*P<0.05,**P<0.01
表14色谱峰质谱鉴定结果
*对照品比对
6.2.2多元线性回归分析结果
如下表15,槐角炭相关色谱峰与其止血作用的2个指标CT、PT所建立的模型相关系数较高,R
表15数学模型建立结果
其中,X2代表保留时间为5.60min色谱峰(色谱峰2)峰面积,X3代表保留时间为6.56min色谱峰(色谱峰3)峰面积,X26代表保留时间为40.90min色谱峰(色谱峰26)峰面积,X28代表保留时间为42.09min色谱峰(色谱峰28)峰面积,X31代表保留时间为44.34min色谱峰(色谱峰31)峰面积;X33代表保留时间为45.31min色谱峰(色谱峰33)峰面积。
综合以上两种分析法的分析结果,基本能够确定槐角炭发挥止血作用的成分为黄酮类和酚酸类小分子化合物。色谱峰2为没食子酸、峰3为对羟基苯甲醇、峰7为没食子酸甲酯、峰12为二氢山柰酚-3-O-葡萄糖苷、峰17为山柰酚-3-O-(2”-O-β-D-葡萄糖基)-β-D-芸香糖苷、峰20为山柰酚-3-O-槐糖、峰22为二氢芹菜素-7-O-葡萄糖苷、峰23为芦丁、峰26芹菜素-7-O-芸香糖苷、峰28异鼠李素-3-O-β-D-芸香糖苷、峰27为山柰酚-3-O-芸香糖苷,峰30为柃木花甙、峰33为染料木素-4′-O-(6″-乙酰基)-葡萄糖苷可能是槐角炭发挥止血作用的物质基础。
6.2.3PCA分析及ward聚类分析结果
槐角生品、炭品色谱图共有峰见图1、2。分析结果如表16所示,保留主成分个数累计贡献率>90%、特征值>1、经过KMO(越接近1越适合PCA分析)和Bartlett检验(P<0.01)的成分。
表16主成分分析结果
由槐角生品和炭品PCA得分图(图8),可以看到槐角生品位于得分图左边,槐角炭品位于右边,各自聚为一类,说明炒炭后对槐角化学成分的总体特征构成了明显影响。从上述主成分分析能够确定4个主要成分,其中第1主成分贡献率为73.386%,第2主成分贡献率为9.995%,第3主成分独立贡献率为5.421%,第4主成分独立贡献率为3.971%,4个主成分的累计贡献率为92.773%,符合要求。表17成分载荷矩阵可说明各个变量对主成分的贡献率,载荷的绝对值越大,对主成分的贡献越大。第1主成分信息量最大,独立贡献率为73.386%,峰27(0.988)、峰16(0.987)、峰30(0.975)、峰19(0.965)、峰6(0.964)、峰21(0.962)、峰4(0.956)载荷值较大。第2主成分独立贡献率为9.995%,峰2(0.748)、峰35(0.735),峰1(0.680),峰34(0.583)、峰33(0.582)载荷值较大。第3主成分独立贡献率为5.421%,峰25(0.820)、峰20(0.353)、峰28(0.337)、峰9(0.330)、峰3(0.329)载荷值较大。第4主成分独立贡献率为3.971%,峰34(0.629)、峰25(0.345)、峰10(0.340)、峰9(0.332)、峰12(0.308)的载荷较大。说明多种化学成分共同引起槐角的质量差异。
表17主成分载荷
随后利用SPSS 18.0软件中的ward法聚类分析法对主成分分析进行验证,如图9所示,槐角生品和炭品也是各自聚为一类,图中显示结果与PCA分析结果基本一致,说明PCA分析结果可靠,并且槐角生品与炭品的化学指纹峰差异,可以用聚类方式进行识别。结合各36个相关峰峰面积和pearson相关分析结果,可确认成分1、3主要是酚酸和黄酮苷类化合物,成分2主要是酚酸类和黄酮苷元类化合物,成分4为黄酮苷类化合物。
实施例2指纹图谱色谱条件的筛选
1、流动相和梯度洗脱
取槐角炭样品t1(即批号20191101的生品炒制20min的槐角炭样品)适量,按实施例1在待测样品溶液的制备方法制得供试品溶液,分别记录以下色谱条件下的色谱图,其他色谱条件同以上实施例1。
色谱条件1:流动相0.05%甲酸水溶液(A)-甲醇(B)梯度洗脱,色谱柱ThermoHypersi1 Gold UPLC柱(100*2.1mm,1.9μm),流速0.2mL/min,紫外波长278nm,柱温35℃,进样量1μL;
采用表18所示的程序进行梯度洗脱:
表18
结果如表19和图10所示。
表19
色谱条件2:流动相0.05%甲酸水溶液(A)-甲醇(B)梯度洗脱,色谱柱ThermoHypersi1 Gold色谱柱(100*2.1mm,1.9μm),流速0.2mL/min,紫外波长278nm,柱温35℃,进样量1μL;
采用表20所示的程序进行梯度洗脱:
表20
结果如表21和图11所示。
表21
色谱条件3:流动相0.05%甲酸水溶液(A)-甲醇(B),色谱柱Thermo Hypersi1Gold色谱柱(100*2.1mm,1.9μm),流速0.2mL/min,紫外波长278nm,柱温35℃,进样量1μL;
采用表22所示的程序进行梯度洗脱:
表22
结果如表23和图12所示。
表23
色谱条件4:流动相0.05%甲酸水溶液(A)-甲醇(B),色谱柱Thermo Hypersi1Gold色谱柱(100*2.1mm,1.9μm),流速0.2mL/min,紫外波长278nm,柱温35℃,进样量1μL;
采用表24所示的程序进行梯度洗脱:
表24
结果如表25和图13所示。
表25
色谱条件5:流动相0.05%甲酸水溶液(A)-乙腈(C),色谱柱Thermo Hypersi1Gold色谱柱(100*2.1mm,1.9μm),流速0.2mL/min,紫外波长278nm,柱温35℃,进样量1μL;
采用表26所示的程序进行梯度洗脱:
表26
结果如表27和图14所示。
表27
色谱条件6:流动相0.05%甲酸水溶液(A)-乙腈(C),色谱柱Thermo Hypersi1Gold色谱柱(100*2.1mm,1.9μm),流速0.2mL/min,紫外波长278nm,柱温35℃,进样量1μL;
采用表28所示的程序进行梯度洗脱:
表28
结果如表29和图15所示。
表29
色谱条件7:流动相0.05%甲酸水溶液(A)-乙腈(C),色谱柱Thermo Hypersi1Gold色谱柱(100*2.1mm,1.9μm),流速0.2mL/min,紫外波长278nm,柱温35℃,进样量1μL;
采用表30所示的程序进行梯度洗脱:
表30
结果如表31和图16所示。
表31
色谱条件8:流动相0.05%甲酸水溶液(A)-乙腈(C),色谱柱Thermo Hypersi1Gold色谱柱(100*2.1mm,1.9μm),流速0.2mL/min,紫外波长278nm,柱温35℃,进样量1μL;
采用表32所示的程序进行梯度洗脱:
表32
结果如表33和图17所示。
表33
/>
2、波长的选择
取槐角炭样品t8(即批号210100881的生品炒制20min的槐角炭样品)适量,按实施例1在待测样品溶液的制备方法制得供试品溶液,分别记录检测波长为230、254、280nm下的色谱图,其他色谱条件同以上实施例1。
检测结果见图18,图中,从下至上波长依次为230、254、280nm,色谱峰差异不大,280nm波长下,色谱峰响应更高,因此优选波长为280nm。
3、色谱柱的选择
取槐角炭样品t8(即批号210100881的生品炒制20min的槐角炭样品)适量,按实施例1在待测样品溶液的制备方法制得供试品溶液,分别采用1-ACQUITY CSH C18色谱柱(150mm×2.1mm,1.7μm)、2-Hypersi1 Gold(100×2.1mm,1.9μm)色谱柱依次测定,其他色谱条件同以上实施例1。
检测结果见表34和图19,图中,从下至上依次为ACQUITY CSH C18色谱柱和Hypersi1 Gold色谱柱。结果显示:Hypersi1 Gold色谱柱(100×2.1mm,1.9μm)中保留时间25.000min和31.508min的色谱峰分离度不好,因此使用ACQUITY CSH C18色谱柱(150mm×2.1mm,1.7μm)。
表34不同色谱柱中色谱峰保留时间和分离度
/>
4、柱温的选择
取槐角炭样品t8(即批号210100881的生品炒制20min的槐角炭样品)适量,按实施例1在待测样品溶液的制备方法制得供试品溶液,分别将柱温调节为30℃、35℃和40℃依次测定,其他色谱条件同以上实施例1。
检测结果见表35和图20,图中,从下至上柱温依次为30℃、35℃和40℃。柱温为30℃时对于黄酮类比如芦丁(t
表35不同柱温条件下色谱峰保留时间和分离度
5、流速的选择
取槐角炭样品t8(即批号210100881的生品炒制20min的槐角炭样品)适量,按实施例1在待测样品溶液的制备方法制得供试品溶液,分别将流速调节为0.12mL/min、0.15mL/min和0.18mL/min依次测定,其他色谱条件同以上实施例1。
检测结果见表36和图21,图中,从下至上依次为0.12mL/min、0.15mL/min和0.18mL/min。色谱峰总体差异不大,只是流速为0.18mL/min时小峰消失(t
表36不同流速条件下色谱峰保留时间和分离度
/>
- 一种穿山龙高效液相色谱指纹图谱的建立方法及其标准指纹图谱和应用
- 一种灵芝孢子油的指纹图谱及其构建方法和应用
- 一种假俭草种质的指纹图谱的构建方法及应用
- 一种天然中蜂荔枝蜜和天然意蜂荔枝蜜指纹图谱的构建方法及鉴别应用
- 一种天然中蜂龙眼蜜和天然意蜂龙眼蜜指纹图谱的构建方法及鉴别应用
- 一种中药复方UPLC指纹图谱的建立、指纹图谱及应用
- 一种五倍子药材的UPLC指纹图谱的构建方法、通过该方法构建的指纹图谱及其应用