一种基于水相的固相多肽合成方法
文献发布时间:2024-05-31 01:29:11
技术领域
本发明属于多肽合成领域,具体涉及一种基于水相的固相多肽合成方法。
背景技术
多肽作为一种内源性的生物活性物质,以激素、神经递质、生长因子等多种形式参与调控生命体的系列生理功能。在医药研究领域,多肽类药物越来越引起人们的关注,在抗菌、抗病毒、抗肿瘤、糖尿病等诸多领域发挥着重要作用。
据统计,目前在研或上市的多肽药物主要以化学合成为主,这得益于美国科学家Merr ifield在1963年提出的固相多肽合成(sol id-phase peptide synthesis,SPPS)方法。固相合成技术为多肽合成领域带来了颠覆性的改变,如今广泛地应用于多肽药物的研究和生产中,尤其以基于Fmoc(9-芴甲氧羰基)化学发展起来的Fmoc-SPPS为主导。然而,从绿色化学的角度而言,固相多肽合成仍然存在诸多可改善的地方。由于氨基酸被固定在树脂上,缩合反应的效率降低,因此往往需要加入大大过量的缩合剂和氨基酸砌块(通常在3-4当量)来保证缩合效率,造成了原料的极大浪费。同时,就目前常用的缩合剂如苯并三氮唑-N,N,N',N'-四甲基脲六氟磷酸盐(HATU)、N,N'-二异丙基碳二亚胺(DIC)和1-羟基苯并三唑(HOBT)等而言,氨基酸的侧链大都需要保护基来抑制可能的副反应发生,从原子经济性的角度而言,是相对较低的。另一方面,反应过程,特别是洗涤过程中,涉及到大量有机溶剂如N,N-二甲基甲酰胺(DMF)和二氯甲烷(DCM)的使用,则给工业生产中的废物处理成本和环境保护方面带来了一定的压力。
近些年来,发展绿色固相多肽合成是多肽合成领域的一大研究兴趣,其中的思路之一是将传统固相合成中大量使用的N,N-二甲基甲酰胺(DMF)和二氯甲烷(DCM)等溶剂由成本更低廉、对环境更友好的溶剂进行替代。
2013年,Watson课题组筛选了一系列溶剂作为媒介来进行传统的酰胺缩合反应;研究表明,碳酸二甲酯(DMC)、乙酸乙酯(EA)、2-甲基四氢呋喃(2-MeTHF)可以取得和N,N-二甲基甲酰胺(DMF)和二氯甲烷(DCM)同等的效果。
2017年,North等人发展了基于丙烯碳酸脂这样一种极性非质子溶剂作为媒介的多肽合成方法,在液相或固相两种模式下均可很好地进行,他们以丙烯碳酸脂为溶剂完成九肽---缓激肽的固相合成验证了这一溶剂的可行性。
同年,Al ber icio等人发现对环境更加友好的2-甲基四氢呋喃和环戊基甲醚用于Fmoc-SPPS中的酰胺缩合步骤,γ-戊内酯用于Fmoc脱除步骤可以取得良好的效果。
2018年,诺华团队John Lopez等人筛选了一系列可能用于替代DMF的相对绿色的溶剂,经过一系列系统地验证,证明1-丁基-2-吡咯烷酮(NBP)是最优的替代DMF用于Fmoc-SPPS放大合成的溶剂,并完成了十肽的合成。
此外,有意思的是,Lamaty等人在2009年发展了一种无溶剂的基于Boc保护的N-羧基环内酸酐砌块的酰胺缩合反应,合成了一系列二肽和三肽。
上述对有机溶剂的优化,为固相多肽合成提供了更多较为绿色的溶剂选择,促进了绿色多肽固相合成的发展。然而,对于溶剂而言,水更符合绿色化学指导原则。另一方面,就目前的报道方法来看,主要是基于HATU或DI C等经典的缩合方法进行的,该类方法不仅对缩合剂的用量高,极大地提高了多肽合成中的生产成本;同时,氨基酸侧链官能团需要全保护来抑制副反应的发生。因此,从生产成本、原子经济性等角度而言,目前的基于水相的固相多肽合成仍存在较大改善的空间。
基于以上原因,发展更加符合绿色化学指导原则的固相多肽合成新方法,仍然具有重要的研究意义和应用价值。
《反向固相多肽合成方法新策略及其应用》(朱玥,[D]南华大学,2021)中报道过基于酰基叠氮化学的N端到C端的固相多肽合成方法,但其并非完全基于水相合成。因此,进一步开发基于水相的固相多肽合成方法仍有必要。
发明内容
本发明提供了一种基于水相的固相多肽合成新方法,进一步完善水相反应体系,加入了亲水性可切割连接子,所涉及到的试剂和无保护的氨基酸砌块在水相中进行反应。通过将首个氨基酸(如天冬氨酸和谷氨酸)的侧链共价连接到树脂上,通过酰基叠氮的方法初步实现了水相固相多肽合成。
本发明一方面提供了一种基于水相的固相多肽合成方法,包括以下步骤:
1)将起始氨基酸共价连接到树脂上;
2)将步骤1)所得树脂滤干,水洗三遍,随后置于20%肼解试剂中,振荡30min进行肼解;
3)将步骤2)所得树脂滤干,水洗三遍,随后将树脂溶胀在缓冲体系中,用HCl将体系pH调至3,置于-15℃的环境下,缓慢加入10当量叠氮化试剂,反应30分钟;
4)将步骤3)所得树脂滤干,将氨基酸酯的缓冲液体系调至碱性后,快速加入到树脂中,反应30分钟;
5)重复步骤2)-4);
6)将步骤5)所得树脂滤干,水洗三遍,向所得的树脂中加入由三氟乙酸、水、三异丙基硅烷组成的切割试剂,反应一小时;过滤除去树脂,将滤液吹干,冻干得到目标多肽。
作为优选,所述树脂选自2-Chorotrityl resin,Wang resin,Sieber resin,PEGAresin,Rink AM MBHA resin,Rink amide resin。
作为优选,所述肼解试剂选自水合肼,醋酸肼。
作为优选,所述叠氮化试剂选自亚硝酸钠,亚硝酸叔丁酯。
作为优选,所述叠氮化试剂的浓度为0.2M。
作为优选,所述氨基酸酯选自氨基酸甲酯、氨基酸乙酯,氨基酸苄酯,氨基酸二苯甲-氨甲酰乙酯,优选氨基酸苄酯和氨基酸二苯甲-氨甲酰乙酯;所述氨基酸包括天然氨基酸和非天然氨基酸。
作为优选,所述切割试剂中,三氟乙酸、水、三异丙基硅烷的体积比为95:2.5:2.5。
作为优选,所述缓冲体系为含有6M盐酸胍和0.2M磷酸氢二钠的溶液。
作为优选,所述步骤4)中的碱性的pH范围为8-10。
发明人在现有技术的基础上,进一步在亲水性树脂(如PEGA树脂)上引入亲水性可切割连接子,通过将首个氨基酸的N端固定在树脂上,即可很好地在水相进行氨基酸的组装,实现多肽的水相固相合成。在以往同类的合成案例中,多肽合成只能通过侧链固定在树脂上,局限在合成含有天冬酰胺和谷氨酰胺的多肽。本次发明引入了亲水性可切割连接子,可以将多肽首个氨基酸(不限种类)的N端(不需要通过侧链)固定在树脂上,可以更加灵活、多样性地合成多肽。
附图说明
图1-27依次为实施例1-27所得目标多肽的色谱图;
图28是实施例28的合成步骤示意图;
图29是实施例28所得目标多肽的色谱图。
具体实施方式
以下将通过实施例来详细说明本申请的实施方式,借此对本申请如何应用技术手段来解决技术问题并达成技术功效的实现过程能充分理解并据以实施。
本申请中所用原料、设备,若无特别说明,均为本领域的常用原料、设备,均来自市售产品。本申请中所用方法,若无特别说明,均为本领域的常规方法。
本申请还存在其它多种可实施的技术方案,在此不做一一列举,本申请权利要求中要求保护的技术方案都是可以实施的。
“包含”或“包括”旨在表示组合物(例如介质)和方法包括所列举的要素,但不排除其他要素。当用于定义组合物和方法时,“基本上由……组成”意味着排除对于所述目的的组合具有任何重要意义的其他要素。因此,基本上由本文定义的元素组成的组合物不排除不会实质上影响要求保护的本申请的基本和新颖特征的其他材料或步骤。“由……组成”是指排除其他组成部分的微量元素和实质性的方法步骤。由这些过渡术语中的每一个定义的实施方案都在本申请的范围内。
下面结合附图对本发明做进一步说明。
实施例1:
本实施例所涉及的反应方程式如下所示:
各步骤的试剂及条件:
i)20% N
ii)10eq NaNO
iii)5eq Gly-OBn,6M Gn·HCl in PB,DIPEA,pH=8,30min;
iv)TFA/H
具体步骤为:
将Cbz-Glu-OBn(N-苄氧羰基谷氨酸苄酯)(1mmol)溶解在0.4MHATU(2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯)(1mmol)和0.8M DIPEA(N,N-二异丙基乙胺)(0.5mmol)中,加入到PEGA(聚[丙烯酰基-双(氨丙基)聚乙二醇])树脂(0.25mmol)中持续反应30分钟后将树脂滤干,洗涤三次。将负载Cbz-Glu-OBn的树脂置于20%水合肼溶液中,肼解30min后将树脂滤干,水洗三次;随后将树脂溶胀在6M盐酸胍的磷酸盐缓冲体系中,用HCl调pH至3,置于-15℃的环境下,缓慢加入200mM NaNO
实施例2:
将Cbz-Glu-OBn(1mmol)溶解在0.4M HATU(1mmol)和0.8M DIPEA(0.5mmol)中,加入到PEGA树脂(0.25mmol)中持续反应30分钟后将树脂滤干,洗涤三次。将负载Cbz-Glu-OBn的树脂置于20%水合肼溶液中,肼解30min后将树脂滤干,水洗三次;随后将树脂溶胀在6MGn·HCl 0.2M Na
将实施例1中的Gly-OBn变为表1所列的替换化合物,其余操作相同,得到实施例3-7。实施例3-7中各目标多肽的色谱图如图3-7所示。
表1
实施例8:
试剂及条件:
i)20% N
ii)10eq NaNO
iii)5eq Gly-OBn,6M Gn·HCl in PB,DIPEA,pH=8,30min;
iv)TFA/H
将Cbz-Glu-OBn(1mmol)溶解在0.4M HATU(1mmol)和0.8MDIPEA(0.5mmol)中,加入到PEGA树脂(0.25mmol)中持续反应30分钟后将树脂滤干,洗涤三次。将负载Cbz-Glu-OBn的树脂置于20%水合肼溶液中,肼解30min后将树脂滤干,水洗三次;随后将树脂溶胀在6M盐酸胍的磷酸盐缓冲溶液中,用HCl调pH至3,置于-15℃的环境下,缓慢加入200mM NaNO
将实施例8中的Ala变为其他如表2所示的替换化合物,其余操作相同,得到实施例9-27。实施例9-27中各目标多肽的色谱图如图9-27所示。
表2
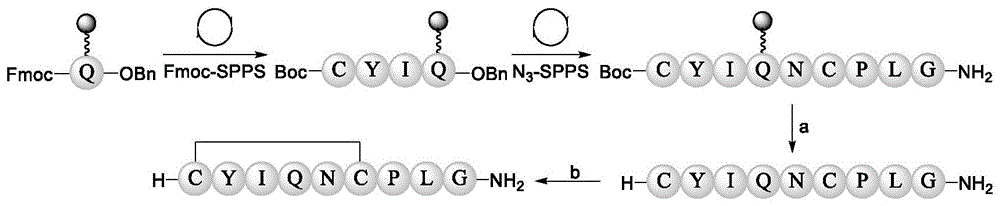
实施例28
催产素(oxytocin),又名“缩宫素”,是一种多肽类激素,具有“产前引产、产中催产、产后催乳、止血”的作用。催产素还具有降低人体内肾上腺酮等压力激素的水平,起到降低血压的作用。
其反应式如图28所示。
Reaction condion.Fmoc-SPPS:
i)20%piperidine in DMF for 15min;
ii)HATU/DIPEA in DMF for 30min.N
iii)20% N
iv)10eq NaNO2,6M Gn·HCl buffer,HCl,pH=3,30min;
iv)5eq Amino benzyl ester,6M Gn·HCl,DIPEA,pH=8,30min.a.Cock tail:TFA/H
如图28所示,催产素的序列为Cys-Tyr-Ile-Gln-Asn-Cys-Pro-Leu-Gly-OH,通过正向和反相结合的方式进行合成。首先,将Fmoc-Glu-OBn的侧链羧基与PEGA树脂(含linker)的氨基缩合进行连接。随后通过经典的Fmoc-SPPS进行氨基酸的组装,具体操作为:1)以20%哌啶将Fmoc脱去,洗净树脂;2)将Fmoc-Xaa-OBn(0.4mmol,4.0equiv),0.4M HATU(1mL,4equiv)和0.8M DIPEA(1mL,8equiv)加入到树脂中,室温反应1.5小时,洗净树脂。重复以上操作,依次连接Ile,Tyr,最后加入Boc-Cys-OH(0.4mmol,4.0equiv)0.4M HATU((2.5mL,4equiv)和0.8M DIPEA(2.5mL,8equiv),加入到树脂中,室温反应1.5小时,洗净树脂,由此完成正相序列的合成。反相固相合成方法的具体操作为:用20%水合肼对氨基酸苄酯进行肼解,肼解30min后将树脂滤干,水洗三次;随后将树脂溶胀在6M Gn·HCl 0.2MNa
本申请说明书中未作详细描述的内容属于本领域技术人员的公知常识。
如在通篇说明书及权利要求当中所提及的“包含”为一开放式用语,故应解释成“包含但不限定于”。“大致”是指在可接收的误差范围内,本领域技术人员能够在一定误差范围内解决所述技术问题,基本达到所述技术效果。
还需要说明的是,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的商品或者系统不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种商品或者系统所固有的要素。在没有更多限制的情况下,由语句“包括一个……”限定的要素,并不排除在包括所述要素的商品或者系统中还存在另外的相同要素。
上述说明示出并描述了本申请的若干优选实施例,但如前所述,应当理解本申请并非局限于本文所披露的形式,不应看作是对其他实施例的排除,而可用于各种其他组合、修改和环境,并能够在本文所述发明构想范围内,通过上述教导或相关领域的技术或知识进行改动。而本领域人员所进行的改动和变化不脱离本申请的精神和范围,则都应在本申请所附权利要求的保护范围。
- 一种以超顺磁纳米颗粒为固相进行多肽合成及同步构建多肽磁纳米探针的方法
- 一种以超顺磁纳米颗粒为固相进行多肽合成及同步构建多肽磁纳米探针的方法