一种无溶剂化合成神经酰胺的方法
文献发布时间:2023-06-19 09:27:35
技术领域
本发明属于有机化学合成技术领域,具体涉及三步反应醇与卤代烃醚化,乙醇胺与环氧开环,以及缩合,利用廉价的原料,除去反应试剂外无利用额外的溶剂来参与反应,发展了一种无溶剂化合成神经酰胺的方法。
背景技术
在传统的有机化合物的合成过程中,有机溶剂是最常见的介质,可以使反应物分散到同相中,降低粘度促进反应的进行,但是溶剂的毒性和试剂的回收对环境有不利影响。近年来绿色合成兴起,产生了许多取代传统有机溶剂的绿色化学方法,其中无溶剂化有机反应就是其中之一,在不加溶剂或加入微量溶剂条件下进行化学反应。
近年来,皮肤研究的成果表明,存在与角质细胞间的脂质对维持健康的皮肤有极大的贡献,其中的主要成分神经酰胺(Ceramide)是保护皮肤的最重要的组分,在表皮角质层中的主要作用有屏障作用,粘合作用,保湿作用,抗衰老作用和抗过敏作用,它可以很好的维持屏障结构的完整性,对于修复受损的保水性具有最大的作用。神经酰胺是毛发脂质的主要成分,可以减小毛皮细胞的粘合力,减少因光等对毛发引起的损伤和装饰毛发表面,神经酰胺是一种新型的生物活性物质,神经酰胺在护肤,护发中的应用具有广阔的开发前景。
CN102351730A公开了一种从魔芋飞粉中制备神经酰胺的方法,是以魔芋飞粉为原料,用体积分数95%的乙醇溶液提取,然后用石油醚萃取,再用硅胶柱层析纯化,最后用中压C18键合硅胶ODS柱层析纯化,得纯度95%以上神经酰胺。但是,该工艺粗提阶段采用体积分数95%的乙醇不适合工业化生产,且后续纯化分离采用硅胶层析柱和中压C18键合硅胶ODS柱层析串联,生产成本较高。
CN102093244A公开了一种神经酰胺的提取方法,是以海葵为原料,用体积分数95%的乙醇溶液提取,然后用乙酸乙酯萃取,再用大孔树脂色谱柱纯化,最后,结晶得到神经酰胺。但是,该工艺粗提阶段采用体积分数95%的乙醇不适合工业化生产,且后续纯化+结晶的方法收率低,一般为0.015~0.03%,生产成本高。
CN102058727A公开了含神经酰胺的茶籽提取物及其制备方法,是用水或低碳醇继续提取。但是,提取液中杂质多,增加了后续纯化处理难度,增加了纯化环节,延长了生产周期。
发明内容
本发明的目的在于发展了一种无溶剂的的反应方法,具体涉及三步反应醇与卤代烃醚化,乙醇胺与环氧开环,以及缩合,利用廉价的原料,除去反应试剂外无利用额外的溶剂来参与反应,实现了很多神经酰胺及类似物的合成。
为了实现上述目的,本发明采用以下技术方案:
一种无溶剂化合成神经酰胺的方法,所述方法包括以下步骤:
1)醇与卤代烃醚化:在长链脂肪醇中加入催化剂,与卤代烃反应得到长链缩水甘油醚;
2)乙醇胺与环氧开环反应:步骤1)得到的长链缩水甘油醚加入至乙醇胺溶液中反应,反应得到中间体;
3)缩合反应:将步骤2)得到的中间体与碱性物质混合,加热后加入脂肪酸甲酯反应,反应完成后产物结晶得到神经酰胺及其其类似物。
在本技术方案中,本发明的反应路线为:
R=C
第一步醚化反应,反应方程式如下:
其中R为C
长链脂肪醇加入催化剂,在20-50℃反应0.5-2h,加入环氧氯丙烷20-80℃反应2-20h后,反应完成先后蒸馏出为反应完的环氧氯丙烷和产物长链缩水甘油醚。
第二步开环反应,反应方程式如下:
其中R为C
第三步反应缩合,反应方程式如下:
将中间体与碱性物质混合,体系加热到20-80℃反应3-20h,反应完成后产物用乙醇结晶得到神经酰胺及其其类似物。
作为优选,步骤1)中,所述长链脂肪醇包括C12-C16的脂肪醇。
作为优选,步骤1)中,所述卤代烃为环氧氯丙烷;所述的催化剂包括NaOH,KOH,K
作为优选,步骤1)中,所述长链脂肪醇和卤代烃的物质的量之比为1:1-1:2,优选的物质的量之比为1:1.5。
作为优选,步骤1)中,所述反应温度为20-80℃,反应时间为2-20h,优选的反应温度为50℃,优选的反应时间为12h。
作为优选,步骤2)中,所述长链缩水甘油醚与乙醇胺的体积比为1:1-1:10,优选的体积比为1:6。
作为优选,步骤2)中,所述反应的温度为20-80℃,反应时间为3-20h,优选的反应温度为60℃,优选的反应时间为8h。
作为优选,步骤3)中,所述中间体与脂肪酸甲酯的物质的量之比为1:1~1:2,优选的中间体与脂肪酸甲酯的物质的量之比为1:1.5。
作为优选,步骤3)中,所述反应的温度为20-80℃,反应时间为3-20h,优选的反应温度为50℃,优选的反应时间为6h。
作为优选,步骤3)中,所述碱性物质包括NaOH,KOH,K
与现有技术相比,本发明的有益效果为:
本发明针对现有的技术特点和不足,发展了一套无需溶剂的反应体系,利用廉价的原料,除去反应试剂外无利用额外的溶剂来参与反应,减少了物料的消耗和浪费,同时减少了后续的后处理流程,节省了时间和人力,绿色环保,高效的完成了神经酰胺及其其类似物的合成。
附图说明
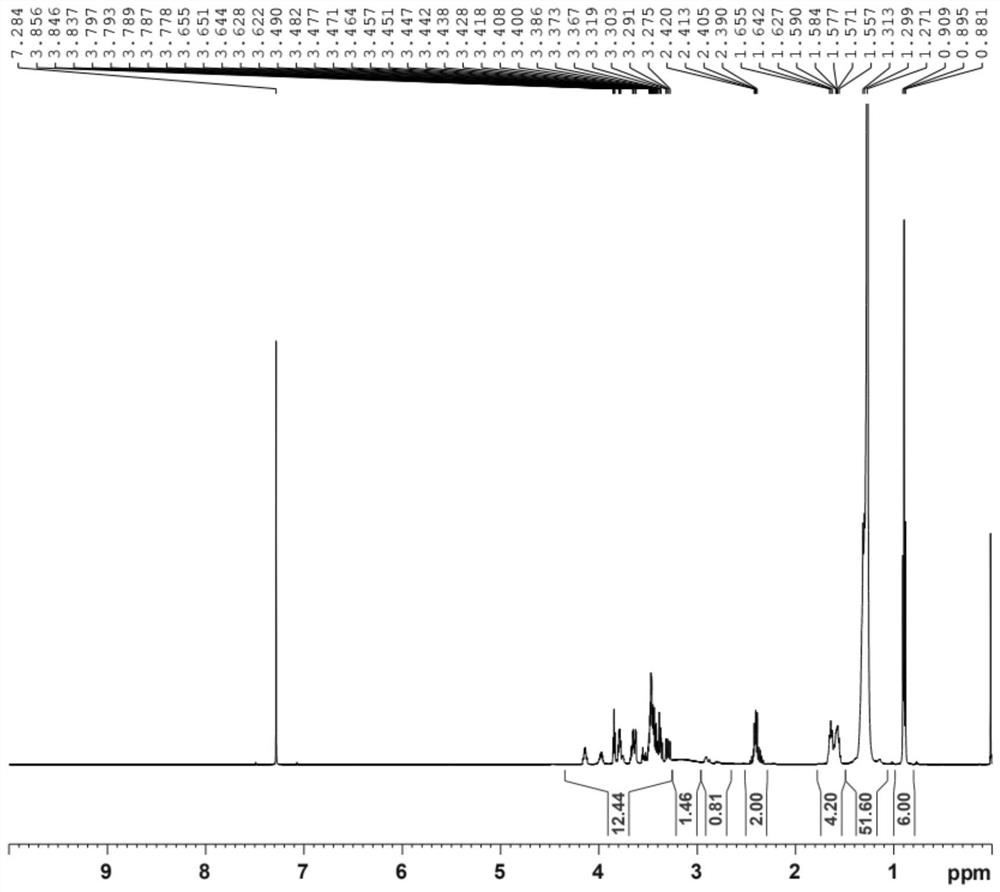
图1是实施例1的氢谱图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
本实施例提供了一种无溶剂化合成神经酰胺的方法,所述方法包括以下步骤:
1)醇与卤代烃醚化:
在四丁基溴化铵(TBAB)640mg存在下,将十六醇48.4g置于圆底烧瓶中,然后将温度升高到50℃溶解,加入甲醇钠16.2g,搅拌20分钟,在剧烈搅拌下的滴加入环氧氯丙烷28g,用薄层色谱(TLC)对反应进行监测),当生成物的量保持不变时,反应混合物降至室温用水淬灭,分液直接用水洗两次,有机相在无水硫酸钠上干燥,然后在真空下蒸发未反应的环氧氯丙烷和缩水甘油醚,最后得到缩水甘油醚液体54g。
2)乙醇胺与环氧开环反应:
在四口烧瓶中加入310g乙醇胺,机械搅拌,50℃下滴加步骤1)得到的十六烷基缩水甘油醚50g,在2h内滴加完毕,在反应2h,反应完全后蒸馏出乙醇胺,产品用乙醇结晶,得到中间体44g,熔点64-65℃。
3)缩合反应:
将步骤2)得到的中间体42g和11.2g的甲醇钾加入四口烧瓶,加热至50℃滴加43g十六酸甲酯1h,继续保温反应1h,反应完全后,然后处理得到的固体用乙醇结晶得到类白色固体神经酰胺68g。
对实施例1得到的类白色固体神经酰胺进行测定,结果见图1与表1。
表1核磁共振谱归属
由表1和图1表明,本实施例制得的类白色固体和预期结构相符,说明本发明通过无溶剂化合成得到了神经酰胺,本发明利用廉价的原料,除去反应试剂外无利用额外的溶剂来参与反应,减少了物料的消耗和浪费,同时减少了后续的后处理流程,节省了时间和人力,绿色环保,高效的完成了神经酰胺及其其类似物的合成。
实施例2
本实施例提供了一种无溶剂化合成神经酰胺类似物的方法,所述方法包括以下步骤:
1)醇与卤代烃醚化:
在四丁基溴化铵(TBAB)640mg存在下,将十四醇42.8g置于圆底烧瓶中,然后将温度升高到50℃溶解,加入甲醇钠16.2g,搅拌20分钟,在剧烈搅拌下的滴加入环氧氯丙烷28g,用薄层色谱(TLC)对反应进行监测),当生成物的量保持不变时,反应混合物降至室温用水淬灭,分液直接用水洗两次,有机相在无水硫酸钠上干燥,然后在真空下蒸发未反应的环氧氯丙烷和缩水甘油醚,最后得到缩水甘油醚液体48g。
2)乙醇胺与环氧开环反应:
在四口烧瓶中加入240g乙醇胺,机械搅拌,50℃下滴加步骤1)得到的十四烷基缩水甘油醚40g,在2h内滴加完毕,在反应2h,反应完全后蒸馏出乙醇胺,产品用乙醇结晶,得到中间体37g,熔点64-65℃。
3)缩合反应:
将步骤2)得到的中间体36g和10g的甲醇钾加入四口烧瓶,加热至50℃滴加37g十四酸甲酯1h,继续保温反应1h,反应完全后,然后处理得到的固体用乙醇结晶得到类白色固体神经酰胺58g。
尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。