C19支架和甾体及其使用和制造方法
文献发布时间:2023-06-19 11:39:06
相关申请的交叉引用
本专利申请要求于2018年9月7日提交的美国临时专利申请号62/728,163,于2019年4月5日提交的美国临时专利申请号62/829,722和于2019年4月12日提交的美国临时专利申请号62/833,291的优先权,将其全部内容完全通过援引并入本文。
联邦资助的研究或开发
本发明是在由美国国立卫生研究院(National Institutes of Health)授予的R01 GM080266下在政府支持下进行的。政府拥有本发明的某些权利。
技术领域
本公开提供了用于获得在一个或多个环稠合处包含季中心的立体定义碳环的简明合成方法,可通过这样的合成方法获得的化合物,以及使用这样的化合物治疗疾病诸如脑肿瘤并且尤其是胶质瘤的方法。
背景技术
甾体和四环萜类化合物(更广泛地)(包括非天然变体)已经对医学和社会产生了变革性的影响,作为口服避孕药,治疗癌症(包括抗血管生成剂)、心力衰竭、炎症、疼痛和创伤性脑损伤等发挥至关重要的作用,并且在此类中天然分子已充当药品发现和开发中的重要化学前体。尽管这有丰富的历史,但仍然存留大量的障碍,这些障碍极大地限制了在可以有效地制备和探索为潜在药物和生物工具/探针的这个广阔的化学空间领域中合成的物质组合物的类型。
目前可获得的合成和半合成这类分子的途径通常是复杂的、低效的和/或完全不能产生通过现代药品开发进展所必需的高度氧化/官能化的目标组合物的有利集合(即,库)。实际上,“甾族”体系或四环萜类化合物启发的物质组合物的有效从头合成仍然是化学中具有挑战性的问题。
在此化学空间领域中,所有超过100种FDA批准的药品都是天然对映异构体(具体参考甾族骨架的C13处的绝对立体化学)——这一事实必定受到制备这样的化合物的方式的影响。实际上,此类中的药剂通常是从天然存在的甾体合成的。与在C13处具有天然绝对立体化学的甾族物质组合物(“nat-甾体”)相比,合成的ent-甾族化合物(由非自然的绝对立体化学定义)具有互补的三维结构,同时提供类似的“似药的”物理特性。因此,合成的ent-甾体是天然产物启发的具有巨大潜在治疗相关性的特权(privileged)支架,并且与它们的天然异构体相比是独特的组合物。(参见,Akwa,Y.,et al.,Proc.Natl.Acad.Sci.U.S.A.,98,14033-14037[2001];Green,P.S.,et al.,Endocrinology,142,400-406[2001];Biellmann,J.F.,Chem.Rev.,103,2019-2033[2003];Covey,D.F.,Steroids,74(7):577-585[2009];以及Petit,G.H.,et al.,Eur.Neuropsychopharmacol.,21,211-215[2011])。然而,由于与制备/获得这样的物质组合物有关的巨大困难,过去一直阻碍对甾体启发的化合物的非天然对映异构体的研究。尽管nat-甾体(即,开始于容易获得的甾体或相关天然产物的那些)的半合成途径难以置信地强大,但这样的制备方法不适合用于生产非天然存在的ent-甾体,因为起始材料具有该类中天然分子固有的镜像主链。总之,ent-甾体是一类重要的特权似药物药品的分子,由于这些分子无法容易从天然来源中可获得,并且现有的化学合成途径低效而且没有足够的灵活性来产生适合用于药品发现和开发的这样的分子的多样性集合,所以这些分子目前无法在生物学和药物研究工作中充分利用。
一种用于有效且立体定向的生产ent-甾体以及其他非天然立体异构体和在四环萜类化合物的广泛类别中的简单独特的物质组合物的实际方法,将使科学家和医师能够更好地开发此药物特权类(包括ent-甾体)中的作为有用的工具和治疗剂的新分子的尚未开发的潜力。实际上,即使在潜在药物的nat-甾体家族中,在很大程度上依赖于半合成(其中合成是从容易可获得的天然产物进行的)的当前的现有技术伴随基于丰富且容易可获得的天然材料的结构(例如,起始材料的不饱和水平、氧合密度和取代度)的显著限制发生。因此,即使采用现有技术的方法,仍然难以探索医用科学的特权化学空间的广大区域。因此,所需要的是合成具有变化的立体化学和取代和/或促进以研究和/或生产规模的随后的分子扰动过程(即,特征四环核的各自环中的官能度操纵)的官能度的合成的nat-和/或ent-甾体的有效且步骤经济的(即简明)、灵活、收敛和对映选择性(enantiospecific)的方法。
甾体和类似甾体的化合物的靶标的一种实例是雌激素受体(ERα和ERβ),两者均可在癌症中发挥作用。
胶质瘤(I-IV级)是致死性原发性脑肿瘤,其占所有恶性脑肿瘤的80%和所有中枢神经系统肿瘤的约30%。低级别胶质瘤(I和II级)随着时间的增长变成高级别胶质瘤(III和IV级)。这些在超过90%的病例中复发,并且具有14个月的中位生存率并且5年的生存率低于10%。它们隐匿多种癌基因和突变的肿瘤抑制基因,它们的改变和表达模式在肿瘤与肿瘤之间变化很大。
然而,大多数胶质瘤均表达升高水平的具体雌激素受体(ER)亚型ERβ。已经鉴定出雌激素受体的两种亚型,ERα和ERβ。尽管它们的显著的序列同源性,但这些受体的分布和功能仍存在值得注意的差异:ERα主要在骨、乳腺、前列腺(基质)、子宫、卵巢(膜细胞)和脑中表达,而ERβ通常存在于卵巢(颗粒细胞)、膀胱、结肠、免疫、心血管和神经系统中。ERα激活负责雌激素的经典功能,包括子宫刺激。ERβ激活可以具有包括在许多癌症类型中的抗增殖作用。的确,ERβ是几种癌症中已确立的肿瘤抑制基因;ERβ的较高的表达与更好的预后相关,并且ERβ激动剂诱导细胞凋亡。虽然如此,尽管ERβ的抑制肿瘤的作用,但17β-雌二醇(一种有效但非选择性的ERα和ERβ激动剂)不用作针对胶质瘤作为长期治疗的治疗剂,因为它可能导致女性生殖系统的癌症和男人中的前列腺癌。因此,选择性ERβ激动剂可以能够抑制癌细胞增殖而不刺激子宫。
然而,雌激素受体的两种亚型几乎相同,只有两个残基在配体结合口袋中不同。因此,在获得亚型选择性配体中存在重大挑战。例如,erteberel是一种非甾族ERβ激动剂,已被研究和/或考虑用于治疗精神分裂症、良性前列腺增生和胶质母细胞瘤。Erteberel具有对ERβ的结合选择性是ERα的14倍(分别为Ki=0.19nM与2.68nM),以及对ERβ激活的功能选择性是ERα的32倍(分别为EC
发明内容
本公开涉及用于通过独特的中间体、合成策略和化学反应生产包括对映异构体定义的(enantiodefined)体系的立体定义的多环的环化合物的方法。更特别地,本公开提供了用于生产天然产物启发的复杂多环四环的合成方法,所述四环包括但不限于具有“C19甾族支架”的化合物。如本文所用,术语“C19甾族支架”不仅包括甾体以及可以被定义为甾族的具有19个碳原子的化合物,而且还包括具有额外的碳原子的化合物,包括但不限于C20、C21、C22、C23、C24、C25、C26、C27、C28、C29、C30或C31化合物。在某些实施方式中,所述方法涉及C9-C10键的立体选择性分子内形成(例如,用弗里德-克拉夫茨(Friedel-Crafts)烷基化、赫克(Heck)反应或自由基环化)。在某些实施方式中,所述方法涉及伴随着1,2-迁移(从甾族C9碳至甾族C10碳)发生的立体定向的氧化脱芳构化。
本公开还涉及甾族化合物,其包括具有天然(“nat-”)绝对立体化学的分子和具有非天然(“ent-”)绝对立体化学的化合物,以及基于这样的骨架的合成变体。在某些实施方式中,所述化合物具有C19甾族支架。在其他实施方式中,具有C19甾族支架的化合物使得能够获得基于或衍生自所述C19支架的另外的化合物,诸如涉及天然萜类化合物(即,甾体、柠檬苦素类化合物、蟾蜍二烯羟酸内酯等)的合成剂的非天然对映体。在某些实施方式中,所述化合物是合成的C9-α和C9-β-烷基以及C10-α和C10-β-烷基甾族四环。
本公开还涉及这样的化合物作为生物活性(例如,治疗性)组分在例如药物组合物中和/或直接作为人和/或动物治疗剂和药物的用途。在某些实施方式中,所述化合物是ERβ选择性激动剂和/或可用于治疗或预防病症、疾患或疾病,诸如癌症(例如,乳腺癌、前列腺癌、卵巢癌、急性髓细胞性白血病和胶质瘤等)或者神经变性。目前,对于原发性脑肿瘤不存在广泛可适用的治愈。本文公开的化合物(例如,化合物100和101)是抑制人胶质母细胞瘤细胞系以及来自患者的原发性胶质瘤肿瘤细胞的生长的雌激素受体β(ERβ)的有效且高度选择性的激动剂。
在一方面,本公开提供了用于通过向有此需要的患者给药本文公开的化合物或其药学上可接受的盐或前药来治疗脑肿瘤的方法。在一些实施方式中,所述化合物是化合物100。在一些实施方式中,所述化合物是化合物101。在一些实施方式中,所述脑肿瘤是胶质瘤。在一些实施方式中,口服给药所述化合物。
本文进一步描述了所述化合物,包括所述化合物的药物组合物,以及用于通过给药所述化合物治疗或预防病症、疾患或疾病的方法。
在以下段落中描述了本发明的这些和其他目的。这些目的不应被视为缩小本发明的范围。
附图说明
为了更好地理解本发明,可以参考以下附图中所示出的实施方式。附图中的部件不一定按比例,并且可以省略相关元件,或者在一些情况下,比例可能已经被放大,以便强调和清楚地说明本文描述的新颖特征。另外,如本领域中已知的,系统部件可以被不同地布置。
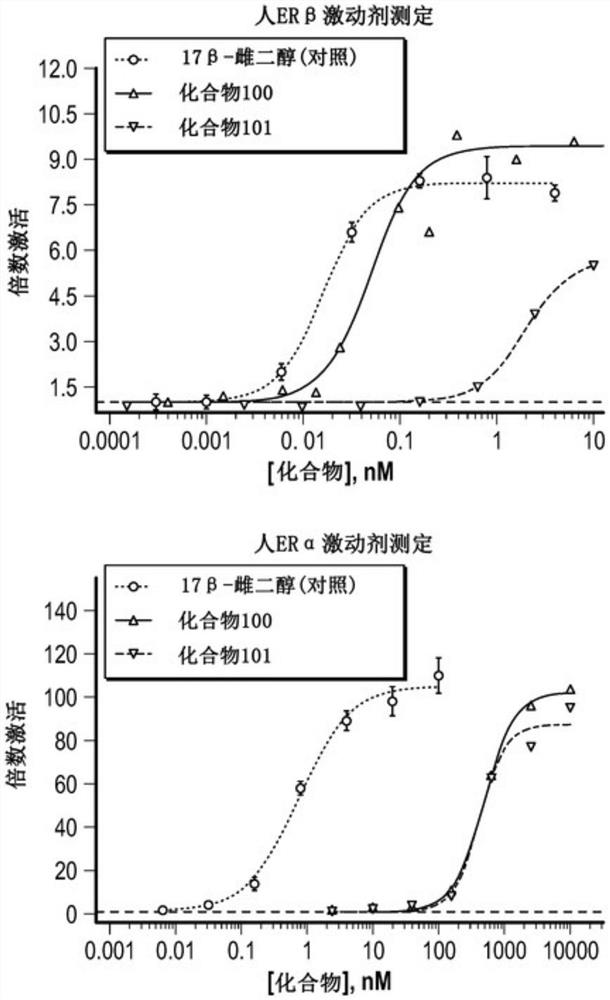
图1是示出化合物100和101的ERβ和ERα激动剂测定的结果的一组线图。
图2是示出在人DU-145前列腺癌细胞中碱性磷酸酶活性的柱状图(在5μM下评估了17β-雌二醇、化合物100和化合物101)。
图3A-3B是描绘以下的线图:(A)用化合物100、化合物101、TMZ和媒介物对照DMSO(n=4)处理的U251和U87细胞的剂量依赖性生长抑制,以及(B)每24小时测定的持续96小时的用化合物100、化合物101、TMZ和媒介物对照DMSO(n=4)处理的U251和U87细胞的活细胞计数。
图3C是用化合物100、化合物101、TMZ和媒介物对照DMSO(n=4)处理后用膜联蛋白V-FITC/7-AAD共染色的U251和U87的流式细胞术分析。
图3D描绘了响应于用化合物100、化合物101、TMZ和媒介物对照DMSO的处理,使用Incucyte对U251和U87细胞进行活细胞追踪的相衬图像。追踪活细胞并对其成像96小时(数据未示出)。比例尺=300μm。图3A-3D中所有数据表示四个独立实验,并且每个实验分析每种处理条件的四个重复。
具体实施方式
此详细描述仅旨在使本领域技术人员熟悉本发明、其原理及其实际应用,使得本领域技术人员可以以其多种形式来适应和应用本发明,因为它们可能最适合于具体用途的要求。此描述及其特定实例仅旨在用于说明的目的。因此,本发明不限于此专利申请中描述的实施方式,并且可以进行不同修改。
在某些方面,本公开涉及包括如下的通用四环甾族(A、B、C、D)环结构的化合物(和制造这样的化合物的方法、包括这样的化合物的组合物以及使用这样的化合物的方法):
更特别地,本公开涉及包括式(I)或式(II)的通用C19甾族核心骨架的化合物(和制造这样的化合物的方法、包括这样的化合物的组合物以及使用这样的化合物的方法),其中,关于这些基本结构的额外的取代旨在在本发明的范围内:
在一方面,本公开提供了组合物,该组合物包括具有包括式(I)的C19甾族核心骨架的化学结构的合成的立体异构体的集合,所述C19甾族核心骨架在碳C9和碳C13中的每一处具有季中心,包括合成的立体异构体的集合中的立体异构体变化;其中,该组合物包括相对于其他C9/C13立体异构体大于约70%,替代地大于约75%,替代地大于约80%,替代地大于约85%,替代地大于约90%,或者替代地大于约95%的单一C9/C13立体异构体。
在某些实施方式中,单一C9/C13立体异构体是天然化合物的非对映异构体。在某些实施方式中,单一C9/C13立体异构体是天然化合物的对映异构体。
在另一方面,本公开提供了组合物,该组合物包括具有包括式(II)的C19甾族核心骨架的化学结构的合成的立体异构体的集合,所述C19甾族核心骨架在碳C10和碳C13中的每一处具有季中心,包括合成的立体异构体的集合中的立体异构体变化;其中,该组合物包括相对于其他C10/C13立体异构体大于约70%,替代地大于约75%,替代地大于约80%,替代地大于约85%,替代地大于约90%,或者替代地大于约95%的单一C10/C13立体异构体。
在某些实施方式中,单一C10/C13立体异构体是天然化合物的非对映异构体。在某些实施方式中,单一C10/C13立体异构体是天然化合物的对映异构体。
在某些实施方式中,单一C10/C13立体异构体具有包括式(A-1)、式(A-2)、式(A-3)或式(A-4)的化学结构:
上面描绘的C19甾族核心骨架尤其包括C20甾族核心骨架,诸如:
其中,C15和C20之间的虚线键表示形成稠合环丙烷的任选的键。
贯穿本公开的编号规则是根据上面编号的结构。
关于通用的四环甾族(A、B、C、D)环结构和通用的C19和C20甾族核心骨架,应当很好地理解,鉴于本文所包含的本公开以及相关技术领域的教导,本公开的化合物、组合物和方法不限于在各种编号的碳原子处的一个或多个任何具体的各自的构成的(R)基团。例如,R基团可以是氢、C
A.定义
如在说明书和所附权利要求中使用的,除非有相反的说明,否则下列术语具有指定的含义:
如本文所用,术语“约”意指大约并且在大多数情况下在所述值的10%内。
如本文所用,术语“脂肪族”包括饱和和不饱和、非芳香族、直链(即,无支链)、支链、无环和环状的(即碳环)烃。在一些实施方式中,脂肪族基团被一个或多个官能团任选地取代。在一些实施方式中,脂肪族的一个或多个单元(例如,亚甲基单元)可以被–O–、–NR
术语“脑肿瘤”包括胶质瘤,诸如星形细胞瘤、胶质母细胞瘤、室管膜瘤(例如间变性室管膜瘤或黏液乳头状型室管膜瘤)、少突胶质细胞瘤(例如间变性少突胶质细胞瘤或间变性少突星形细胞瘤),以及所有归类于WHO1级至4级的胶质瘤。
术语“药学上可接受的”作形容词使用,意指该修饰的名词适合用作人使用的药物产品或用作人使用的药物产品的一部分。术语“前药”是指可以在体内容易地转化(例如代谢)以产生母体化合物的化合物。前药包括但不限于具有在C3(甾体编号)处附连至羟基基团的取代基(诸如酯部分)的化合物,其在体内转化时产生具有酚A环的母体化合物。合适的C3取代基在US2007/0015740A1中鉴定,将其通过援引以其全文并入本文。示例性的酯部分包括但不限于烷基酯(例如,–O-C
术语“治疗(treat)”、“治疗(treating)”和“治疗(treatment)”是指减轻或消除病症、疾患或疾病和/或其伴随症状的方法。
B.合成方法和中间体化合物
在一方面,本公开提供了:(1)立体选择性分子内反应,以形成甾族C9-C10键(例如,通过弗里德-克拉夫茨反应、赫克反应或自由基环化)和/或(2)氧化脱芳构化反应,该反应通过同面1,2-转移终止,在一些情况下还包括质子损失,以递送在环稠合处(在甾族C10碳处)包含季中心的稠合的多环体系。在某些实施方式中,本公开提供了允许优选地从廉价的、可商购的起始材料诸如环氧氯丙烷开始对映选择性构建C19甾族核心骨架的合成方法。在某些实施方式中,本公开提供了合成方法,其使得能够获得涉及天然萜类化合物(即,甾体、柠檬苦素类化合物、蟾蜍二烯羟酸内酯等)的合成剂的非天然对映体。
在一方面,本公开提供了用于制造四环化合物的方法。在某些实施方式中,该方法包括通过分子内形成C9–C10键(例如,通过弗里德-克拉夫茨、赫克或自由基环化反应)将不饱和茚烷(hydrindane)转化为甾族四环的步骤。在一些这样的实施方式中,转化步骤在手性催化剂或试剂存在下进行。在某些实施方式中,该方法包括将甾族四环的取代基从C9转移至C10。
在一方面,本公开提供了用于将不饱和茚烷转化为甾族四环的方法。在某些实施方式中,不饱和茚烷具有对应于以下的结构:
在某些实施方式中,通过赫克反应或自由基环化将不饱和茚烷转化为甾体四环。
在另一方面,本公开提供了用于将硅烷基取代的茚烷转化为甾族四环的方法。以下通用方案表示该方法的具体的实施方式:
在某些实施方式中,M表示有机硅或替代地有机锡或有机锗取代基。在某些实施方式中,M是–Si(R
在某些实施方式中,采用串联酸介导的反应在C11(甾体编号)处诱导原脱硅基化(protodesilylation)并引发位点-和立体选择性分子内弗里德-克拉夫茨烷基化,以生成甾族四环。应当理解,如果不是优选的话,可以容许在C6或C7处的取代,不仅是为了改变产生的分子的生物学特性,而且还为了控制成环事件的立体选择性。
在某些实施方式中,该方法在催化剂或试剂诸如布朗斯特
在某些实施方式中,催化剂或试剂是手性催化剂或试剂。在一些这样的实施方式中,手性催化剂或试剂是1,1’-联-2-萘酚(联萘酚)或联萘酚衍生物。在一些这样的实施方式中,手性催化剂或试剂是(R)-联萘酚或(S)-联萘酚,与包括SnCl
使用一种联萘酚的对映异构体作为手性催化剂或试剂可以使反应偏向于有利于具体的对映异构体或非对映异构体四环。例如,如实施例1-1中,当(R)-联萘酚与茚烷起始材料的具体的对映异构体一起使用时,反应进行以递送具有非常高水平的非对映选择性(≥20:1)的甾体4。
在某些实施方式中,通过利用手性催化剂或试剂的单一对映异构体和/或保护/去除存在于环化底物中的游离羟基基团来调节该方法的立体选择性。
在某些实施方式中,该方法(例如,包括如本文所述的适当的环化步骤)产生具有高选择性(即dr>10:1,并且甚至>20:1)的立体异构体。在一些这样的实施方式中,该方法产生具有至少80%、至少85%、至少90%、至少95%、至少97%或至少99%非对映异构体纯度的组合物。在一些这样的实施方式中,该方法不包括手性纯化步骤(例如,通过结晶或色谱法拆分)。在一些这样的实施方式中,该方法产生具有至少85%的一种非对映异构体和不超过15%的任何其他非对映异构体的组合物。在一些这样的实施方式中,该方法产生具有至少90%的一种非对映异构体和不超过10%的任何其他非对映异构体的组合物。在一些这样的实施方式中,该方法产生具有至少95%的一种非对映异构体和不超过5%的任何其他非对映异构体的组合物。在一些这样的实施方式中,该方法产生具有至少97%的一种非对映异构体和不超过3%的任何其他非对映异构体的组合物。在一些这样的实施方式中,该方法产生具有至少99%的一种非对映异构体和不超过1%的任何其他非对映异构体的组合物。在某些实施方式中,基于所用手性中间体的光学纯度,可以按需要采用手性纯化步骤来获得对映异构体富集的/纯的产物。在某些实施方式中,该方法采用光学纯的起始材料。因此,在某些实施方式中,无需使用手性纯化步骤即可获得期望的纯度。
在某些实施方式中,该方法用对映异构体富集的茚烷(例如,由廉价的手性起始材料如环氧氯丙烷制备)进行,并且产生具有至少80%、至少85%、至少90%、至少95%、至少97%或至少99%的对映异构体纯度的组合物。在一些这样的实施方式中,该方法不包括手性纯化步骤(例如,通过结晶或色谱法拆分)。在一些这样的实施方式中,该方法在不采用手性纯化步骤下产生具有至少85%的一种对映异构体和不超过15%的其他对映异构体的组合物。在一些这样的实施方式中,该方法在不采用手性纯化步骤下产生具有至少90%的一种对映异构体和不超过10%的其他对映异构体的组合物。在一些这样的实施方式中,该方法在不采用手性纯化步骤下产生具有至少95%的一种对映异构体和不超过5%的其他对映异构体的组合物。在一些这样的实施方式中,该方法在不采用手性纯化步骤下产生具有至少97%的一种对映异构体和不超过3%的其他对映异构体的组合物。在一些这样的实施方式中,该方法在不采用手性纯化步骤下产生具有至少99%的一种对映异构体和不超过1%的其他对映异构体的组合物。在某些实施方式中,可以采用手性纯化步骤以去除残留的对映异构体杂质或以拆分外消旋产物(例如,非对映异构体富集的或纯的衍生自外消旋起始材料的产物)。
以下通用方案表示该方法的具体的实施方式,并允许“C19”四环化合物的简明和立体选择性合成:
步骤(i)是在容易可获得的烯炔(a)与任选取代的炔烃(例如,在Ti(Oi-Pr)
步骤(ii)是非对映选择性环化,其可以包括酸介导的原脱硅基化,然后是二烯的第二区域选择性质子化以递送假定的瞬时完全取代的烯丙基阳离子中间体(未描述),以及分子内区域选择性和立体选择性弗里德-克拉夫茨烷基化以建立在该方案中命名为“甾体(a)-anti”和“甾体(a)-syn”的稠合C19四环化合物。
在一方面,本公开提供了用于将甾族四环的取代基从C9转移至C10的方法。以下通用方案表示该方法的具体的实施方式:
在某些实施方式中,采用通过将1,2-烷基从C9转移至C10为特征的氧化重排。在一些这样的实施方式中,获得了A环二烯酮的伴随建立。
在某些实施方式中,该方法在氧化剂存在下进行。在某些实施方式中,氧化剂是羧酸芳基碘(III),诸如二乙酸碘(III)苯(PIDA)或(双(三氟乙酸)碘)苯(PIFA)。在一些这样的实施方式中,氧化剂是二乙酸碘(III)苯(PIDA)。
在一方面,本公开提供了用于制造四环化合物,诸如具有本文所述的通用C19甾族核心骨架的化合物的方法。该方法包括将硅烷基取代的茚烷转化为甾族四环。该方法进一步包括将甾族四环的取代基从位置9的碳原子(C9)转移至位置10的碳原子(C10)。以下通用方案表示该方法的具体的实施方式:
在一方面,本公开包括用于制备尤其是本文公开的甾族四环的中间体化合物。
在一个具体方面,本公开提供了具有对应于以下的结构的中间体化合物:
在本文描述的任何方面或实施方式中,通用方案和中间体中示出的变量可具有以下含义:
R
X是卤素或适合于赫克反应或自由基环化的其他官能度。
在另一个具体方面,本公开提供了具有对应于以下的结构的中间体化合物:
在某些具体的实施方式中,中间体化合物具有对应于以下的结构:
C.示例性化合物
在一方面,本公开提供了化合物、其中间体或盐,其中,该化合物具有对应于式(I-A)或式(II-A)的结构,或者可以通过合成有机化学的本领域技术人员众所周知的方法将其转化为这样的结构:
在某些实施方式中,该化合物具有对应于式(I-A’)或式(II-A’)的结构,或者可以通过合成有机化学的本领域技术人员众所周知的方法将其转化为这样的结构:
在某些实施方式中,通过
在某些实施方式中,通过
在一些这样的实施方式中,该化合物具有对应于式(I-A1)、(I-A2)、(I-A3)或(I-A4)的结构,或者可以通过合成有机化学的本领域技术人员众所周知的方法将其转化为这样的结构:
在一些这样的实施方式中,该化合物具有对应于式(I-A1.1)、(I-A2.1)、(I-A3.1)或(I-A4.1)的结构,或者可以通过合成有机化学的本领域技术人员众所周知的方法将其转化为这样的结构:
在一些这样的实施方式中,该化合物具有对应于式(II-A1)、(II-A2)、(II-A3)或(II-A4)的结构,或者可以通过合成有机化学的本领域技术人员众所周知的方法将其转化为这样的结构:
在一些这样的实施方式中,该化合物具有对应于式(II-A1.1)、(II-A2.1)、(II-A3.1)或(II-A4.1)的结构,或者可以通过合成有机化学的本领域技术人员众所周知的方法将其转化为这样的结构:
在另一方面,本公开提供了化合物、其中间体或盐,其中,该化合物具有对应于式(III-A)的结构,或者可以通过合成有机化学的本领域技术人员众所周知的方法将其转化为这样的结构:
在某些实施方式中,通过
在一些这样的实施方式中,式(III)的化合物是合成的雄甾烷或孕甾烷,及其变体,通过合成有机化学的本领域技术人员众所周知的方法从这样的中间体中可获得。
在某些实施方式中,该化合物具有对应于式(III-B)的结构,或者可以通过合成有机化学的本领域技术人员众所周知的方法将其转化为这样的结构:
式(III-B)的化合物包括碳C8和碳C9之间的双键(即8,9-不饱和),和任选地,(i)碳C14和碳C15之间的双键(即8,9,14,15-不饱和)或(ii)碳C15和碳C16之间的双键,前提是如果碳C14和碳C15之间的键是双键,则R
在某些实施方式中,通过
在一些这样的实施方式中,该化合物具有对应于式(III-B1)或式(III-B2)的结构,或者可以通过合成有机化学的本领域技术人员众所周知的方法将其转化为这样的结构:
在某些实施方式中,该化合物具有对应于式(III-C)的结构,或者可以通过合成有机化学的本领域技术人员众所周知的方法将其转化为这样的结构:
在某些实施方式中,通过
在某些实施方式中,该化合物具有对应于式(III-D)或式(III-E)的结构,或者可以通过合成有机化学的本领域技术人员众所周知的方法将其转化为这样的结构:
式(III-D)或式(III-E)的化合物包括碳C8和碳C9之间的双键(即8,9-不饱和),和任选地,碳C14和碳C15之间的双键(即8,9,14,15-不饱和)。另外,式(III-D)或式(III-E)的化合物任选地包括碳C1和碳C2之间的双键和/或碳C4和碳C5之间的双键。
在某些实施方式中,通过
在一种具体的实施方式中,式(III-D)或式(III-E)的附连至碳C10和碳C13的取代基均在相同的取向上(例如,两个
在某些实施方式中,化合物可以具有对应于式(III-D1)、式(III-D2)、式(III-D3)、式(III-D4)、式(III-E1)、式(III-E2)、式(III-E3)或式(III-E4)的结构,其中,R
在本文所述的任何方面或实施方式中,虚线半圆(例如,表示A环)表示包含5或6个碳环原子的饱和或不饱和碳环或杂环。在一些这样的实施方式中,A环是任选取代的苯。在其他这样的实施方式中,A环是饱和或部分不饱和的任选取代的6-元碳环。在仍其他这样的实施方式中,A环是5-或6-元杂环,诸如噻吩或呋喃。
在本文所述的任何方面或实施方式中,通用结构中示出的变量可具有以下含义:
n为选自由0、1、2、3、4、5、6、7和8组成的组的整数;
m为选自由0、1、2和3组成的组的整数;
R
其中,R
其中,R
其中,R
R
R
R
R
R
R
其中,R
R
R
R
R
R
R
R
其中,任何C
在某些优选的实施方式中,n是1或2。在一些这样的优选的实施方式中,n是1。在一些这样的优选的实施方式中,n是2。
在某些优选的实施方式中,m是0或1。在一些这样的优选的实施方式中,m是0。在一些这样的优选的实施方式中,m是1。
在某些优选的实施方式中,R
在某些优选的实施方式中,n是2并且一个R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
在某些优选的实施方式中,R
应当理解,变量(例如,n、R
在一方面,本公开提供了化合物或其盐或前药,其中,该化合物具有对应于以下化合物之一的结构:
D.使用方法
在至少一个方面,本公开包括用于在需要这样的治疗或预防的受试者中治疗或预防增生性疾病的方法。示例性的增生性疾病包括癌症(即“恶性赘生物”)、良性赘生物、血管生成、炎性疾病和自身免疫性疾病。尤其,可以被治疗或预防的示例性癌症包括乳腺癌、前列腺癌、卵巢癌、急性髓细胞性白血病和胶质瘤。
因此,本公开的一方面包括用于治疗脑肿瘤的方法。该方法包括向有此需要的患者给药治疗有效量的本文所述的化合物(包括但不限于化合物100或101)或其药学上可接受的盐或前药。在一些这样的实施方式中,化合物是选择性的ERβ激动剂。在一些实施方式中,该化合物是化合物100。在一些实施方式中,该化合物是化合物101。在一些实施方式中,口服给药化合物(或其药学上可接受的盐)。在一些实施方式中,脑肿瘤是胶质瘤,诸如胶质母细胞瘤。在一些这样的实施方式中,脑肿瘤选自由以下组成的组:星形细胞瘤、胶质母细胞瘤、室管膜瘤(例如,间变性室管膜瘤或黏液乳头状型室管膜瘤)和少突胶质细胞瘤(例如,间变性少突胶质细胞瘤或间变性少突星形细胞瘤)。在至少一个方面,本公开包括本文公开的化合物或其药学上可接受的盐或前药,用于在用于治疗癌症尤其是脑肿瘤的方法中。在某些实施方式中,该化合物是化合物100。在某些实施方式中,该化合物是化合物101。在某些实施方式中,两种化合物(100和101)可以与彼此或与其他药物活性剂组合使用。
本公开的另一方面包括用于在需要这样的治疗或预防的受试者中治疗或预防精神分裂症的方法。
本公开的仍另一方面包括用于在需要这样的治疗或预防的受试者中治疗或预防神经变性的方法。在一些这样的实施方式中,人受试者患有神经系统变性疾病或有神经系统变性疾病的风险,诸如脊髓损伤(SCI)、多发性硬化(MS)、帕金森病(PD)和阿尔茨海默病(AD)。
本公开的还另一方面包括用于在需要这样的治疗或预防的受试者中治疗或预防神经性疼痛的方法。
本公开的一个方面包括用于在需要这样的治疗或预防的受试者中治疗或预防至少部分受ERβ介导或影响的疾病或病症的方法。
本公开的另一方面包括用于在需要这样的治疗或预防的受试者中通过选择性调节ERβ来治疗或预防可治疗或可预防的疾病或病症的方法。
在某些实施方式中,对于任何上述方面,受试者是哺乳动物。在一些这样的实施方式中,哺乳动物是人。
在某些实施方式中,对于任一上述方面,方法包括向受试者给药治疗有效量的本文所述的化合物(包括但不限于,化合物100或101)或其药学上可接受的盐或前药,作为单一剂或者与另一化学治疗化合物组合。在一些这样的实施方式中,方法包括向受试者给药治疗有效量的化合物100或其药学上可接受的盐或前药,优选化合物100。在其他这样的实施方式中,方法包括向受试者给药治疗有效量的化合物101或其药学上可接受的盐或前药,优选化合物101。在某些实施方式中,口服给药化合物。
化合物或盐的优选总日剂量(以单剂量或分开剂量给药)通常为约0.001至约100mg/kg、更优选约0.001至约30mg/kg以及甚至更优选约0.01至约10mg/kg(即mg的化合物或盐/kg体重)。在某些实施方式中,剂量单位组合物包含构成日剂量的这样的量或其多个亚剂量(submultiples)。在许多情况下,化合物或盐的给药将重复多次。在某些实施方式中,如果需要,每天通常可以使用多剂量来增加总日剂量。
影响优选剂量方案的因素包括患者的类型、年龄、体重、性别、饮食和病症;病理病症的严重性;给药途径;药理考量,诸如使用的具体化合物或盐的活性、功效、药物代谢动力学和毒理学特征;是否利用了药品递送系统;以及化合物或盐是否作为药品组合的一部分给药。因此,实际采用的剂量方案可以变化很大,并且因此可以从上述优选的剂量方案中得出。
化合物的活性可以使用各种已知方法来测定。例如,可以使用各种已知方法来测定化合物的抗增殖活性,包括使用癌细胞系诸如U251和/或U87(人胶质母细胞瘤来源的细胞系)、DU-145(前列腺癌细胞系)、MDA-MB-231(人乳腺腺癌)、AsPC-1(人胰腺腺癌腹水转移)和A549(肺癌)的体外和体内抗增殖测定。
E.组合物
在至少一个方面,本公开包括含有本文所述化合物(包括但不限于化合物100或101)或其药学上可接受的盐或前药的组合物。在某些实施方式中,该组合物包括一种或多种常规的药学上可接受的赋形剂。
在至少一个方面,本公开包括含有本文所述对映异构体化合物的组合物。在某些实施方式中,组合物是对映异构体纯的或富集的。例如,组合物可以包括至少85%的一种对映异构体和不超过15%的其他对映异构体;替代地,至少90%的一种对映异构体和不超过10%的其他对映异构体;替代地,至少95%的一种对映异构体和不超过5%的其他对映异构体;替代地,至少97%的一种对映异构体和不超过3%的其他对映异构体;或替代地,至少99%的一种对映异构体和不超过1%的其他对映异构体。在某些实施方式中,组合物基本上不含对映异构体杂质。在一些这样的实施方式中,组合物不含任何可检测量的对映异构体杂质。
本文公开的药物组合物包括本文公开的化合物或其药学上可接受的盐或前药,优选化合物100或化合物101。在一些实施方式中,药物组合物是口服剂型,优选固体口服剂型(例如片剂)。在一些这样的实施方式中,固体口服剂型可以包括药学上可接受的赋形剂,诸如起粘合剂、助流剂、润滑剂和填充剂作用的赋形剂。因此,包括本文公开的化合物或其药学上可接受的盐的固体口服剂型进一步任选地包括一种或多种常规药学上可接受的赋形剂。
在一些实施方式中,化合物与化学治疗剂共同给药。在一些这样的实施方式中,化学治疗剂是用于治疗脑肿瘤的剂,诸如替莫唑胺(temozolomide)(TMZ)。
在一些实施方式中,化学治疗剂和本公开的化合物以基本上同时的方式(例如,或在彼此的约5min内),以顺序的方式或两者向患者共同给药。例如,预期的是,这样的联合治疗可以包括在给药其他之间多次给药一种治疗剂。在每种剂的给药之间的时间段的范围可以是几秒钟(或更短)至几个小时或几天,并且将取决于例如每种组合物和活性成分的特性(例如效力、溶解度、生物利用度、半衰期和动力学特征)以及患者的病症。在一些实施方式中,化学治疗剂和本公开的化合物以单独的药物组合物给药。在一些实施方式中,化学治疗剂和本公开的化合物以同一药物组合物给药。
在至少一个方面,本公开包括用于治疗脑肿瘤的药物组合物,该组合物包括本文公开的化合物或其药学上可接受的盐以及药学上可接受的赋形剂。在某些实施方式中,该化合物是化合物100。在某些实施方式中,该化合物是化合物101。
对于本领域技术人员而言将显而易见的是,在不脱离本发明或本文公开的实施方式的范围的情况下,可以使用合适的等同物对本文所述的本发明的组合物和方法进行其他合适的修改和改编。
通过参考以下实施例,将更好地理解本文所述的化合物、组合物和方法,包括这些实施例作为对本发明的范围的说明并且不是对本发明的范围的限制。
F.实施例
材料和方法.
除非另有说明,否则所有反应均在氮气气氛下用干燥溶剂在火焰干燥的玻璃器皿中进行。除非另有指明,否则所有试剂和起始材料均购自商业来源并且按供应原样使用。
通过Glass Contour溶剂纯化系统获得无水二乙醚(Et
中间体的制备.
烯炔1的合成
烯炔1:在–78℃下在N
1的光谱数据:
茚烷2的合成
茚烷2:在–78℃下在N
2的光谱数据:
实施例1-1.
化合物100——(9S,13R,16S)-9,13-二甲基-7,9,11,12,13,15,16,17-八氢-6H-环戊二烯并[a]菲-3,16-二醇
化合物101——(9R,13S,16R)-9,13-二甲基-7,9,11,12,13,15,16,17-八氢-6H-环戊二烯并[a]菲-3,16-二醇
化合物100和101是对映异构体——彼此镜像。
化合物100是由ent-烯炔1的对映异构体以3个步骤制备的,ent-烯炔1的对映异构体从环氧氯丙烷以仅3个步骤可获得。
由ent-烯炔1制备化合物100:
第一步骤是如上通常所述的钛介导的成环反应,以提供立体定义的ent-茚烷2。
在第二步骤中,可使ent-茚烷2(其为硅烷基取代的二烯)与(R)-联萘酚或(S)-联萘酚和SnCl
ent-茚烷2的环化的匹配的双不对称反应:在–78℃下在N
ent-茚烷2的环化的错配的双不对称反应:在–78℃下在N
甾体3的光谱数据:
在第三步骤中,使用DIBAL进行脱甲基化以提供化合物100。
化合物101以相同的方式制备,除开始于烯炔1(也容易可从环氧氯丙烷获得)之外。
由烯炔1制备化合物101:
第一步骤是如上通常所述的钛介导的成环反应,以提供立体定义的茚烷2。
在第二步骤中,使茚烷2(其为硅烷基取代的二烯)与(R)-或(S)-联萘酚和SnCl
茚烷2的环化的错配的双不对称反应:
在–78℃下在N
5的光谱数据:
茚烷2的环化的匹配的双不对称反应:
在–78℃下在N
6的光谱数据
当使用(R)-联萘酚诱导茚烷2的环化时,非对映异构体四环的等摩尔比率导致~60%的产率(dr~1:1),但是当采用(S)-联萘酚时,该反应进行以递送具有非常高水平的非对映选择性(dr≥20:1)的非对映异构体6。
在第三步骤中,使用DIBAL进行脱甲基化以提供化合物101。
在室温下在N
化合物100和101在C9处具有季中心,并且在C8与C14之间具有不饱和。化合物101具有“非天然”的绝对立体化学(相对于雌二醇,是指在C13处的绝对立体化学)。
评价化合物100和101对雌激素受体(ERα和ERβ两者)的功能活性。将17β-雌二醇用作对照,因为它被认为是ERα和ERβ两者的有效激动剂。
如图1中说明,发现对映异构体四环化合物100和化合物101是ERβ的令人印象深刻的激动剂。
化合物100是ERβ的独特选择性和高效的完全激动剂,具有0.05nM的EC
化合物101是ERβ的部分激动剂,具有1.9nM的EC
实施例1-2.
化合物102–(9R,13S,16R)-3-甲氧基-4,9,13-三甲基-7,9,11,12,13,15,16,17-八氢-6H-环戊二烯并[a]菲-16-醇
化合物103–(9S,13S,16R)-3-甲氧基-4,9,13-三甲基-7,9,11,12,13,15,16,17-八氢-6H-环戊二烯并[a]菲-16-醇
化合物102和103如上制备,除开始于烯炔7之外。
如下示出,由环氧化物7a和炔烃7b合成烯炔7:
烯炔7:在–78℃下在N
茚烷8:在-78℃下在N
实施例1-3.
化合物104–(9R,13S,16R)-3,16-二甲氧基-4,9,13-三甲基-7,9,11,12,13,15,16,17-八氢-6H-环戊二烯并[a]菲
按照本文概述的程序,将茚烷9(C16甲基醚)以67%的产率平稳转化为立体定义的四环(化合物104),并且具有极度水平的立体控制(ds≥20:1),表明在酸介导的环化反应中,立体控制不需要C16(D-环)醇。
实施例1-4.
合成A环芳香族四环的通用程序:金属环状化合物介导的成环交叉偶联,然后为双不对称布朗斯特酸介导的弗里德-克拉夫茨环化和脱甲基化。
同时,将n-BuLi(2.5M,在己烷中,1.0当量)添加至包含烯炔S10(1.0g,1.0当量)在无水甲苯(0.3M)中的溶液的预冷却的单独的烧瓶(在–78℃干冰/丙酮浴中冷却)中。将所得的醇锂溶液(在室温下)经由插管转移至由上述程序(“TMS-炔烃的活化”)产生的溶液中。将所得混合物搅拌,逐渐温热至室温过夜(大约15hr)。然后通过添加苯甲醛(3.3当量),然后引入饱和NaHCO
按照上述通用程序合成以下示例性化合物105–162:
实施例2-1.
化合物200–(10S,13S,16R)-16-羟基-10,13-二甲基-6,7,10,11,12,13,16,17-八氢-3H-环戊二烯并[a]菲-3-酮
从甾体6开始,借助于具有立体定向的烷基转移和质子的区域选择性损失的氧化脱芳构化来制备化合物200。
化合物200可以是用于合成非天然或天然萜类化合物或萜类化合物启发的(合成的新颖的物质组合物)剂的通用中间体。
本公开的一方面是将四环像甾体6发展为具有C10季中心的甾族产物。发现通过用二异丁基氢化铝(DIBAL)处理进行脱保护,并且使酚类产物暴露于氧化剂[在该实施例中,二乙酸碘(III)苯(PIDA)]导致C9甲基取代基立体定向迁移至C10。这种氧化迁移过程大概生成高度稳定的叔和烯丙基碳正离子中间体,然后通过质子的区域选择性损失将其转化为产物。这种类型的氧化重排是新颖的,因为众所周知,PIDA介导的氧化脱芳构化化学过程对甲基基团的迁移存在问题/无效。
将甾体6转化为甾族四环化合物200的具有立体定向的1,2-迁移和区域选择性质子损失的氧化脱芳构化是从根本上对此类靶标进行实验室构建的新方法,并将此过程确立为用于合成在C10处包含角形(angular)季中心的稠合碳环。
实施例2-2.
由化合物101替代合成化合物200。
在0℃下在N
化合物200的光谱数据:
实施例2-3.
化合物201–(10S,13R,16S)-16-羟基-10,13-二甲基-6,7,10,11,12,13,16,17-八氢-3H-环戊二烯并[a]菲-3-酮
如上制备化合物201,除从甾体3开始之外。
用二异丁基氢化铝将对映异构体定义的甾体3还原脱甲基化,递送高产率的相应的苯酚。然后发现通过用PIDA处理,氧化重排以高度受控的方式进行。如上,从C9至C10的立体定向的1,2-甲基转移被认为递送高度稳定的叔烯丙基碳正离子中间体,其通过从C15选择性损失质子而终止,从而导致产生立体定义的二烯酮化合物201——在C10和C13处安置季中心的四环产物。
实施例2-4.
化合物202–(10R,13S,16R)-16-羟基-4,10,13-三甲基-6,7,10,11,12,13,16,17-八氢-3H-环戊二烯并[a]菲-3-酮。
如上制备化合物202,除从化合物102开始之外。
在室温下在N
202的光谱数据:
实施例3.
在甾体四环骨架内的战略性位置处引入不饱和单元具有挑战性。应当理解,尽管在化学和生物学文献中存在具有这样的不饱和的结构的实例,但是由于适用于制备这样的剂的现有技术,对此领域的探索仍然相当繁琐。
本文描述的合成方法提供了获得在甾体四环(例如,C19)骨架内的战略性位置处带有不饱和单元的分子,与饱和变体相比,提供了三维结构的微扰,而且具有不同的溶解度特征。
此实施例表明将额外的不饱和引入示例性的孕甾烷骨架(孕酮)可对预测的水性溶解度具有明显的影响(如由本文报道的CLogP值所指示,其由ChemDraw Professional计算),并且有利于在该类中设计更水溶性的分子。
值得注意的是,剂的额外不饱和诱导分子整体形状的细微变化,这可能赋予不同的物理特性,诸如分子识别。这样的细微变化可利用于旨在鉴定表现出独特效力和选择性特征的合成化合物的项目,这些化合物作为以下的调节剂:医学相关的靶标,诸如GABAA受体(GABAAR)、孕甾烷X受体(PXR)、雄激素受体(AR)、法尼醇(farnesoid)X受体(FXR)、肝X受体(LXR)和雌激素受体,以及其他医学相关的生物学靶标(即,其他激素核受体或激酶靶标,如CDK8和CDK19)。
对比化合物300(孕酮)。测定CLogP为4.0(使用ChemDraw估算)。
化合物301(8,9-不饱和)。测定化合物301的CLogP为3.5(使用ChemDraw估算),并且因此,化合物301相对于孕酮可能将表现出增加的水溶解度。
化合物302(8,9,14,15-多不饱和)。测定CLogP为3.1(使用ChemDraw估算),并且因此,化合物302相对于孕酮可能将表现出增加的水溶解度。
在甾体四环骨架内的战略性位置处引入不饱和单元对预测的水性溶解度具有明显的影响。相对于孕酮,化合物301和化合物302两者在三维结构上均表现出较小但可能可利用的变化。ent-孕酮的结构的类似的扰动是明显的,该ent-孕酮为已被描述为与创伤性脑损伤和其他病症的治疗的医学相关性的分子。
实施例4.
此实施例为获得药学上相关的甾族化合物提供了化学进展。在步骤(a)中将甾体6用氧化性裂解剂处理以产生二酮B。使二酮B经受脱水和闭环反应以产生四环二烯酮D,该四环二烯酮D是用于生产更大医学价值的另外独特的化合物的关键中间体。反应的整体顺序限定了在形式上使甾体6的C13的立体化学反转的化学手段,同时在C15-C17处引入医学相关的官能度(对随后的官能化有价值的烯酮)。
二酮B:在–78℃下,向甾体6(0.25g,0.84mmol,1.0当量)在5:1的MeOH(50mL)和二氯甲烷(10mL)的混合物中的搅拌溶液中引入O
四环二烯酮D:向二酮B(0.36g,1.1mmol,1.0当量)在36mL PhMe中的搅拌溶液中添加p-TsOH(0.27g,1.6mmol,1.5当量)。将所得混合物在60℃油浴中温热,搅拌45min,并且然后冷却至室温。然后将反应混合物用40mL二氯甲烷以及然后50mL水稀释。分离有机层,并且用3×30mL的二氯甲烷萃取水性层。将合并的有机层经Na
值得注意的是,四环二烯酮D已示出是通过化学顺序制备的,该化学顺序涉及包括C13的形式反转和C17酮的设置/建立的概念上新颖的重排。
从关键的四环二烯酮D,可以制备许多另外的化合物。
例如,化合物400,一种新颖的孕甾烷,用本文描述的化学技术以简明且不对称的方式独特地可获得。
化合物400——(10S,13R,17S)-17-((S)-1-羟基乙基)-10,13-二甲基-6,7,10,11,12,13,16,17-八氢-3H-环戊二烯并[a]菲-3-酮
如下示出,从四环二烯酮D开始制备化合物400。
四环酮E:向威尔金森(Wilkinson)催化剂(45mg,0.049mmol,0.1当量)和D(145mg,0.49mmol,1当量)在干燥的苯(5mL)中的搅拌溶液中鼓入N
四环苯酚H:在N
H的光谱数据:
化合物400(孕甾烷I):将四环苯酚H(50mg,0.16mmol,1当量)溶解在1.6mL HFIP中,在N
用于制备化合物400的通用方案总结如下:
同样地,化合物401,一种雄甾烷,用本文描述的化学技术以简明且不对称的方式独特地可获得。
化合物401——(10S,13S,17S)-17-羟基-10,13-二甲基-6,7,10,11,12,13,16,17-八氢-3H-环戊二烯并[a]菲-3-酮
如下示出,由四环二烯酮D制备化合物401。尤其,将四环二烯酮D在步骤(c)(1.威尔金森催化剂,H
该实施例建立在本文先前描述的化学过程上,尤其是在实施例1中,通过提供开环、脱水和闭环的新颖顺序来改变C13出的立体化学以及在C17处设置酮官能团。
本文描述的过程和中间体,尤其是四环二烯酮D,作为通过简单的反应顺序获得多不饱和孕甾烷和雄甾烷骨架两者的手段是有价值的。
总之,本文描述的技术极大地扩展了可以容易地制备的四环萜类化合物骨架的类型。由于用此技术可获得的化合物处于药物特权空间,这样的化合物可用作活性药物剂(API)或用于API的合成/生产。
实施例5.
此实施例描述了错配的双不对称弗里德-克拉夫茨环化中增强水平的立体选择性,其对于匹配的双不对称反应也有效。
用不具有游离羟基基团的底物,已经观察到错配的双不对称弗里德-克拉夫茨环化的最高选择性。据信,在这种双不对称环化中,带有适当酸性官能团的底物可能是有问题的,因为事实是底物本身可以充当阳离子环化的布朗斯特酸的来源。在下面示出的实施例中,茚烷底物14在甾族C16位置处具有甲醚,而不是游离羟基基团。
化合物501的分析数据:
化合物500的分析数据:
实施例6.
在缺乏ERβ的小鼠中,前列腺上皮增生是明显的,这表明ERβ可能在限制前列腺的增殖中发挥作用。这些数据表明ERβ的药理学激活可能在前列腺癌的治疗中是有用的。DU-145前列腺癌细胞系仅表达ERβ,并且在不存在由ERα驱动的任何潜在混杂信号下,可以用作检查ERβ激活的模型。碱性磷酸酶(ALP)表达以ER依赖的方式被诱导,并且已被用来表征合成的ER激动剂和拮抗剂的活性。
为了检查化合物100和化合物101在表达内源性ERβ的细胞系情况下的激活ERβ的能力,用17β-雌二醇、化合物100或化合物101处理DU-145细胞24小时,并且检查源自细胞的ALP活性。如图2所示,17β-雌二醇如预期的有效地诱导ALP活性。此外,化合物100和化合物101两者均有效诱导ALP至与17β-雌二醇相当的水平。这些数据与起ERβ激动剂作用的化合物100和化合物101一致。
实施例7.为了测定化合物100和化合物101的生物学作用,将人胶质母细胞瘤来源的细胞系U251和U87用不同浓度的化合物100、化合物101、替莫唑胺(temazolomide)(TMZ;护理标准的化学治疗剂)或媒介物毒性对照二甲亚砜(DMSO)进行处理,以与达到化合物100和化合物101的所有测试浓度所使用的最终体积相同的最终体积使用。值得注意的是,24小时后,化合物100和化合物101以相比于TMZ的50分之一的浓度显示出对胶质瘤细胞生长的抑制(IC
为了阐明该细胞死亡的机理,通过FACS分析分析了用化合物100、化合物101、TMZ或DMSO对照处理24小时的胶质母细胞瘤细胞的早期和晚期细胞凋亡标志物(膜联蛋白V和7-氨基放线菌素D)。这些实验揭示了,用化合物100或化合物101处理的U251和U87细胞的细胞凋亡诱导是相比于用TMZ或者DMSO对照处理的细胞的7倍(图3C)。此外,在五天的时间段内,使用Incucyte活细胞成像系统追踪了用化合物100和化合物101处理的U251和U87细胞中的形态变化。用化合物100和化合物101处理U251和U87后,观察到粘附和细胞完整性的损失增加,这与先前观察到的生存力降低和细胞死亡增加有关;对于人神经干细胞以及人星形胶质细胞,未看到类似的行为(图3D)。
24小时后,化合物100和化合物101各自以相比于TMZ的50分之一的浓度显示出对胶质瘤细胞生长的抑制(IC
本文呈现的数据示出,化合物100和化合物101各自降低胶质母细胞瘤细胞的细胞生存力,降低存活率并选择性诱导细胞凋亡。与17β-雌二醇相比,化合物100和化合物101对于ERβ的选择性激动具有独特的特征(图1)。而且,用化合物100和化合物101的初始体内毒性研究表明,(i)两种化合物均无毒,并且(ii)每种化合物均降低已建立肿瘤的生长。
数据支持使用化合物100和化合物101作为对所有当前可用的治疗方式均具有抵抗力的抗胶质瘤的治疗剂。
应当理解,前述详细描述和所附实施例仅是说明性的,并且不应被视为对本发明范围的限制,本发明的范围仅由所附权利要求及其等同物来限定。所公开的实施方式的各种改变和修改对于本领域技术人员将是显而易见的。在不脱离本发明的精神和范围的情况下,可以进行这样的改变和修改,包括但不限于涉及化学结构、取代基、衍生物、中间体、合成、制剂、或方法的那些,或本发明的使用的这样的改变和修改的任何组合。
上面引用的所有参考文献(专利和非专利)都通过援引并入到本专利申请中。这些参考文献的讨论仅旨在总结其作者做出的主张。不承认任何参考文献(或任何参考文献的一部分)是相关的现有技术(或完全是现有技术)。申请人保留质疑所引用参考文献的准确性和相关性的权利。
- C19支架和甾体及其使用和制造方法
- HDD用玻璃基板的制造方法、HDD用玻璃基板、HDD用磁记录介质以及HDD用玻璃基板的制造方法中所使用的清洗支架